DOI:
10.1039/C6RA26245G
(Paper)
RSC Adv., 2017,
7, 1016-1025
Synthesis of di(ethylene glycol)-functionalized diketopyrrolopyrrole derivative-based side chain-conjugated polymers for bulk heterojunction solar cells†
Received
3rd November 2016
, Accepted 18th November 2016
First published on 4th January 2017
Abstract
Polythiophenes (PTs) featuring di(ethylene glycol)-substituted 2,5-thienyl diketopyrrolopyrrole (DG-TDPP) moieties as conjugated units in the polymer backbone and tert-butyl-substituted triphenylamine (tTPA)-containing moieties as pendant units have been synthesized through Stille coupling. Incorporating the electron-deficient DG-TDPP moieties within the polymer backbone and appending the tTPA units promoted charge balance and efficient conjugation within the extended conjugated frameworks of the polymers, resulting in lower band gap energies and red-shifting the maximum UV-Vis absorption wavelength. The influence of the DG-TDPP content on the optical, electrochemical, and photovoltaic (PV) properties of the polymers has been studied. Incorporating a suitable content of DG-TDPP moieties in the polymer backbone enhanced the solar absorption ability and conjugation length of the PTs. The PV properties of bulk-heterojunction solar cells based on PT/fullerene derivatives improved after incorporating DG-TDPP units in the backbones of the side chain-conjugated PTs.
1. Introduction
Optoelectronic devices fabricated from conjugated polymers have attracted much attention recently because they can be processed simply and inexpensively over large areas with flexible shapes and light weight.1–3 In the case of polymer solar cells (PSCs), major advances in solar energy conversion efficiencies have been accomplished by replacing the double-layer cell with a bulk-heterojunction (BHJ) blend for the photoactive layer.4–6 For most BHJ cells, the photoactive layer is based on a blend of an electron-donor polymer and an electron-acceptor fullerene derivative, either [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) or [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM).7 An electron-donor polymer having a low band gap is crucial when attempting to harvest large amounts of solar radiation. Using this approach, PSCs have been obtained with high short-circuit current densities (JSC).8,9 Moreover, high charge mobility is also required of the polymer in PSCs to minimize charge recombination in the photoactive layer, and to achieve high degrees of charge collection and large values of JSC.10 In addition, the energy level of the lowest unoccupied molecular orbital (LUMO) of the polymer must be appropriately offset from that of the electron-acceptor fullerene derivative.11 A polymer having a deep highest occupied molecular orbital (HOMO) energy level favors a PSC having a large open-circuit voltage (VOC).12,13 On the other hand, a phase-separated interpenetrating network featuring sizable fullerene derivative-based domains in the photoactive layer facilitates the efficient formation of free carriers, thereby providing PSCs displaying optimal photovoltaic (PV) properties.14–16
PSCs providing good PV performance have been studied widely in which the conjugated polymers contain p-type electron-donating groups and electron-donating and -withdrawing donor–acceptor (D–A)-type structures.10,12,17 The bipolar characteristics of conjugated polymers containing electron- and hole-transporting moieties are responsible for the lower-energy band gaps and higher charge mobilities of the photoactive layers that can enhance the PV efficiencies of PSCs. In addition, polythiophene (PT) derivatives bearing electron-donating and -withdrawing pendant groups, so-called two-dimensional low band gap conjugated polymers, have been proposed by several groups for PSC applications.18–26 PT derivatives bearing conjugated pendant units absorb broadly in the UV and visible regions and can, therefore, harvest greater amounts of solar light. Moreover, the pendant conjugated moieties can enhance the charge mobilities of the polymers. Recently, we synthesized a series of PTs presenting pendant conjugated triphenylamine (TPA)- or carbazole-containing moieties as side chains.27 Although the TPA- or carbazole-based conjugated pendant moieties enhanced the conjugation of the PTs, the cutoff wavelength of the absorption of these p-type conjugated polymers remained less than 750 nm. In addition, electron-deficient moieties, including benzothiadiazole and 2,5-thienyl diketopyrrolopyrrole (TDPP) units, have been incorporated into the backbones of polymers presenting electron-donating conjugated pendant units.28–32 The presence of TDPP units in the polymer backbone enhanced the absorption edge to wavelengths of up to 1000 nm.33–35 Unfortunately, increasing the TDPP content decreased the solubility of the polymers, due to increased degrees of π–π stacking among the polymer chains. The power conversion efficiencies (PCEs) of such polymer-based PSCs are not high.33,34 Therefore, precise control over the molar ratio of electron-donating and -withdrawing units in a polymer chain is necessary to optimize the solubility and the optical and PV performance of their devices.
To validate these ideas, we synthesized a series of PTs (PEGA11, PEGA12, PEGA13) featuring di(ethylene glycol)-substituted 2,5-thienyl diketopyrrolopyrrole (DG-TDPP) moieties as conjugated units in the polymer backbones and tert-butyl-substituted triphenylamine (tTPA)-containing moieties as pendant units. We expected the conjugated DG-TDPP units with di(ethylene glycol) segments as flexible chains would improve the solubility of the PTs. We used UV-Vis absorption spectroscopy and cyclic voltammetry (CV) to study the effect of the DG-TDPP content on the photophysical and electrochemical properties of the polymers. We also employed atomic force microscopy (AFM) and transmission electron microscopy (TEM) to study the morphologies of thin films prepared from PT/PC61BM blends. We then fabricated PSCs having the conventional structure indium tin oxide (ITO)-coated glass/hole-transporting medium (HTM)/photoactive layer/LiF (0.5 nm)/Al (100 nm), by spin-coating a blend of each PT with PC61BM (or PC71BM) to form a composite film-type photoactive layer on the HTM layer deposited on the ITO-coated glass. Based on the evaluated PV performance of these PSCs, we determined the influence of the DG-TDPP content of our PTs.
