
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free synthesis of 5-(aryl/alkyl)amino-1,2,4-triazines and α-arylamino-2,2′-bipyridines with greener prospects†‡
Dmitry S. Kopchukab,
Nikolay V. Chepchugova,
Igor S. Kovaleva,
Sougata Santra *a,
Matiur Rahmana,
Kousik Giric,
Grigory V. Zyryanovab,
Adinath Majee*d,
Valery N. Charushinab and
Oleg N. Chupakhinab
*a,
Matiur Rahmana,
Kousik Giric,
Grigory V. Zyryanovab,
Adinath Majee*d,
Valery N. Charushinab and
Oleg N. Chupakhinab
aDepartment of Organic and Biomolecular Chemistry, Chemical Engineering Institute, Ural Federal University, 19 Mira Street, 620002 Yekaterinburg, Russian Federation. E-mail: sougatasantra85@gmail.com; ssantra@urfu.ru
bI. Ya. Postovskiy Institute of Organic Synthesis, Ural Division of the Russian Academy of Sciences, 22 S. Kovalevskoy Street, 620219 Yekaterinburg, Russian Federation
cCentre for Computational Sciences, School of Basic and Applied Sciences, Central University of Punjab, City Campus, Mansa Road, Bathinda-151001, India
dDepartment of Chemistry, Visva-Bharati (A Central University), Santiniketan, 731235, India. E-mail: adinath.majee@visva-bharati.ac.in
First published on 31st January 2017
Abstract
A green and highly efficient method has been developed for the synthesis of 5-(aryl/alkyl)amino-1,2,4-triazines and α-arylamino-2,2′-bipyridines according to the principles of atom economy. It has been performed by two consecutive solvent-free reaction pathways: the ipso-substitution of a cyano-group in 5-cyano-1,2,4-triazines and the aza-Diels-Alder reaction of the resulting 5-arylamino-1,2,4-triazines with 1-morpholinocyclopentene used as a dienophile. Solvent and catalyst-free conditions, operational simplicity, the compatibility with various functional groups, nonchromatographic purification technique, and high yields are the notable advantages of this procedure. The present methodology possesses a low E-factor.
Introduction
In recent decades development of environmentally benign and clean synthetic techniques has become the goal of organic synthesis.1–4 The concept of sustainable development now plays an important role in deciding strategies for chemical synthesis and, consequently, the search for efficient, economic and ecofriendly synthetic methods has become a major concern. It has been well accepted that solvents are the main reason for an insufficient E-factor, especially in the synthesis of fine chemicals and pharmaceutical industries.5–7 Now various syntheses without using any solvent after reaction, for extraction, chromatography or recrystallization, have been developed because they yield pure products in the absence of any auxiliaries.8,9 In order to modernize classical procedures, making them more clean, safe and easy to perform, solvent-free methods are of fundamental and growing interest for the reason of economy and the pollution prevention. Based on the current working practice with special emphasize on Green Chemistry, it is now often claimed that “the best solvent is no solvent”. The crucial advantages of solvent-free reactions are cost saving, by easy work-up, purification, and remarkable rate acceleration with less energy consumption. As a result, it has become imperative both in academia and industry to design catalyst and solvent-free organic reactions popularly known as neat reaction.10,11 Owing these importances, a library of organic reactions has been carried out under solvent and catalyst-free conditions in the last decade for the synthesis of various biologically active compounds.121,2,4-Triazines having the aromatic amine residue at C5 position are of interest because it act as enzyme inhibitors13–15 and receptor gene expression promoters.16 Some Zn-complexes of chiral α,α-bis-(aminobinaphthyl)-2,2′-bipyridines promote asymmetric Mukaiyama aldol reactions.17–19 Few other α-aminoaryl-substituted 2,2′-bipyridines are of interest due to their prospective photophysical properties.20
The traditional methods for the synthesis of 1,2,4-triazines involve the ipso-substitution of the chlorine atom with an amine residue.21–23 Other approaches are the substitution of trichloromethyl-,24 thiomethyl25 group or the fluorine atom26 and the direct cyclocondensation reactions.27 Synthesis of α-arylamino-2,2′-bipyridine was carried out by the use of various combinations of Pd-catalyzed hetero-coupling reactions with the formation of a new C–N bond,17,20,28 as well as homo-coupling reaction between two properly substituted pyridine rings and step-wise construction of properly substituted 2,2′-bipyridine systems.29 Formation of this 2,2′-bipyridine moiety by the reaction of (4H-pyrido[1,2-a]pyrazin-4-ylidene)anilines with various dienophiles30,31 has also been reported. In the case of 1,10-phenanthrolines the direct ipso-substitution of chlorine atoms in the α-position under the reaction of aniline has been described.32 By means of step-wise approach a number of 2,2′-bipyridines having the residues of carboranes,33 acetylenes,34 polynuclear aromatics,35 cyano-group36 etc. have been obtained. This strategy is of considerable interest because the 1,2,4-triazines are good electron acceptors, therefore they are extremely reactive in SNH reactions37 with a diversity of nucleophiles. In this article, we wish to report our synthetic approach towards an easy and efficient synthesis of 5-(aryl/alkyl)amino-1,2,4-triazines under a solvent and catalyst-free conditions using this approach (Scheme 1). In addition, we also developed a convenient pathway for the synthesis of α-arylamino-2,2′-bipyridine without using any solvent and catalyst.
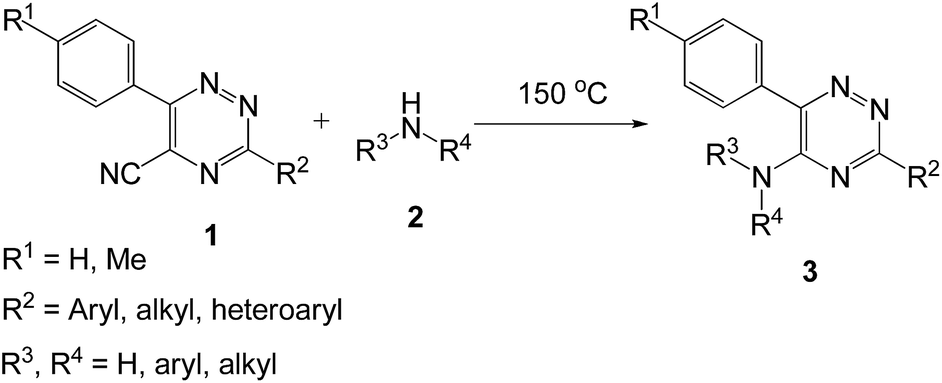 | ||
| Scheme 1 Synthesis of 5-(aryl/alkyl)amino-1,2,4-triazines under solvent and catalyst-free conditions. | ||
The effective introduction of aromatic amines in the 5-position of 1,2,4-triazines, having 2-pyridyl residues in the 3-position, was the major task. It is obvious that the previously reported synthetic approaches in the series of 1,2,4-triazines may not be used for that purpose due to the relatively large number of steps in the synthesis and, thereby, low availability of starting materials. Therefore, we considered some alternative approaches for this strategy based on the current working practice of green chemistry. For instance, several examples of direct amination of C5 position in the series 1,2,4-triazines and their N-oxides have been reported earlier.38–41 However, these procedures usually require the use of additional reagents, in particular, oxidizing agents for the in situ aromatization of the corresponding sigma-adducts afforded by the reaction between 1,2,4-triazines and amines33 or strong bases, such as n-butyllithium or potassium tert-butoxide for the amine activation.34 So far ammonia or aliphatic amines were used for the ipso-substitution of cyano-group in the C5 position of 1,2,4-triazine ring.42–45 These processes underwent smoothly probably due to their greater nucleophilicity. To the best of our knowledge there is no such method for the ipso-substitution of cyano-group in 1,2,4-triazine ring using arylamines.