2. Experimental details
2.1 Chemicals
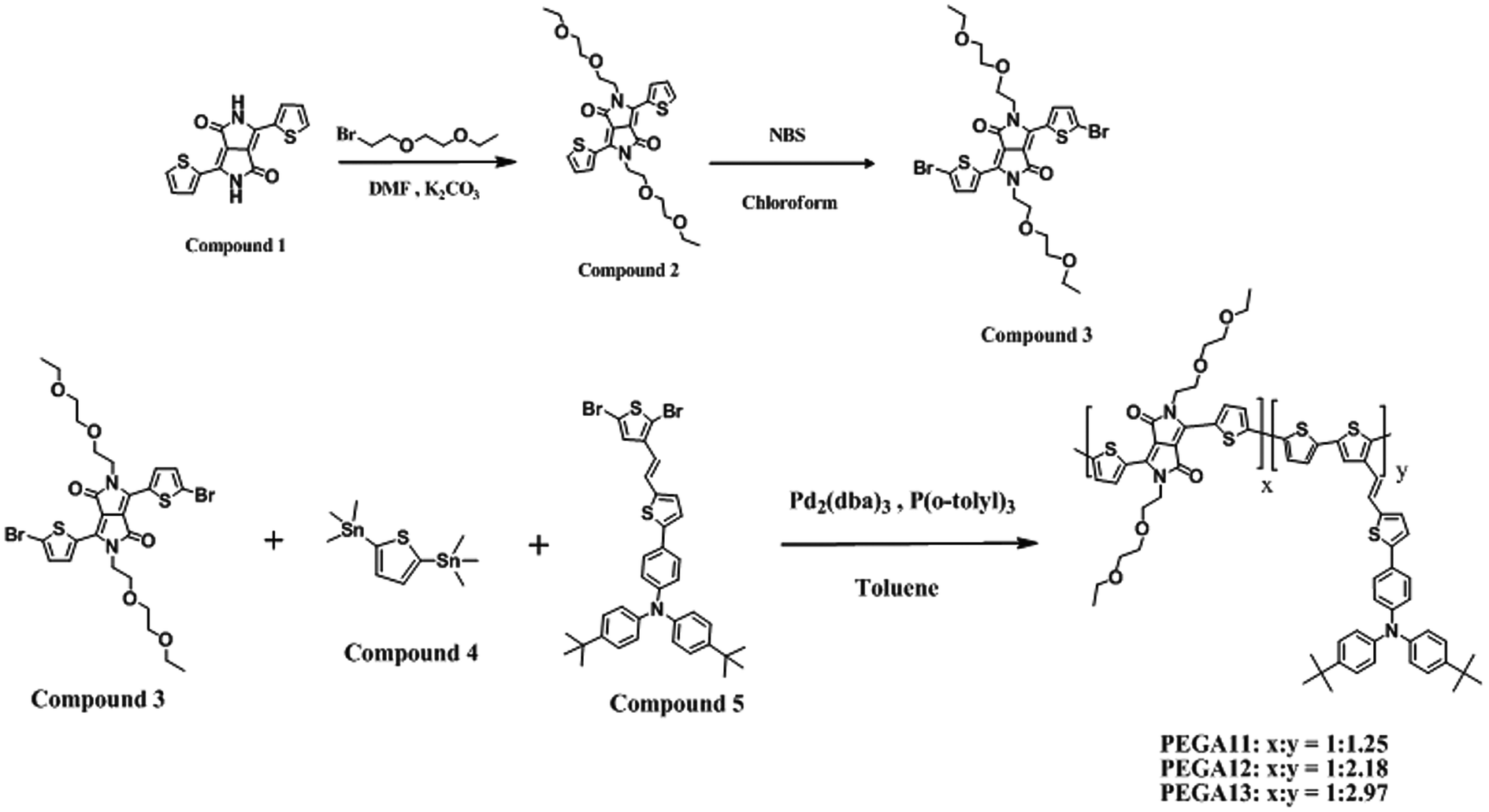
Potassium carbonate (K2CO3) N-bromosuccinimide (NBS), tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba)3], tri(o-tolyl)phosphine [P(o-tolyl)3], 2-(2-ethoxyethoxy)ethyl bromide, and other reagents and chemicals were purchased from Aldrich, Alfa, Acros, and TCI Chemical, and used as received. Chloroform (CHCl3), dimethylformamide (DMF), toluene, and o-dichlorobenzene (o-DCB) were freshly distilled over appropriate drying agents, then purged with N2, prior to use. 3,6-Di(thien-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1), 2,5-bis(trimethylstannyl)thiophene (4), and (E)-4-(5-(2-(2,5-dibromothiophen-3-yl)vinyl)thien-2-yl)-N,N-((4,40-di-tert-butyl)diphenyl)aniline (5) were synthesized according to the literature.27,35,36 The syntheses of 3,6-di(thien-2-yl)-2,5-di(2-(2-ethoxyethoxy)ethyl)-pyrrolo[3,4-c]pyrrole-1,4-dione (2) and 3,6-di(5-bromothien-2-yl)-2,5-di(2-(2-ethoxyethoxy)ethyl)pyrrolo[3,4-c]pyrrole-1,4-dione (3) are illustrated in Scheme 1. The syntheses of PTs (PEGA11, PEGA12, and PEGA13) featuring DG-TDPP moieties as conjugated units in the polymer backbones and tTPA-containing moieties as side chains are also illustrated in Scheme 1.
 |
| | Scheme 1 Synthesis of compounds 2, 3, PEGA11, PEGA12, and PEGA13. | |
2.2 Synthesis
3,6-Di(thien-2-yl)-2,5-di(2-(2-ethoxyethoxy)ethyl)pyrrolo[3,4-c]pyrrole-1,4-dione (2). A solution of compound 1 (2.50 g, 8.33 mmol), K2CO3 (3.83 g, 27.80 mmol), and [18]-crown-6 (0.250 g, 0.95 mmol) in dry DMF (50 mL) was stirred at 120 °C under N2 for 10 min and then a solution of 2-(2-ethoxyethoxy)ethyl bromide (4.17 mL, 27.5 mmol) in dry DMF (30 mL) was added dropwise. The resulting solution was heated with stirring for 20 h. After cooling to room temperature, the mixture was partitioned between CH2Cl2 and water; the organic phase was dried (MgSO4), filtered, and evaporated to dryness. The residue was purified chromatographically (SiO2; EtOAc/hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) to provide a dark-red solid (1.82 g, 41.9%). 1H NMR (δ/ppm, 600 MHz, CDCl3): 1.15 (t, 6H), 3.44 (q, 4H), 3.52 (t, 4H), 3.62 (t, 4H), 3.79 (t, 4H), 4.27 (t, 4H), 7.25 (t, 2H), 7.63 (d, 2H), 8.73 (d, 2H). Anal. calcd for C26H32N2O6S2: C, 58.67; H, 6.02; N, 5.26; O, 18.05; S, 12.0. Found: C, 58.65; H, 6.07; N, 5.24; O, 18.04; S, 11.97.
:4) to provide a dark-red solid (1.82 g, 41.9%). 1H NMR (δ/ppm, 600 MHz, CDCl3): 1.15 (t, 6H), 3.44 (q, 4H), 3.52 (t, 4H), 3.62 (t, 4H), 3.79 (t, 4H), 4.27 (t, 4H), 7.25 (t, 2H), 7.63 (d, 2H), 8.73 (d, 2H). Anal. calcd for C26H32N2O6S2: C, 58.67; H, 6.02; N, 5.26; O, 18.05; S, 12.0. Found: C, 58.65; H, 6.07; N, 5.24; O, 18.04; S, 11.97.
3,6-Di(5-bromothien-2-yl)-2,5-di(2-(2-ethoxyethoxy)ethyl)pyrrolo[3,4-c]pyrrole-1,4-dione (3). A solution of 4 (0.53 g, 1.0 mmol) in CHCl3 (30 mL) was stirred at room temperature for 10 min and then a solution of NBS (0.45 g, 2.3 mmol) in CHCl3 (30 mL) was added dropwise. The resulting solution was stirred at room temperature for 12 h. The mixture was then partitioned between CH2Cl2 and water; the organic phase was dried (MgSO4), filtered, and evaporated to dryness. The residue was recrystallized in MeOH to provide a purple solid (0.29 g, 42.0%). 1H NMR (δ/ppm, 600 MHz, CDCl3): 1.16 (t, 6H), 3.45 (q, 4H), 3.51 (t, 4H), 3.62 (t, 4H), 3.78 (t, 4H), 4.17 (t, 4H), 7.20 (d, 2H), 8.48 (d, 2H). Anal. calcd for C26H30N2O6S2Br2: C, 45.22; H, 4.35; N, 4.06; O, 13.91; S, 9.28. Found: C, 45.28; H, 4.31; N, 4.03; O, 13.86; S, 9.31.
PEGA11. A solution of 5 (0.35 g, 0.50 mmol), 6 (0.41 g, 1.0 mmol), and 7 (0.35 g, 0.50 mmol) in dry toluene (30 mL) was stirred at room temperature under N2 for 10 min and then a solution of Pd2(dba)3 (36.6 mg, 0.04 mmol) and P(o-tolyl)3 (48.7 mg, 0.16 mmol) in dry toluene (10 mL) was added dropwise. The resulting solution was heated with stirring at 90 °C for 48 h. After cooling to room temperature, the solution was poured into MeOH (100 mL). The precipitate was filtered off, placed in a Soxhlet thimble, and extracted sequentially with MeOH, hexane, acetone, CHCl3, and o-DCB. The polymer PEGA11 was recovered from the o-DCB fraction through rotary evaporation. Drying under vacuum for 24 h provided a black solid (0.24 g, 21.4%). 1H NMR (δ/ppm, 600 MHz, CD2Cl2): 0.75–1.39 (m, 28H), 3.15–3.71 (m, 20H), 6.35–7.62 (m, 28H). Anal. calcd for [(C26H30N2O6S2)(C40H37NS3)1.25]: C, 69.42; H, 5.80; N, 3.46; O, 7.31; S, 14.01. Found: C, 69.55; H, 5.75; N, 3.53; O, 7.37; S, 13.80.
PEGA12. Using the procedure described above for the synthesis of PEGA11, the reaction of 5 (0.23 g, 0.33 mmol), 6 (0.41 g, 1.0 mmol), and 7 (0.47 g, 0.67 mmol) provided PEGA12 as a black solid (0.16 g, 14.4%). 1H NMR (δ/ppm, 600 MHz, CD2Cl2): 0.75–1.40 (m, 45H), 3.10–3.75 (m, 20H), 6.30–7.60 (m, 46H). Anal. calcd for [(C26H30N2O6S2)(C40H37NS3)2.18]: C, 71.61; H, 5.83; N, 3.09; O, 5.06; S, 14.41. Found: C, 71.59; H, 5.89; N, 3.05; O, 5.10; S, 14.37.
PEGA13. Using the procedure described above for the synthesis of PEGA11, the reaction of 5 (0.17 g, 0.25 mmol), 6 (0.41 g, 1.0 mmol), and 7 (0.53 g, 0.75 mmol) provided PEGA13 as a black solid (0.26 g, 23.4%). 1H NMR (δ/ppm, 600 MHz, CD2Cl2): 0.65–1.40 (m, 60H), 3.05–3.75 (m, 20H), 6.30–7.42 (m, 60H). Anal. calcd for [(C26H30N2O6S2)(C40H37NS3)2.97]: C, 72.64; H, 5.85; N, 2.90; O, 4.01; S, 14.64. Found: C, 72.60; H, 5.91; N, 2.88; O, 4.10; S, 14.51.
2.3 Characterization of copolymers
1H NMR (600 MHz) spectra were recorded using a Varian Unity Inova spectrometer. The average molecular weights of the polymers were measured by means of gel permeation chromatography (GPC), using a Waters chromatography system (717 plus Autosampler) equipped with two Waters Styragel linear columns, employing polystyrene (PS) standards and THF as the eluent. The glass transition temperatures (Tg) and thermal decomposition temperatures (Td; temperatures at which weight loss reached 5%) of the copolymers were determined through differential scanning calorimetry (TA Instruments, DSC-2010) and thermogravimetric analysis (TA Instruments, TGA-2050), respectively. Both analyses were performed under a N2 atmosphere at scanning (both heating and cooling) rates of 10 °C min−1. The temperatures at the intercepts of the curves in the thermograms (endothermic, exothermic, or weight loss) with the leading baseline were taken as estimates of the values of Tg and Td. Absorption spectra were recorded using a Hitachi U3010 UV-Vis spectrometer; dilute o-DCB solutions of the PTs were filtered through a 0.45 μm filter to remove insoluble materials prior to spectral measurement. The redox potentials of the polymers were determined using a CHI 611D electrochemical analyzer (scanning rate: 50 mV s−1) equipped with Pt electrodes and an Ag/Ag+ (0.10 M AgNO3 in MeCN) reference electrode in an anhydrous, N2-saturated solution of 0.1 M Bu4NClO4 in MeCN. Bu4NClO4 (98%, TCI) was recrystallized three times from MeOH and water (1:1) and then dried at 100 °C under reduced pressure. A Pt plate coated with a thin polymer film was used as the working electrode; a Pt wire and an Ag/Ag+ electrode were used as the counter and reference electrodes, respectively. The electrochemical potential was calibrated against ferrocene/ferrocenium. The morphologies of films prepared from PT/PC61BM blends were studied using an atomic force microscope (AFM, Seiko SII SPA400) operated in the tapping mode and a transmission electron microscope (TEM, JEOL JEM-1400).
2.4 Fabrication and characterization of PSCs
The PSCs fabricated in this study had the structure ITO-coated glass/HTM/photoactive layer/LiF (0.5 nm)/Al (100 nm), where the photoactive layer comprised an interpenetrating network of a PT and a fullerene derivative (PC61BM or PC71BM). ITO-coated glass (sheet resistance: 20 Ω per square) was purchased from Applied Film Corp. PC61BM and PC71BM were purchased from Nanocarbon Corp. and used as received. The PSCs were fabricated as follows: glass substrates with patterned ITO electrodes were washed well and then cleaned through O2 plasma treatment. A thin film of the HTM, poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS, AI4083, Heraeus Clevios GmbH), was deposited on the ITO layer through spin-casting. The sample was dried at 150 °C for 30 min in a glove box. A solution of a mixture of PT and PC61BM (or PC71BM) (30 mg mL−1 in o-DCB) was stirred overnight, then filtered through a 0.2 μm polytetrafluoroethylene (PTFE) filter and spin-coated (1500 rpm, 30 s) onto the HTM layer to prepare the PT/PC61BM (or PC71BM) composite film-based photoactive layer. The sample was dried at 110 °C for 10 min in a glove box. In a high-vacuum chamber, the Ca/Al-based cathode was thermally deposited onto the PT/PC61BM (or PC71BM) composite layer. The active area of the PSC was 0.04 cm2. After electrode deposition, the PSC was encapsulated. The cathode deposition rate was determined using a quartz thickness monitor (STM-100/MF, Sycon). The thicknesses of the thin films were determined using a surface texture analysis system (3030ST, Dektak). The PV properties of the PSCs were measured using a programmable electrometer equipped with current and voltage sources (Keithley 2400) under illumination with solar-simulating light (100 mW cm−2) from an AM1.5 solar simulator (NewPort Oriel 96000).