In this manuscript we wish to report this approach for obtaining 5-arylamino-1,2,4-triazines starting from 5-cyano-1,2,4-triazines 1, which are readily available and can be prepared by the direct high-yield cyanation of 5-H-1,2,4-triazine-4-oxides with acetone cyanohydrin in the presence of triethylamine.36,45 Here in this approach the reaction conditions do not require the high excess of the corresponding amines and any solvent as reported previously.44,45
Results and discussion
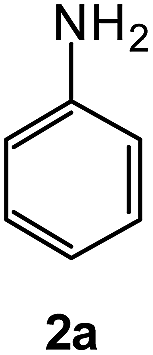
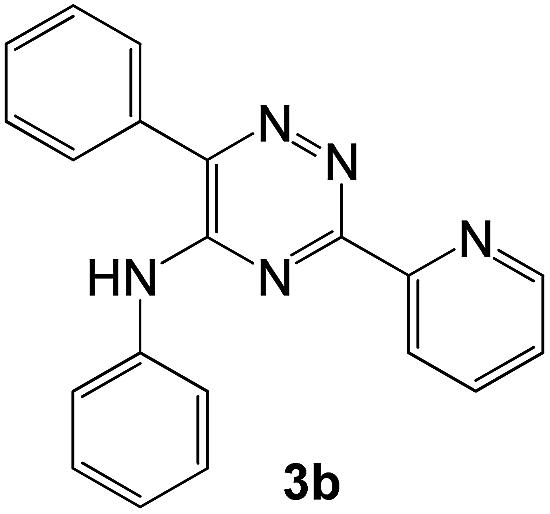
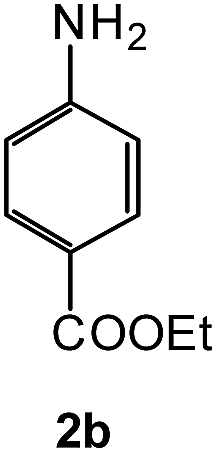
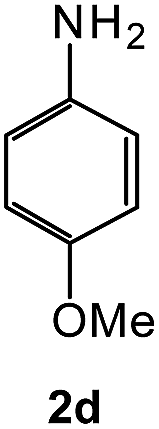
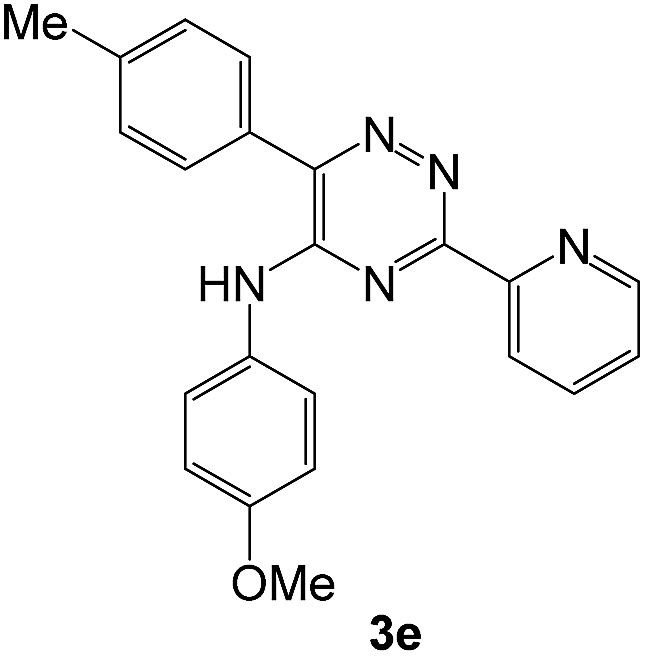
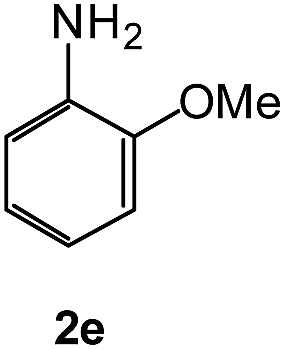
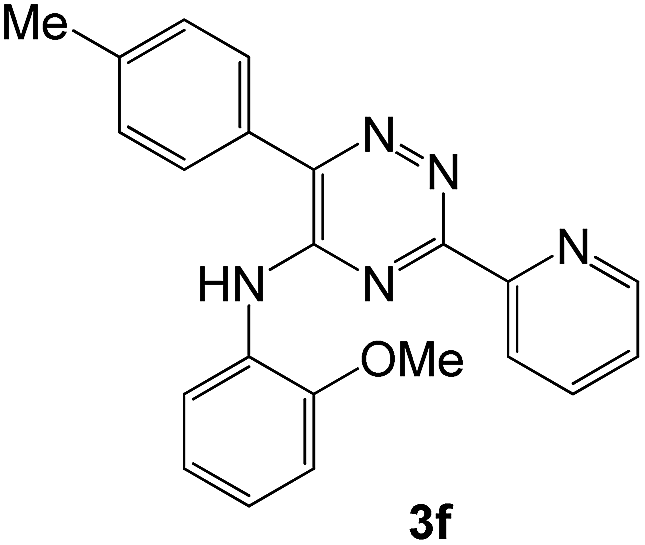
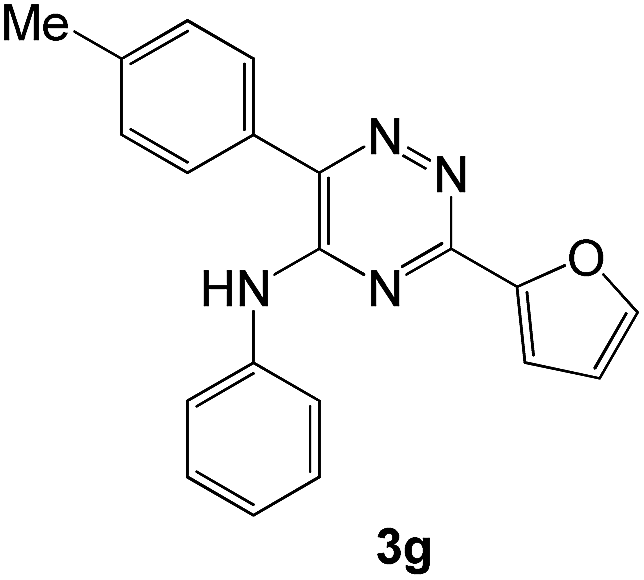
To explore the scope of the reaction, we observed that cyano-group can easily be replaced by amines at 150 °C temperature to afford the desired products 3 in almost quantitative yields. The results are summarized in Table 1. In the course of our study we did not observe any influence on the yields of the products 3 by the different substituents at C3 position of the 1,2,4-triazine ring. Various heteroaryl-substituted 1,2,4-triazines (such as 2-pyridyl-, 2-furyl-, 2-thienyl-) afforded the desired products with excellent yields (3a–3h, 3o). Other substituted 1,2,4-triazines like phenyl- and methyl-1,2,4-triazines have been isolated in more than 90% yields without any additional purification of the crude products (3i–3n). Then our attention was turned to the use of various amines to prove the general applicability of the present methodology. Anilines with electron-donating –OMe substituent afforded the corresponding 5-arylamino-1,2,4-triazines in excellent yields (3e and 3f). The ester and –NO2 substituted anilines also reacted smoothly (3c and 3d). Other primary and secondary amines such as N,N-dimethyl-1,3-propanediamine, morpholine, pyrrolidine and 4-methylpiperazin were well tolerated under the present reaction conditions to afford the desired products (3j–3m).