3. Results and discussion
3.1 Characterization of PTs
The conjugated PTs were synthesized through Stille coupling of DG-TDPP (3), 2,5-bis(trimethylstannyl)thiophene (4), and tTPA-containing thiophene derivative (5). The chemical structures of the monomers were confirmed using 1H NMR spectroscopy and elemental analysis. 1H NMR spectra of compounds 3–5 and PTs are shown in Fig. S1 and S2,† respectively. Chemical shifts and relative intensities of the signals in the 1H NMR spectra were in agreement with the proposed structures of compounds 3–5 and PTs. The repeat unit ratios (x/y; see Scheme 1) of the PTs (PEGA11, PEGA12, and PEGA13) were modulated by controlling the feed ratio of the DG-TDPP and conjugated pendant tTPA-containing thiophene derivative 5. For the copolymers PEGA11, PEGA12, and PEGA13, the actual values of x/y were determined from the relative integral areas of their peaks at 6.20–7.60 ppm (representing protons of the vinylene, phenyl, and thiophene groups) and 3.10–3.80 [representing protons of di(ethylene glycol) groups] in the 1H NMR spectra. The values of x/y of PEGA11, PEGA12, and PEGA13 were approximately 1:1.25, 1:2.18, and 1:2.97, respectively. Using GPC with THF as the eluent and PS internal standards, we determined the number-average molecular weights (Mn) and weight-average molecular weights (Mw) of the conjugated PTs. As summarized in Table 1, the values of Mn and Mw of the conjugated polymers were in the ranges 6.5–13.5 and 14.5–28.4 kg mol−1, respectively. The average molecular weights of PEGA11 and PEGA12 were much lower than that of PEGA13. The early termination of the propagation of the polymer chains resulted from poor solubility of the polymers in the reaction solution and, accordingly, their precipitation. As a result, we observed relatively lower average molecular weights for PEGA11 and PEGA12. Relatively poorer solubility of PEGA11 and PEGA12 was due to their higher contents of DG-TDDP units. In fact, relatively poorer solubility of PT with higher DG-TDDP contents was attributed to the strong intermolecular hydrogen-bonding interactions between the lactam units.37 Nevertheless, these three conjugated copolymers were all soluble in common organic solvents, including THF, CH2Cl2, CHCl3, and o-DCB. Excellent solubility is favorable for the formation of the solution-processed PTs based photo-active layers.
Table 1 Average molecular weights and thermal properties of the PTs
| PT |
Mn (kg mol−1) |
Mw (kg mol−1) |
Tga (°C) |
Tdb (°C) |
| Tg: measured at a heating rate of 10 °C min−1. Td: temperature at which weight loss reached 5%. |
| PEGA11 |
6.5 |
14.5 |
113.4 |
319.8 |
| PEGA12 |
7.8 |
16.4 |
112.8 |
347.1 |
| PEGA13 |
13.5 |
28.4 |
120.9 |
398.5 |
The operational stability of a PSCs is directly related to the thermal stability of its component conjugated polymer. Thus, high values of Tg and Td are desirable. We measured the values of Tg and Td of our conjugated copolymers by means of thermal analyses using DSC and TGA (Table 1). The values of Td for PEGA13, PEGA12, and PEGA11 were 398.5, 347.1, and 319.8 °C, respectively. The thermal stability of the PTs decreased upon increasing the DG-TDPP content. Moreover, we determined glass transition temperatures from the second round of DSC heating scans. The values of Tg for PEGA13, PEGA12, and PEGA11 were 120.9, 112.8, and 113.4 °C, respectively, making them suitable for PSC applications. Relatively low values of Tg resulted from the polymers having higher contents of flexible di(ethylene glycol)-substituted diketopyrrolopyrrole units. In addition, we observed a single endothermic glass transition for each conjugated copolymer, implying that the diketopyrrolopyrrole conjugated units and electron-donating bulky tTPA-containing pendant moieties were distributed homogeneously.
3.2 Optical properties of PTs
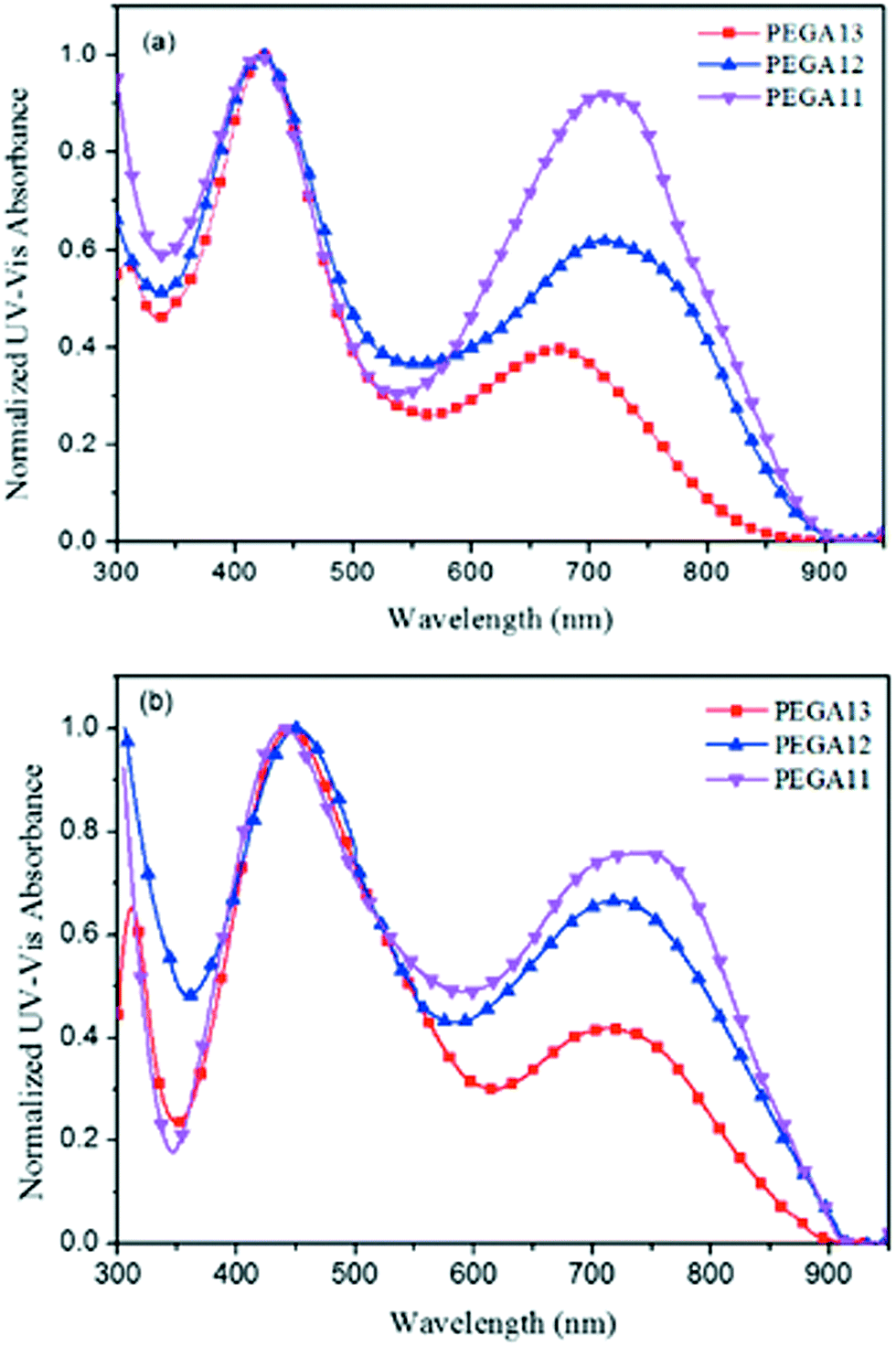
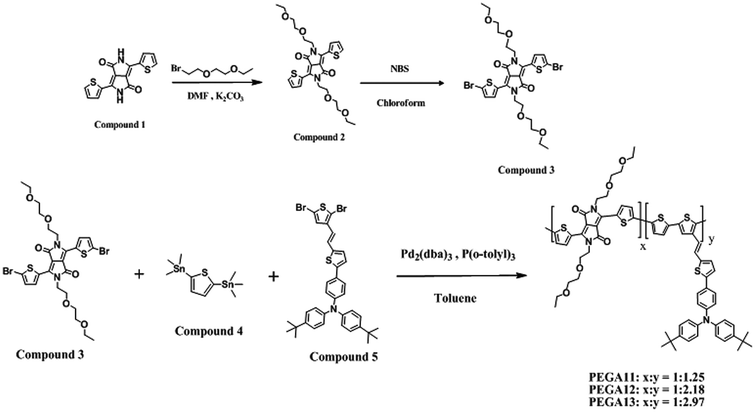
Fig. 1 presents normalized UV-Vis absorption spectra of the DG-TDPP-containing PTs as solutions in o-DCB and as solid films. Table 2 summarizes their photophysical properties. The absorptions of the DG-TDPP-containing PTs in o-DCB ranged from 325 to 900 nm; each polymer displayed two broad absorption bands. We attribute the first absorption band, in the range 350–550 nm, to the n–π* transitions of the conjugated side chains and the π–π* transitions of the conjugated main chains. The second absorption band, in the range 550–900 nm, originated from intramolecular charge transfer (ICT) between the thiophene-based donor units and DG-TDPP-based acceptor units.33,34 The absorption intensities of the second absorption band increased upon increasing the content of DG-TDPP units in the copolymers, both in solution and as thin films. Moreover, the red-shifts and full widths at half-maximum of the absorption bands of the copolymers in their film forms were greater than those in solution, presumably because of strong noncovalent interactions (e.g., π-stacking) between the polymer backbones and conjugated pendant units.27,36
 |
| | Fig. 1 Normalized UV-Vis absorption spectra of PTs (a) in o-DCB solution and (b) as thin films. | |
Table 2 Optical properties, electrochemical onset potentials, and electronic energy levels of the PTs
| PT |
λabsmaxa (nm) |
λabsmaxb (nm) |
Eoptgc (eV) |
Eoxon (V) |
HOMO (eV) |
LUMO (eV) |
| Maximum absorption wavelength of polymer in solution. Maximum absorption wavelength of polymer as thin film. Calculated value of Eg from the onset absorption (λabsonset) of polymer thin film: Eg = 1240/λonset. |
| PEGA11 |
420, 717 |
445, 740 |
1.40 |
0.40 |
−5.11 |
−3.71 |
| PEGA12 |
423, 715 |
450, 722 |
1.41 |
0.38 |
−5.09 |
−3.68 |
| PEGA13 |
426, 673 |
447, 720 |
1.47 |
0.26 |
−4.97 |
−3.50 |
We determined the band gap energies (Eg) of the conjugated polymers in their thin film states from the onset wavelengths of their absorption bands. Table 2 reveals that the values of Eg for PEGA11, PEGA12, and PEGA13 were 1.40, 1.41, and 1.47 eV, respectively. The lower values of Eg were observed for the copolymers having higher contents of DG-TDPP units. In general, a broader absorption range and a lower band gap energy will improve the efficiency of solar light absorption of the photo-energy conversion layer in a PSC, resulting in the generation of a larger photocurrent.