|
|---|
| a Reaction conditions: a mixture of 0.5 mmol of 5-cyano-1,2,4-triazine 1 and 0.5 mmol of corresponding amine 2 was stirred at 150 °C for 10 h under argon atmosphere. All are isolated yields.b Reaction was carried out in the presence of 1 mL of toluene as solvent at 100 °C for 10 h under argon atmosphere.c Reaction was carried out at 180 °C for 25 h. |
 |
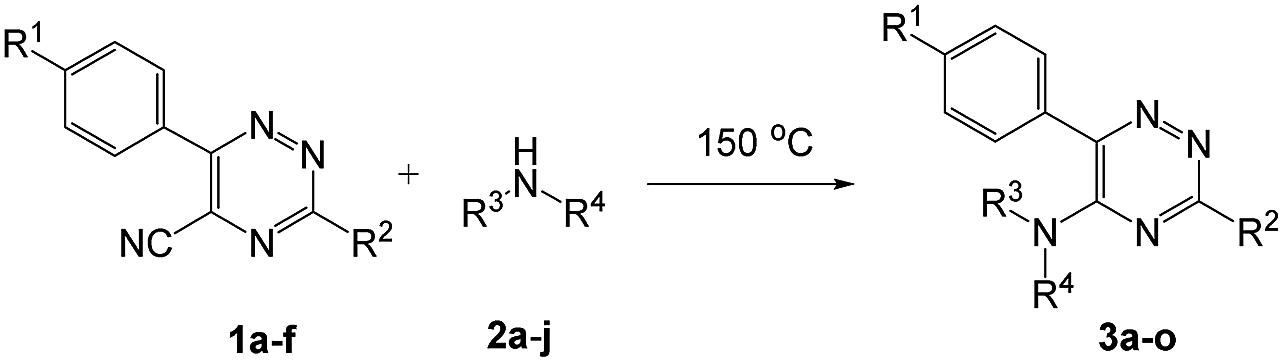
In case of more bulky secondary aromatic amine such as N,N′-diphenylamine the reaction proceeded only at higher temperature (180 °C and above) at longer reaction time. Further increase in temperature led to a considerable tarring of the reaction mixture. In a typical example the reaction of compound 1a with N,N′-diphenylamine (2j) at 180 °C afforded product 3o in 30% isolated yield after performing column chromatography. However, obtaining such compounds by alternative approaches is extremely challenging.
We also examined the solvent and temperature effects of the reactions. In cases of aromatic amines no desired products (3a, 3b, 3c, 3e and 3g) were obtained in the presence of toluene at lower temperature (100 °C). However, moderate yields were obtained for aliphatic amines (3j and 3k) in the presence of toluene at 100 °C.
It should be noted that a very few examples have been reported for the substitution of cyano-group by reaction of amines under solvent-free conditions in some other heterocycles, for instance, in pyrazines,46 4H-imidazole,47 1-arylethene-1,2,2-tricarbonitriles,48 quinazolinone (in an aqueous media),49 and benzonitrile.50 In addition, the intermolecular cyclization reaction of tetrazine followed by the ipso-substitution of cyano-group has been reported.51
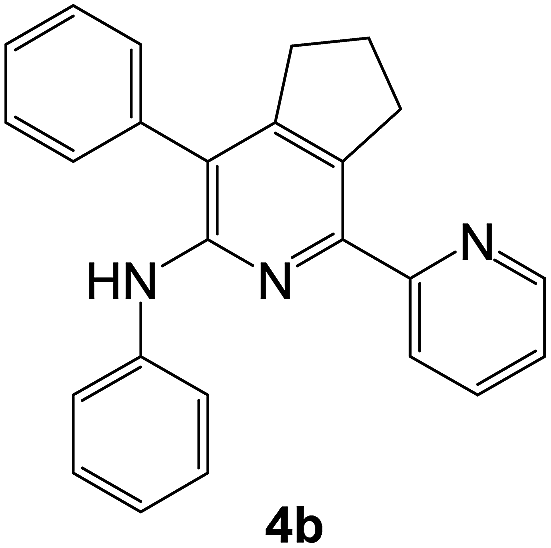