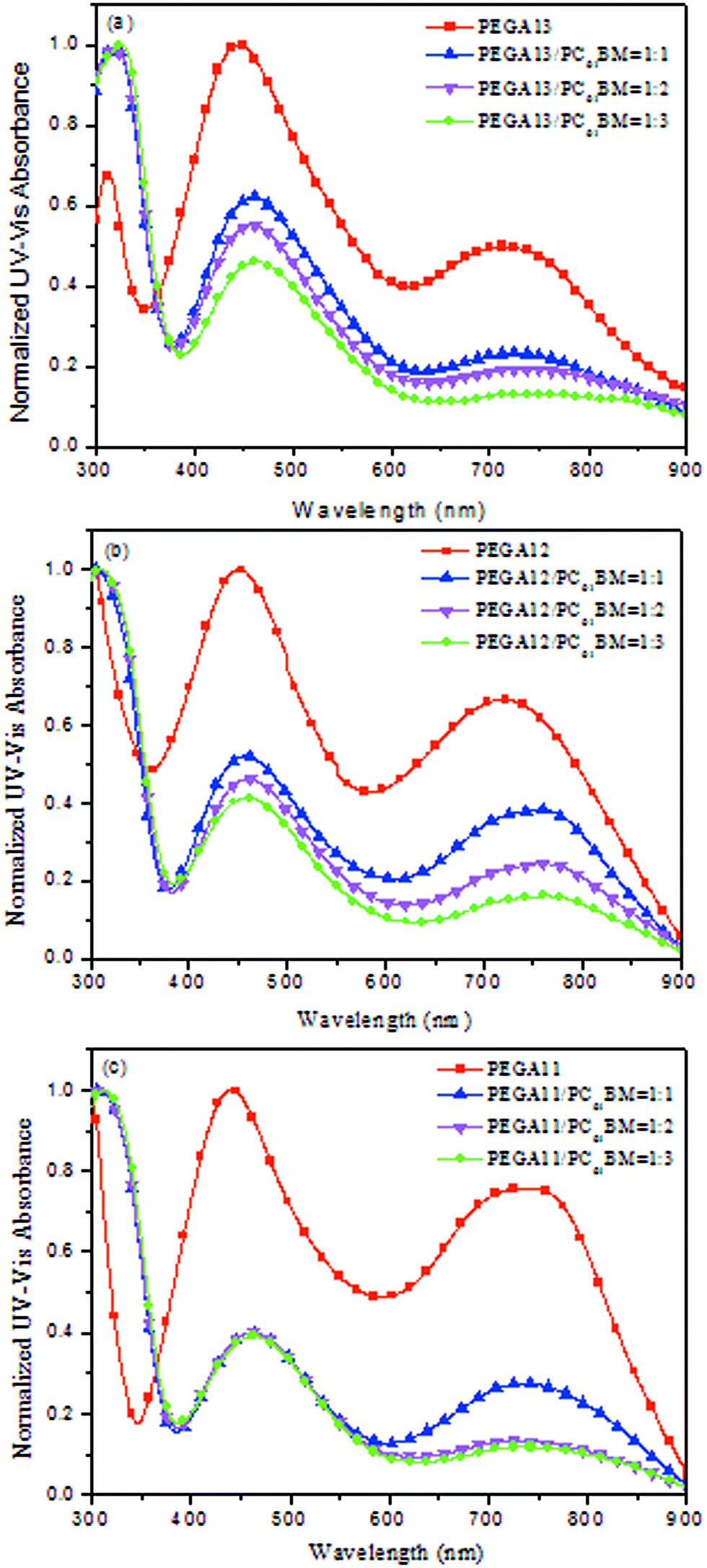
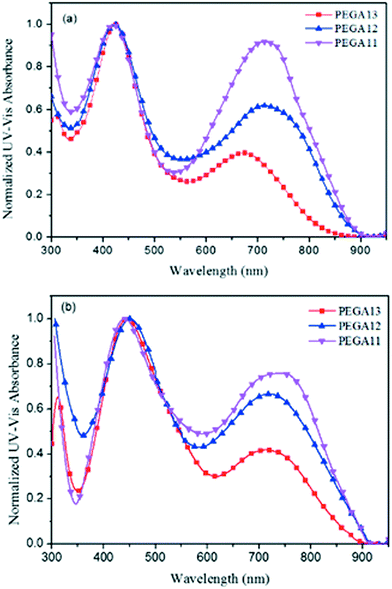
Fig. 2 displays normalized UV-Vis absorption spectra of the PT/PC61BM composite films. The absorption bands of the DG-TDPP-containing copolymers ranged from 375 to 900 nm, while the absorption band of PC61BM ranges from 300 to 375 nm. The maximum absorption peaks of the conjugated copolymer and PC61BM appeared near 425 and 325 nm, respectively. Moreover, the absorption intensity of the conjugated polymer decreased as the PC61BM content increased for these PT/PC61BM composite films. From the viewpoint of photon absorption, employing less PC61BM in the photoactive layer would be preferred. Nevertheless, the typical stoichiometry of polymer/PC61BM blends, optimal for devices in several PSC systems, ranges from 1:1 to 1:4 by weight.38 A high proportion of PC61BM limits the optical absorption in the composite layer because the absorption of PC61BM is quite inefficient in the visible region.
 |
| | Fig. 2 Normalized UV-Vis absorption spectra of (a) PEGA13/PC61BM, (b) PEGA12/PC61BM, and (c) PEGA11/PC61BM blend films. | |
3.3 Electrochemical properties of PTs
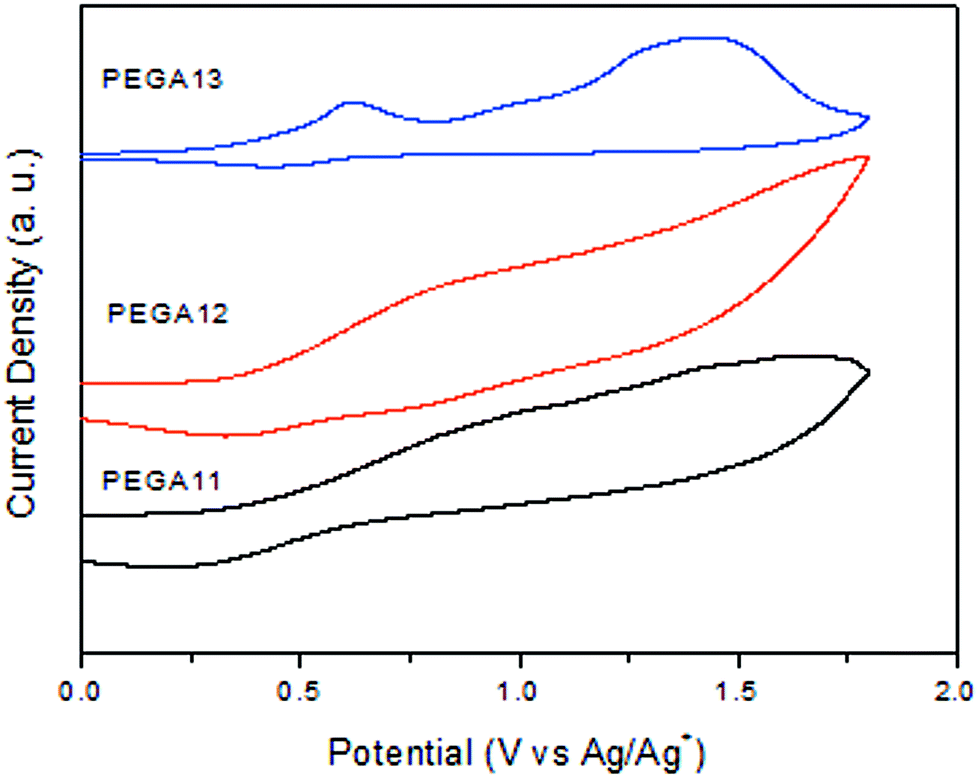
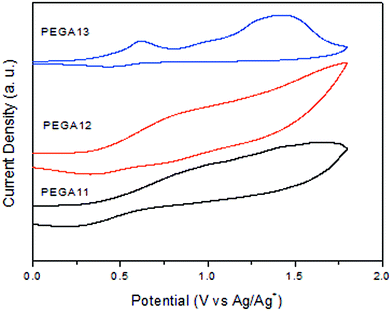
Because the PV performance of a PSC is related to the electrochemical behavior of its conjugated polymer, we employed CV to investigate the electrochemical behavior of our PTs and estimate the energy levels of their HOMOs. Fig. 3 presents the oxidation behavior in the CV curves of the PTs. Table 2 summarizes the electrochemical properties of the copolymers. The oxidation potentials (Eoxon) of PEGA11, PEGA12, and PEGA13 were 0.40, 0.38, and 0.26 V, respectively. From those values, we calculated the HOMO energy levels of the copolymers according to the equation
| HOMO = −e(Eoxon − Eoxon, ferrocene + 4.71) (eV) |
where 4.71 eV is the energy level of ferrocene below the vacuum level and the value of Eoxon of ferrocene/ferrocene+ is 0.09 V in 0.1 M Bu4NClO4/MeCN. The HOMO energy levels obtained for PEGA11, PEGA12, and PEGA13 were −5.11, −5.09, and −4.97 eV, respectively. Lower HOMO energy levels were observed for the PTs having lower contents of tTPA-containing conjugated pendant units, suggesting lower electron-donating abilities for these polymers.39 In addition, because no reversible n-doping process was evident in the CV spectra, we estimated the LUMO energy levels from the HOMO energy levels and the values of Eg determined from the UV-Vis absorption spectra, using the equation
 |
| | Fig. 3 Cyclic voltammograms of PEGA11, PEGA12, and PEGA13. | |
The calculated LUMO energy levels were −3.71 eV for PEGA11, −3.68 eV for PEGA12, and −3.50 eV for PEGA13. Lower LUMO levels were obtained for the PTs having higher contents of electron-deficient (i.e., DG-TDPP) units. Thus, the electrochemical properties of these PTs could be tuned by incorporating electron-accepting DG-TDPP units into the polymer backbone. In general, the HOMO energy level of a p-type conjugated polymer is an important parameter affecting the performance of BHJ-type cells. High open-circuit voltages (VOC) are typically obtained for PSCs fabricated from conjugated polymers having low HOMO energy levels.12
3.4 Morphology of thin films of PT/PC61BM blends
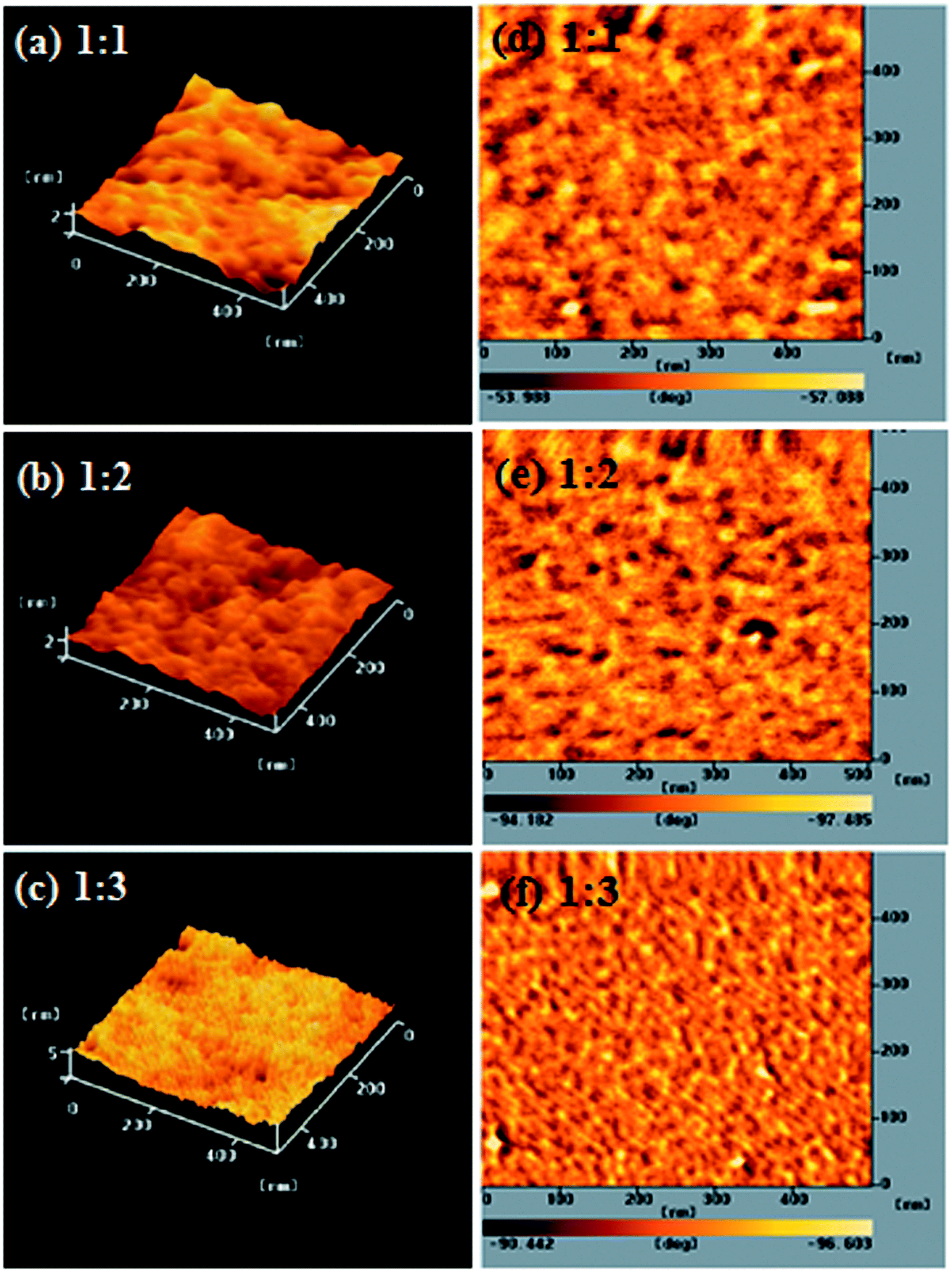
The performance of a PSC is strongly dependent on the morphology of the film of its conjugated polymer/fullerene derivative composite. To avoid recombination of excitons, the P/N heterojunction phase must be controlled at the nanoscale level.27,36 We used AFM microscopy to investigate the compatibility and morphology of our conjugated polymer/PC61BM composite films. Fig. 4 displays topographic and phase-contrast images of PEGA13/PC61BM composite films (1:1, 1:2, and 1:3, w/w) after annealing at 110 °C for 10 min; Fig. S3 and S4 (ESI†) present the corresponding the topographic and phase-contrast images of the PEGA11/PC61BM and PEGA12/PC61BM composite films (1:1, 1:2, and 1:3, w/w). The phase-contrast images indicated that the distribution of the PC61BM units in the polymers was uniform for the DG-TDPP-containing PT/PC61BM composite films. In each case, we observed a phase-separated interpenetrating network with sizable PC61BM domains. Some degree of phase separation is critical for efficient formation of free carriers to provide PSCs with optimal PV properties. Moreover, the sizes of the phase separation domains decreased upon increasing the PC61BM content. The nanoscale phase separation was more obvious for the PT/PC61BM composite film having the higher PC61BM content (1:3, w/w). Larger interface area will be obtained for PT/PC61BM composite films having larger PC61BM contents and smaller domain sizes.