The structures of the products 3 have been confirmed based on the 1H and 13C NMR, mass-spectrometry and elemental analysis. In particular, in case of compounds 3 the dramatic downfield shift for the protons of N–H-moieties of amine residues up to 8.66–8.99 ppm (except for compound 3o) as well as for the protons of phenyl residues of the aniline fragments were observed. For the compound 3o with the diphenylamine moiety introduced the up-field for the resonance signals for all the protons in comparison with compound 1a. In case of 2,2′-bipyridine 4 the resonance signal of proton N–H group of the aniline moiety can be observed in a stronger field as compared to compound 3a along with the appearance of resonance signals of protons of fused cyclopentene fragment in the aliphatic proton resonance region. The structures of the products were well characterized by the spectral data and the X-ray crystallographic analysis of 5-(morpholine-4-yl)-3,6-diphenyl-1,2,4-triazine (3k) was performed to confirm the structure of the product as shown in Fig. 1.52
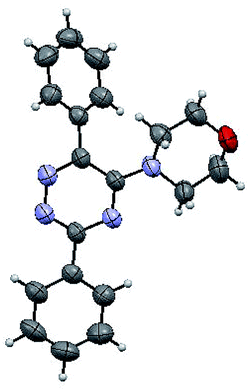 | ||
| Fig. 1 X-ray crystal structure of 5-(morpholine-4-yl)-3,6-diphenyl-1,2,4-triazine (3k). | ||
In addition, we have developed a greener reaction condition bearing lower E-factors5–7 in cases of synthesizing the desired products 3 (Table 2, see also ESI‡).
| # | 5-Cyano-1,2,4-triazine (1) | Amines (2) | Products (3) | Yields (%)b | E-Factor (kg waste per kg product)c |
|---|---|---|---|---|---|
| a All reactions were performed on 0.5 mmol scale at 150 °C for 10 h.b Isolated yields.c Otherwise not mentioned no solvent was used for further purification.d 10 mL toluene was used as eluent for flash chromatography. | |||||
| 1 | 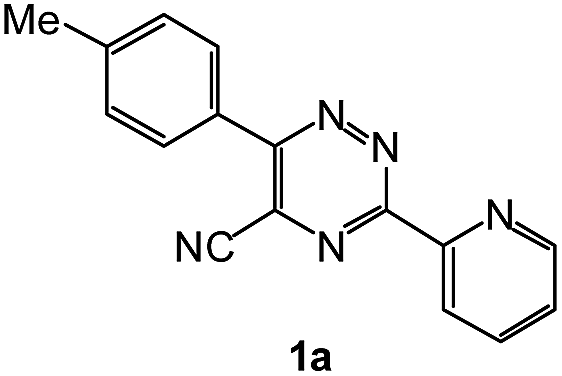 |
 |
 |
94 | 0.15 |
| 2 | 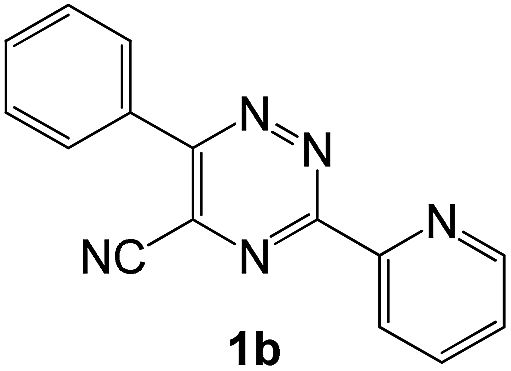 |
 |
 |
93 | 0.17 |
| 3 | 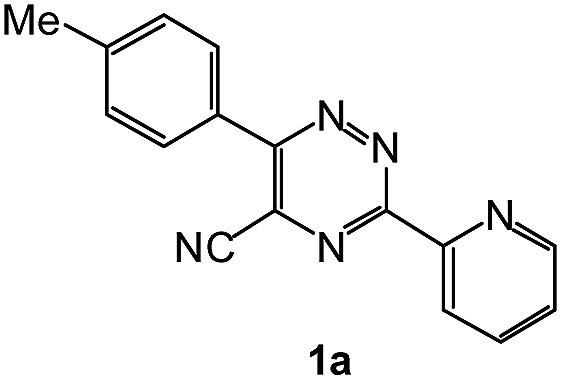 |
 |
 |
92 | 0.15 |
| 4 | 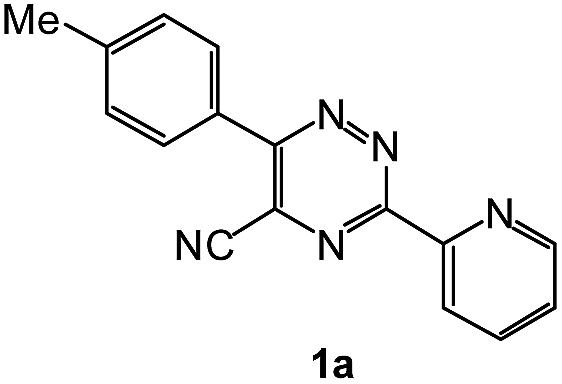 |
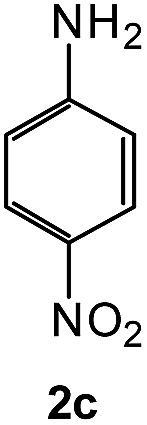 |
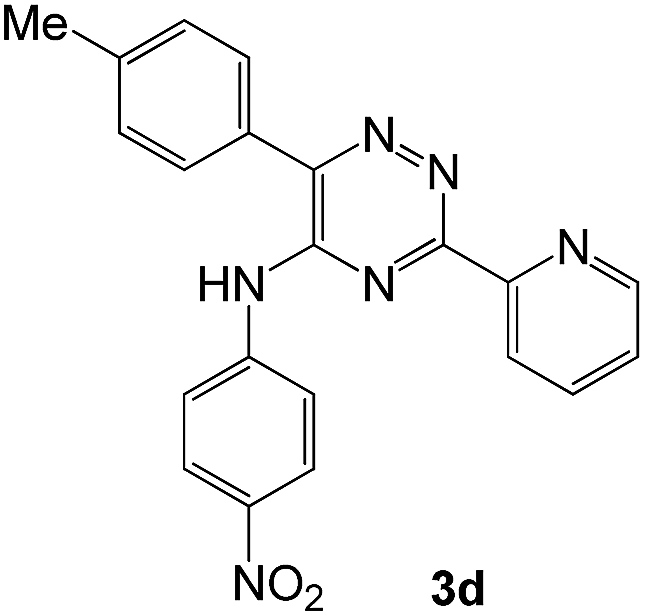 |
93 | 0.15 |
| 5 | 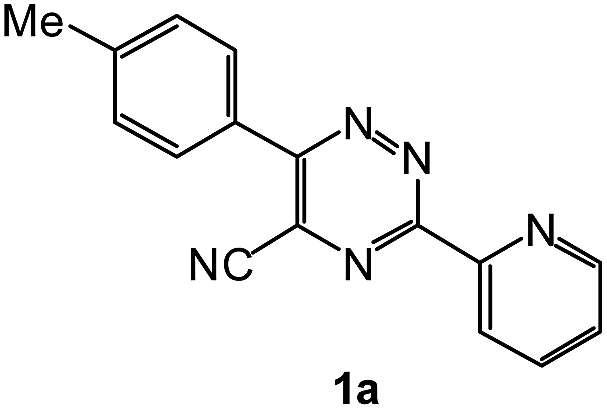 |
 |
 |
95 | 0.14 |
| 6 | 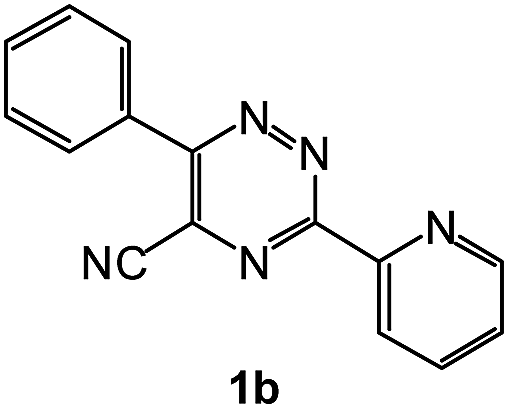 |
 |
 |
95 | 0.14 |
| 7 |  |
 |
 |
90 | 58d |
| 8 | 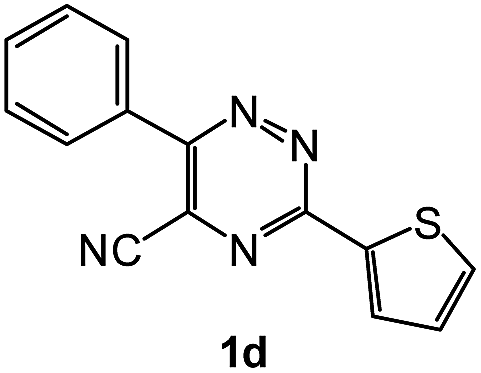 |
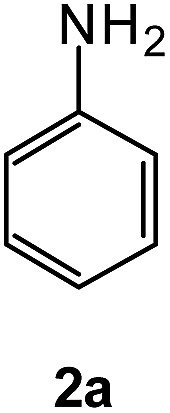 |
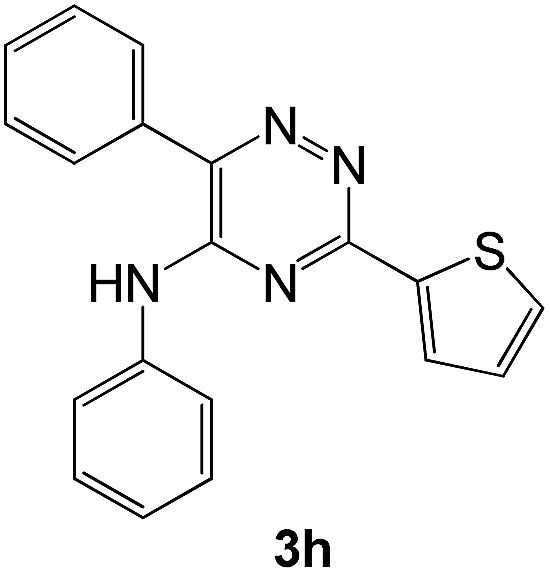 |
90 | 58d |
| 9 | 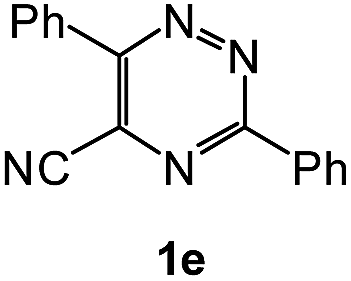 |
 |
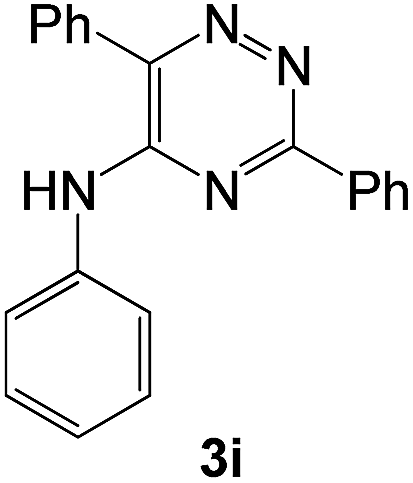 |
98 | 0.10 |
| 10 | 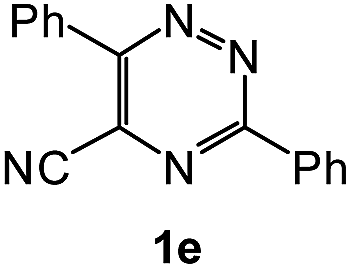 |
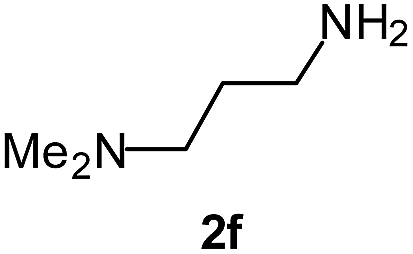 |
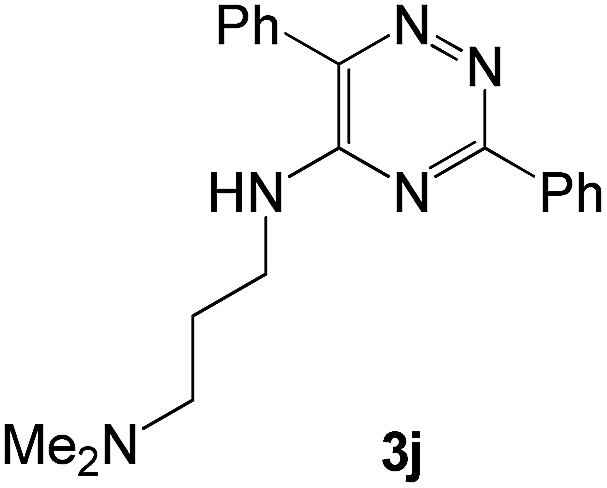 |
88 | 59d |
| 11 | 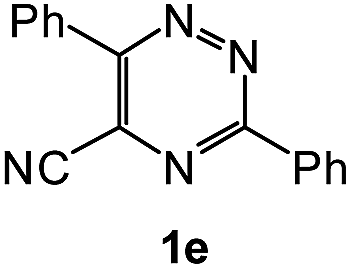 |
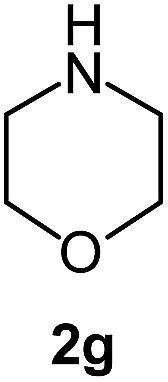 |
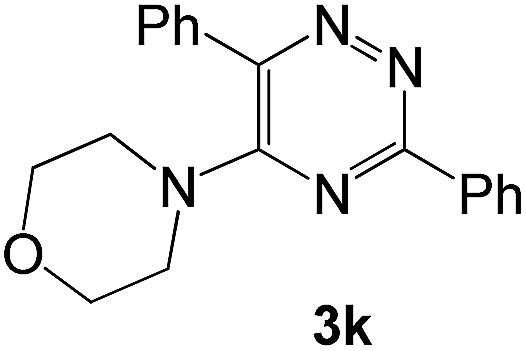 |
90 | 61d |
| 12 | 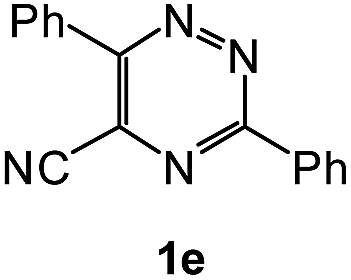 |
 |
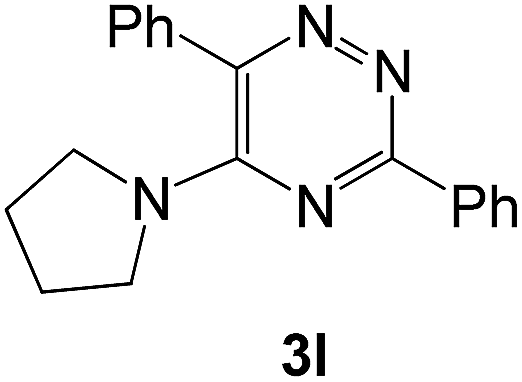 |
91 | 0.20 |
| 13 |  |
 |
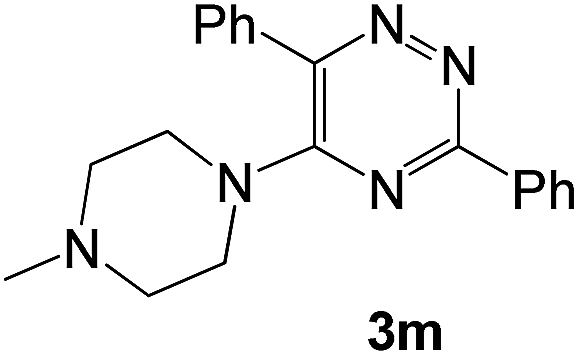 |
90 | 0.20 |
| 14 | 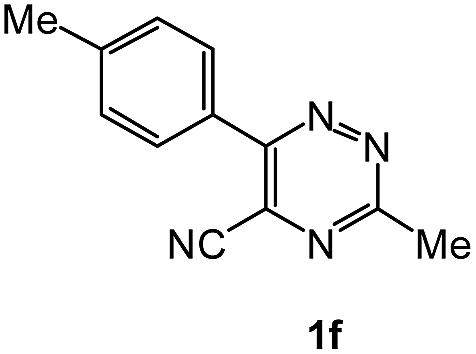 |
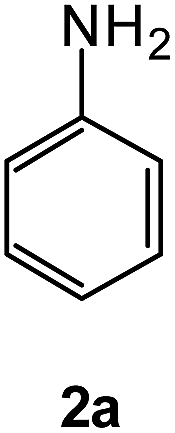 |
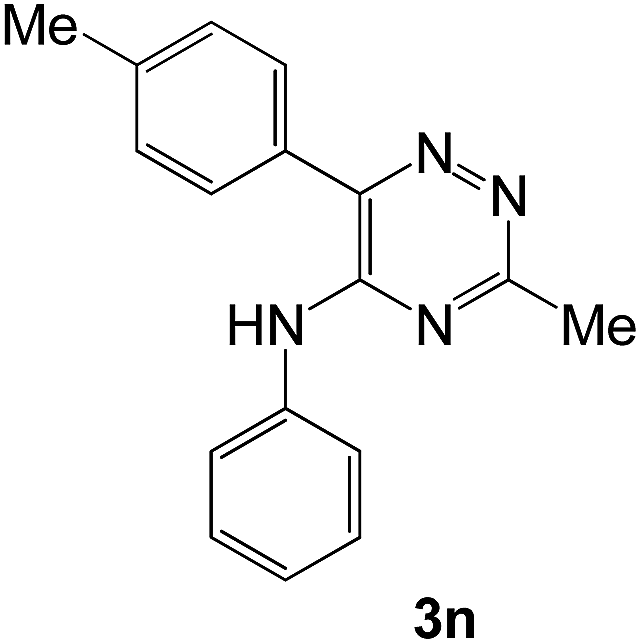 |
87 | 72d |
| 15 | 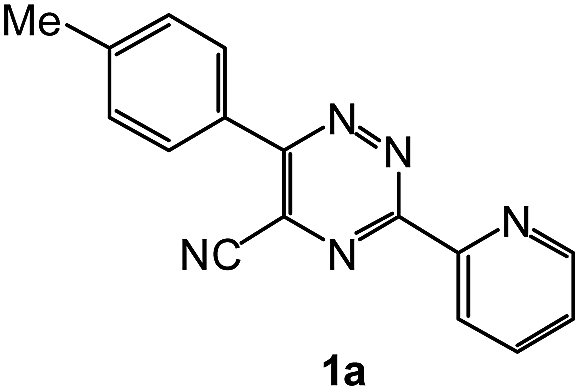 |
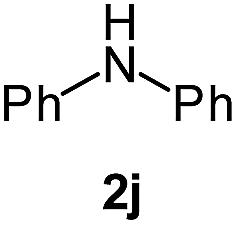 |
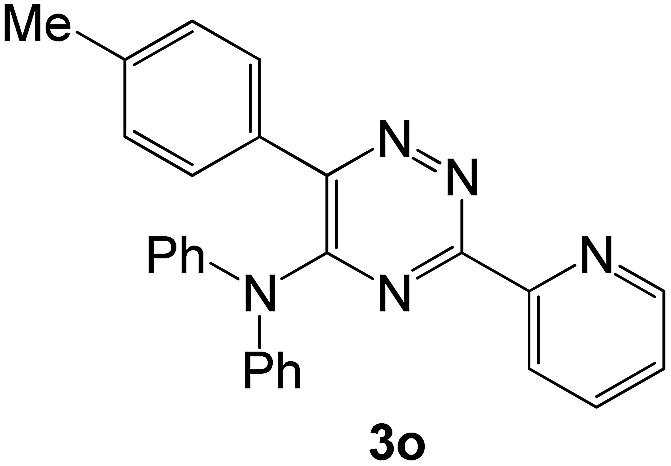 |
30 | 166d |
A plausible mechanistic pathway is outlined in Scheme 2. Though the mechanism of this reaction has not been experimentally established but the probable pathway is a simple nucleophilic ipso-substitution into the aromatic ring. The presence of the –CN group makes the possible electron deficiency in the ring where the attack takes place by amine. The intermediates are stabilized by resonance (A–D) and followed by elimination of –CN− produces the desired product (3).
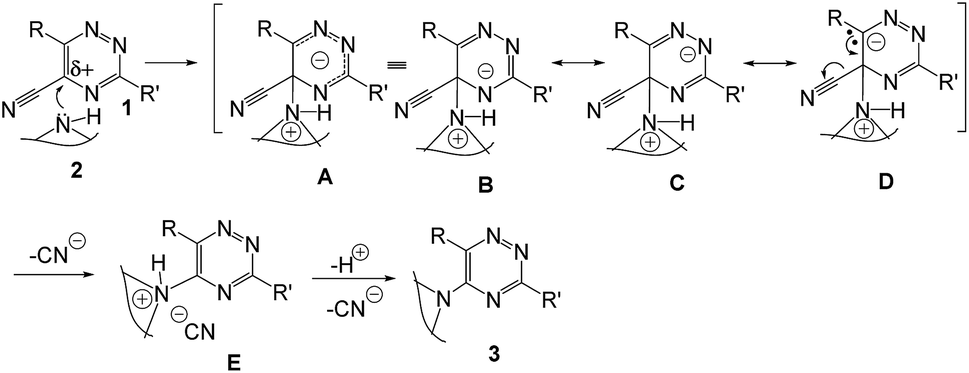 | ||
| Scheme 2 Plausible reaction pathway. | ||
It is noteworthy to mention that to consume the released HCN on a green chemistry point of view, it can be performed the successive reaction by obtaining potassium ferrocyanide by employing the reported method.55
Synthesis of α-arylamino-2,2′-bipyridines
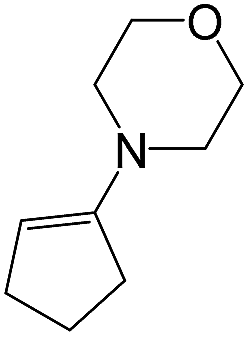
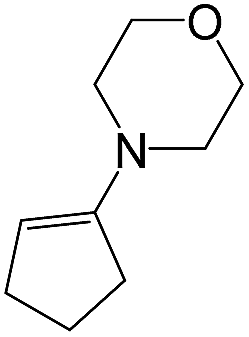
As a next step, we demonstrated the possibility of obtaining of novel 2,2′-bipyridine ligands 4 bearing the residual aniline derivatives in α-position starting from 5-arylamino-1,2,4-triazines 3 (Scheme 3). Remarkably, this step can be again carried out under the solvent-free conditions using slight excess (ca. 1.2–1.5 equiv.) of 1-morpholinocyclopentene dienophile according to a reported method.53 Moreover, the corresponding compound 3 was introduced in the reaction without further purification just after the reaction of ipso-substitution of cyano group, and the aza-Diels-Alder products 4 have been isolated in 63–70% yields. It is worthy to mention that the introduction of a fused cyclopentene fragment into the 2,2′-bipyridine core dramatically increases the solubility of both the resulting ligand and its metal chelates in organic solvents, which may be practically useful.54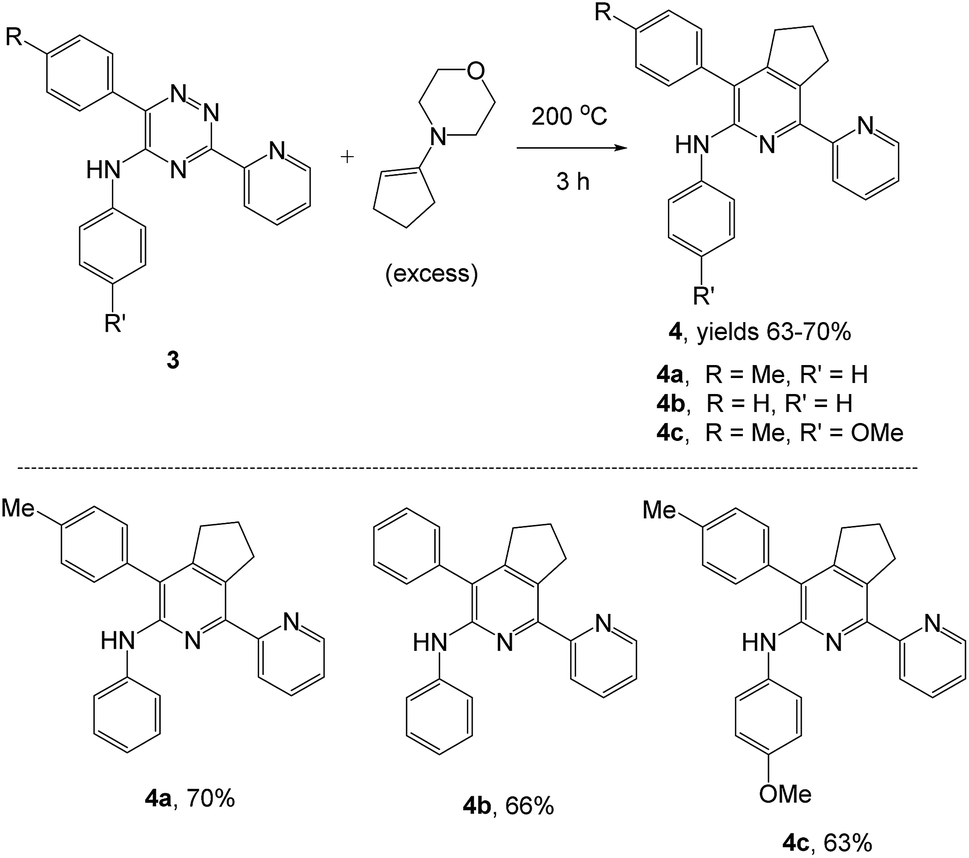 | ||
| Scheme 3 Synthesis of α-arylamino-2,2′-bipyridine under solvent-free condition. | ||
We have also calculated the E-factors for the synthesized α-arylamino-2,2′-bipyridines (4a–4c) and summarized in Table 3 (see also ESI‡).
| # | 5-Arylamino-1,2,4-triazine (3) | 1-Morpholinocyclopentene | Products (4) | Yieldsa (%) | E-Factorb (kg waste per kg product) |
|---|---|---|---|---|---|
| a Isolated yields.b 5 mL ethanol was used for recrystallization.c A mixture of 3a (0.28 mmol) and 1-morpholinocyclopentene (0.42 mmol) was stirred at 200 °C for 3 h under argon atmosphere.d A mixture of 3b (0.26 mmol) and 1-morpholinocyclopentene (0.39 mmol) was stirred at 200 °C for 3 h under argon atmosphere.e A mixture of 3e (0.25 mmol) and 1-morpholinocyclopentene (0.38 mmol) was stirred at 200 °C for 3 h under argon atmosphere. | |||||
| 1c | 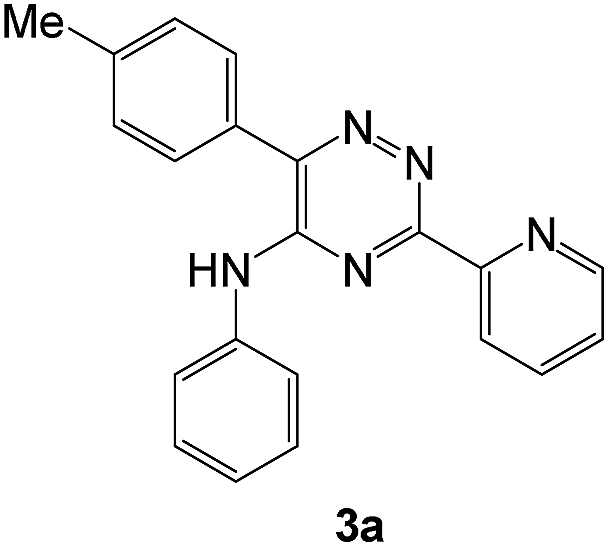 |
 |
 |
70 | 40 |
| 2d | 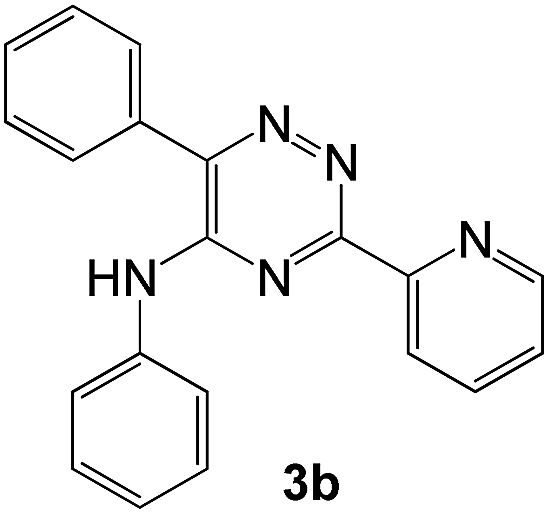 |
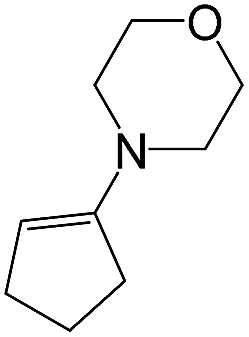 |
 |
66 | 41 |
| 3e | 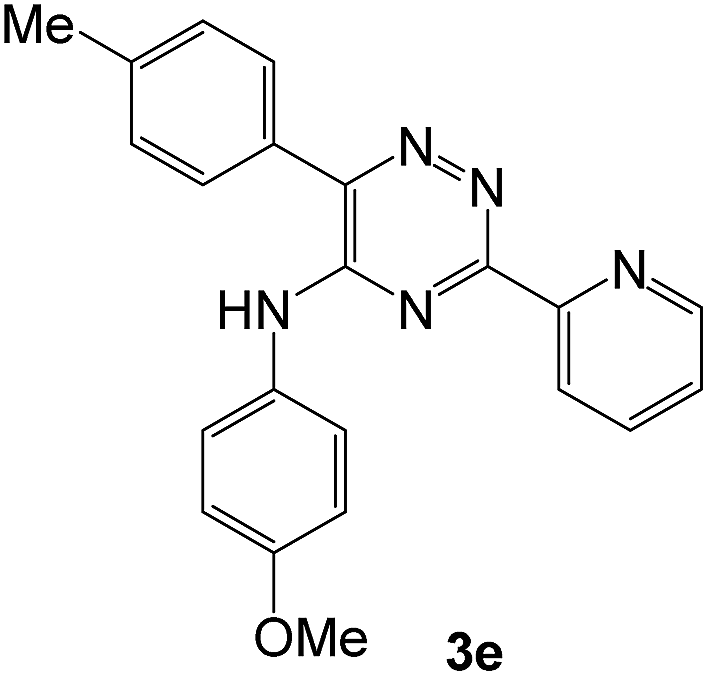 |
 |
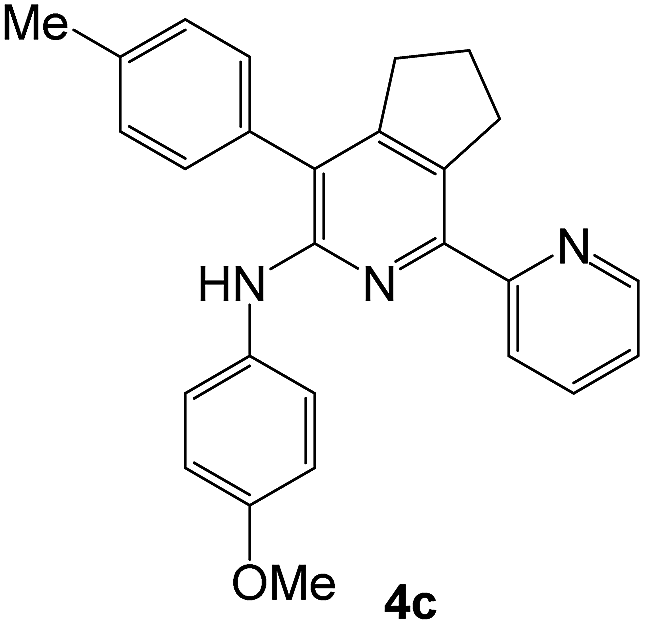 |
63 | 40 |
Conclusions
In summary, in this article we reported a convenient synthetic approach towards functionalized 5-(aryl/alkyl)amino-1,2,4-triazines and α-arylamino-2,2′-bipyridines. This approach involves the direct introduction of the various amines in the C5-position of 3-R-1,2,4-triazines (via SNH reaction), following ipso-substitution of cyano-group by the amino groups under the solvent-free conditions, and, finally, the aza-Diels-Alder reaction with 1-morpholynocyclopentene. Solvent and catalyst-free conditions, operational simplicity, the compatibility with various functional groups, non-chromatographic purification technique, and high yields are the notable advantages of this procedure. The proposed approach is consistent with the principles of the atom economy, and also allows varying the nature of the substituents in the final compounds in a fairly wide range.Experimental section
General experimental methods
1H NMR spectra were determined on a Bruker Avance-400 spectrometer, 298 K, digital resolution ±0.01 ppm. Chemical shifts are expressed in parts per million (d) and are referenced to tetramethylsilane (TMS) as internal standard and the signals were reported as s (singlet), d (doublet), t (triplet), m (multiplet) and coupling constants J were given in Hz. 13C NMR spectra were recorded at 100 MHz. TLC was done on silica gel coated glass slide (Merck, Silica gel G for TLC). Silica gel (60–120 mesh, SRL, India) was used for column chromatography. Melting points were measured on the instrument Boetius. Mass-spectra were recorded on MicrOTOF-Q II (Bruker Daltonics), electrospray as a method of ionization. Microanalyses (C, H, N) were performed using a Perkin-Elmer 2400 elemental analyzer. All solvents were dried and distilled before use. Commercially available substrates were freshly distilled before the reaction. Solvents, reagents and chemicals were purchased from Aldrich, Fluka, Merck, SRL, Spectrochem and Process Chemicals. All reactions involving moisture sensitive reactants were executed using oven dried glassware. Starting compounds 1a, 1b36 and 1e45 were synthesized as described in literature.General method for the synthesis of 5-arylamino-1,2,4-triazines 3
The mixture of corresponding 5-cyano-1,2,4-triazine 1 (0.5 mmol) and corresponding amine 2 (0.5 mmol) was stirred at 150 °C for 10 h under argon atmosphere. Then the resulting mixture was cooled to room temperature and the analytical samples were obtained without additional purification. Also, for the synthesis of α-amino-2,2′-bipyridines 4 the products 3 were used without additional purification. Analytical samples were obtained for compounds 3g, 3h, 3j, 3k, 3n and 3o by flash chromatography using toluene (10 mL) as eluent.General method for the synthesis of N-aryl-1-(pyridine-2-yl)-6,7-dihydro-5H-cyclopenta[c]pyridin-3-amines 4
A mixture of corresponding 5-arylamino-1,2,4-triazine 3 (0.4 mmol) and 1-morpholinocyclopentene (96 μL, 0.6 mmol) was stirred at 200 °C for 3 h under argon atmosphere. The reaction mass was cooled to room temperature. Ethanol (5 mL) was added to the reaction mixture to obtain the analytical samples by recrystallization.Abbreviations
| CCR2 | CC chemokine receptor 2 |
| CCL2 | CC chemokine ligand 2 |
| CCR5 | CC chemokine receptor 5 |
| TLC | Thin layer chromatography |
Acknowledgements
We are pleased to acknowledge the Russian Science Foundation (Ref. # 16-43-02020) for funding. K. Giri is grateful to the UGC, New Delhi for Start-up Project Funding. A. Majee is grateful to BRNS-DAE, Govt. of India (Grant No. 37(2)/14/35/2014-BRNS/563) for financial support.Notes and references
- M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602–1634 CrossRef PubMed.
- M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- Y. Gu, Green Chem., 2012, 14, 2091–2128 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 29, 3352–3365 RSC.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- A. Kamal, V. Srinivasulu, B. N. Seshadri, N. Markandeya, A. Alarifi and N. Shankaraiah, Green Chem., 2012, 14, 2513–2522 RSC.
- K. Tanaka, Solvent-free Organic Synthesis, Wiley-VCH, Cambridge, 2003 Search PubMed.
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS PubMed.
- M. S. Singh and S. Chowdhury, RSC Adv., 2012, 2, 4547–4592 RSC.
- M. Ješelnik, R. S. Varma, S. Polanc and M. Kocevar, Chem. Commun., 2001, 1716–1717 RSC.
- A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, Green Chem., 2016, 18, 4475–4525 RSC.
- Z. J. Jia, W. D. Thomas and W. Chen, Patent US2015158865A1, 2015.
- P. A. J. Janssen, G. J. C. Hoornaert, A. Kilonda, J. Heeres, P. J. Lewi, M. R. De Jonge, F. F. D. Daeyaert, H. M. Vinkers and L. M. H.Koymans, Patent WO2006015985A1, 2006.
- H. Koyama, R. Baiazitov, W. Du, C.-S. Li, Y.-C. Moon, S. D. Paget, H. Ren and N. Sydorenko, Patent WO2015076801A1, 2015.
- Y. Ueno, K. Morishita, M. Muraoka and N. Ohashi, Patent US6159974A, 1998.
- R. Gotoh and M. Yamanaka, Molecules, 2012, 17, 9010–9022 CrossRef CAS PubMed.
- A. W. Garner, C. F. Harris, D. A. K. Vezzu, R. D. Pike and S. Huo, Chem. Commun., 2011, 47, 1902–1904 RSC.
- C. F. Harris, D. A. K. Vezzu, L. Bartolotti, P. D. Boyle and S. Huo, Inorg. Chem., 2013, 52, 11711–11722 CrossRef CAS PubMed.
- S. Huo, C. F. Harris, D. A. K. Vezzu, J. P. Gagnier, M. E. Smith, R. D. Pike and Y. Li, Polyhedron, 2013, 52, 1030–1040 CrossRef CAS.
- G. Thoma, A. B. Smith, M. J. van Eis, E. Vangrevelinghe, J. Blanz, R. Aichholz, A. Littlewood-Evans, C. C. Lee, H. Liu and H.-G. Zerwes, J. Med. Chem., 2015, 58, 1950–1963 CrossRef CAS PubMed.
- E. Garnier, F. Suzenet, D. Poullain, B. Lebret and G. Guillaumet, Synlett, 2006, 472–474 CAS.
- Y. Sanemitsu, Y. Nakayama, Y. Tanabe, H. Matsumoto and S.-i. Hashimoto, Agric. Biol. Chem., 1990, 54, 3367–3369 CAS.
- S. Konno, M. Sagi, M. Yokoyama and H. Yamanaka, Heterocycles, 1990, 31, 1933–1935 CrossRef CAS.
- M. M. Eid, A. M. K. El-Zanaty and Y. A. Ibrahim, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1990, 29, 435–439 Search PubMed.
- M. G. Barlow, R. N. Haszeldine, C. Simon, D. J. Simpkin and G. Ziervogel, J. Chem. Soc., Perkin Trans. 1, 1982, 1251–1254 RSC.
- Z. E. Lu, D. Q. Sun, T. L. Xu, J. Wan, L. C. Xu and K. Q. Chen, Org. Prep. Proced. Int., 1992, 24, 358–362 CrossRef CAS.
- J.-D. Cheon, T. Mutai and K. Araki, Tetrahedron Lett., 2006, 47, 5079–5082 CrossRef CAS.
- S. Tu, B. Jiang, Y. Zhang, R. Jia, J. Zhang, C. Yao and F. Shi, Org. Biomol. Chem., 2007, 5, 355–359 CAS.
- T. Billert, R. Beckert, P. Fehling, M. Doering and H. Goerls, Tetrahedron, 1997, 53, 5455–5462 CrossRef CAS.
- J. Brandenburg, R. Beckert, P. Fehling, M. Döring and H. Görls, Adv. Synth. Catal., 1996, 338, 430–435 CAS.
- W.-J. Wang, A. Sengul, C.-F. Luo, H.-C. Kao and Y.-H. Cheng, Tetrahedron Lett., 2003, 44, 7099–7101 CrossRef CAS.
- A. M. Prokhorov, D. N. Kozhevnikov, V. L. Rusinov, O. N. Chupakhin, I. V. Glukhov, M. Y. Antipin, O. N. Kazheva, A. N. Chekhlov and O. A. Dyachenko, Organometallics, 2006, 25, 2972–2977 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, A. M. Prokhorov, M. M. Ustinova, V. L. Rusinov, O. N. Chupakhin, G. G. Aleksandrov and B. König, Tetrahedron Lett., 2006, 47, 869–872 CrossRef CAS.
- I. S. Kovalev, D. S. Kopchuk, A. F. Khasanov, G. V. Zyryanov, V. L. Rusinov and O. N. Chupakhin, Mendeleev Commun., 2014, 24, 117–118 CrossRef CAS.
- V. N. Kozhevnikov, D. N. Kozhevnikov, T. V. Nikitina, V. L. Rusinov, O. N. Chupakhin, M. Zabel and B. König, J. Org. Chem., 2003, 68, 2882–2888 CrossRef CAS PubMed.
- D. N. Kozhevnikov, V. L. Rusinov and O. N. Chupakhin, Adv. Heterocycl. Chem., 2002, 82, 261–305 CrossRef CAS.
- A. Rykowski and H. C. Van Der Plas, Synthesis, 1985, 884–886 CrossRef CAS.
- E. Garnier, J. Audoux, E. Pasquinet, F. Suzenet, D. Poullain, B. Lebret and G. Guillaumet, J. Org. Chem., 2004, 69, 7809–7815 CrossRef CAS PubMed.
- W. R. Carling, A. R. Guiblin, K. W. Moore, C. R. Moyes, M. Rowley and L. J. Street, Patent US6608062 B1, 2003.
- S. Konno, S. Ohba, M. Sagi and H. Yamanaka, Chem. Pharm. Bull., 1987, 35, 1378–1382 CrossRef CAS.
- J. J. Huang, J. Org. Chem., 1985, 50, 2293–2298 CrossRef CAS.
- A. Rykowski, D. Branowska, M. Makosza and P. Van Ly, J. Heterocycl. Chem., 1996, 33, 1567–1571 CrossRef CAS.
- V. N. Kozhevnikov, D. N. Kozhevnikov, O. V. Shabunina, N. N. Kataeva, S. A. Yushchuk, V. L. Rusinov and O. N. Chupakhin, Russ. Chem. Bull., 2005, 54, 2187–2196 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, I. S. Kovalev, V. L. Rusinov, O. N. Chupakhin and G. G. Aleksandrov, Russ. J. Org. Chem., 2002, 38, 744–750 CrossRef CAS.
- A. Nakamura, M. Ono, H. Segawa and T. Takematsu, Agric. Biol. Chem., 1984, 48, 1009–1016 CAS.
- D. I. Potapenko, M. A. Foster, D. J. Lurie, I. A. Kirilyuk, J. M. S. Hutchison, I. A. Grigor'ev, E. G. Bagryanskaya and V. V. Khramtsov, J. Magn. Reson., 2006, 182, 1–11 CrossRef CAS PubMed.
- Y. Shigemitsu, B.-C. Wang, Y. Nishimura and Y. Tominaga, Dyes Pigm., 2011, 92, 580–587 CrossRef.
- K.-I. Ozaki, Y. Yamada and T. Oine, Chem. Pharm. Bull., 1983, 31, 2234–2243 CrossRef CAS.
- A. Gilbert, S. Krestonosich and D. L. Westover, J. Chem. Soc., Perkin Trans. 1, 1981, 295–302 RSC.
- E. M. Beccalli, M. D. Puppo, E. Licandro and A. Marchesini, J. Heterocycl. Chem., 1981, 18, 685–686 CrossRef CAS.
- Further information can be found in the CIF file. This crystal was deposited in the Cambridge Crystallographic Data Centre and assigned as CCDC 1505314.
- V. N. Kozhevnikov, M. M. Ustinova, P. A. Slepukhin, A. Santoro, D. W. Bruce and D. N. Kozhevnikov, Tetrahedron Lett., 2008, 49, 4096–4098 CrossRef CAS.
- A. P. Krinochkin, D. S. Kopchuk and D. N. Kozhevnikov, Polyhedron, 2015, 102, 556–561 CrossRef CAS.
- E. Gail, S. Gos, R. Kulzer, J. Lorösch, A. Rubo, M. Sauer, R. Kellens, J. Reddy, N. Steier and W. Hasenpusch, Cyano Compounds, Inorganic, Ullmann's Encyclopedia of Industrial Chemistry, 2011 Search PubMed.
Footnotes |
| † This article is dedicated to the memory of Prof. Yuri Yu. Morzherin, Ural Federal University. His spirit will remain with us and in our hearts forever. |
| ‡ Electronic supplementary information (ESI) available: Scanned copies of 1H and 13C NMR spectra of the synthesized compounds, crystallographic data for compound 3k (CIF). CCDC 1505314. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26305d |
| This journal is © The Royal Society of Chemistry 2017 |