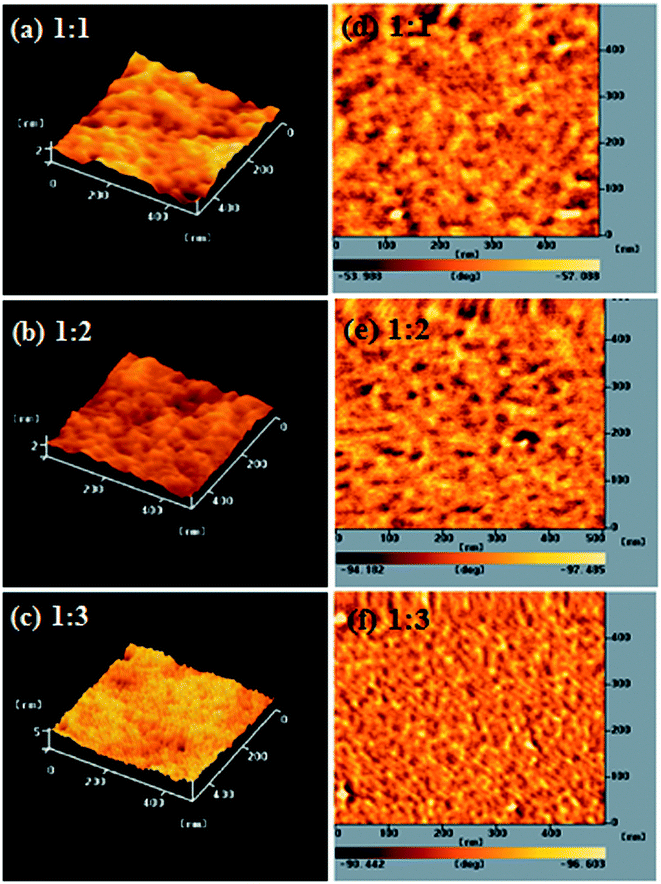 |
| | Fig. 4 AFM (tapping mode) (a–c) topographic and (d–f) phase images of PEGA13/PC61BM blend films [(a and d) 1:1, w/w; (b and e) 1:2, w/w; (c and f) 1:3, w/w] after annealing at 110 °C for 10 min. | |
3.5 PV properties of PSCs incorporating PT/PC61BM films
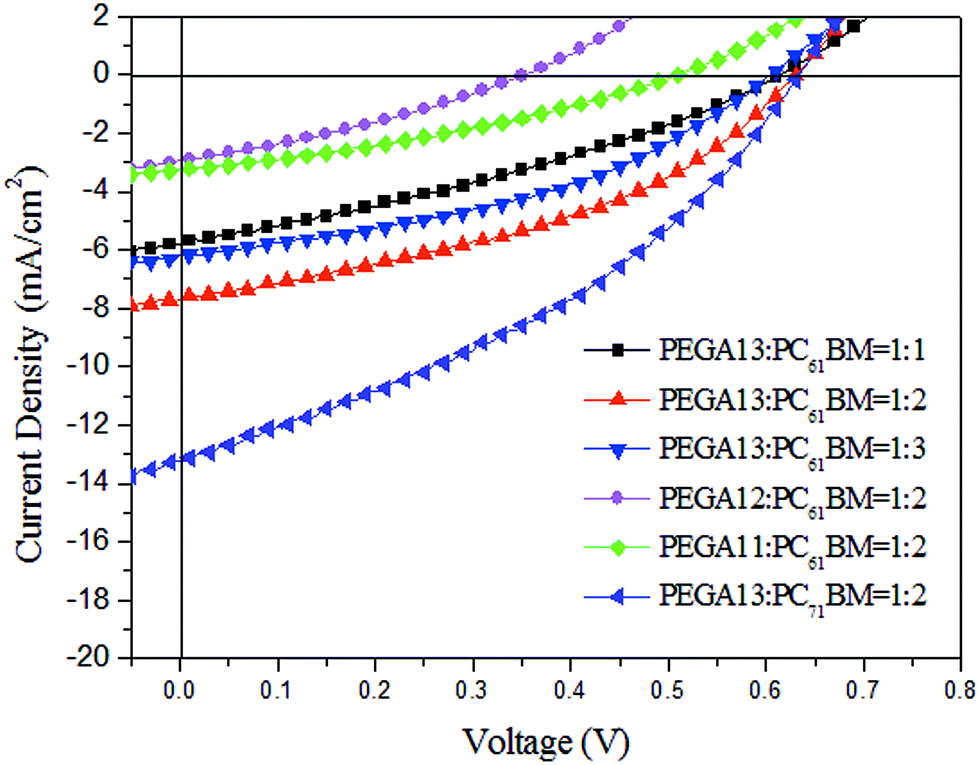
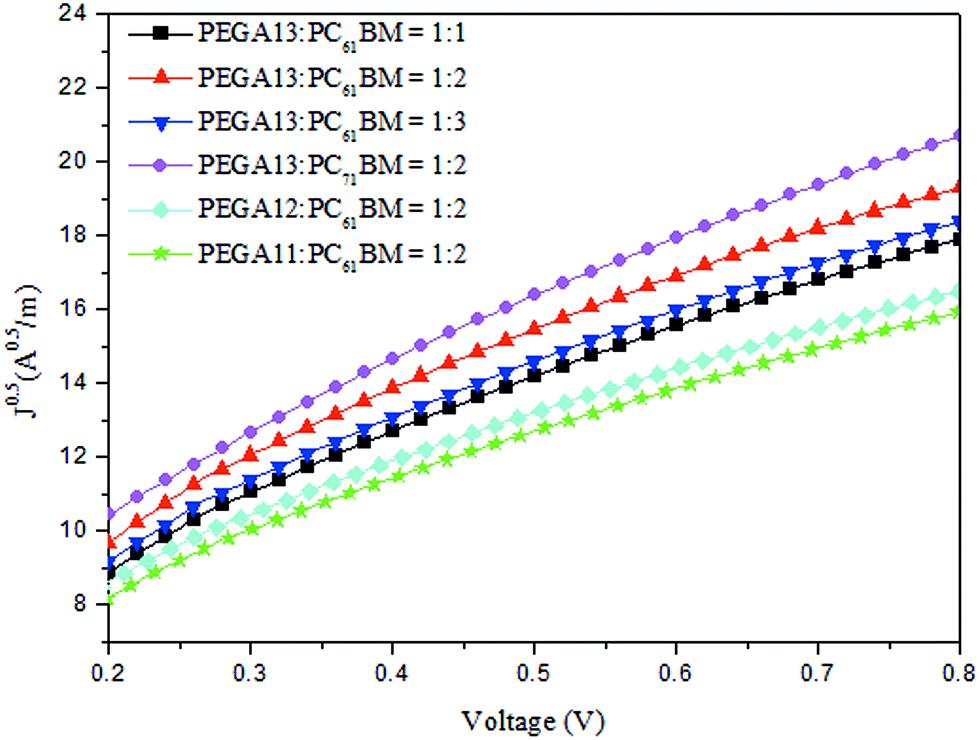
We used an optimized spin-coating procedure to fabricate PSCs incorporating PT/PC61BM blends as photoactive layers. Fig. 5 displays the photocurrent density–voltage plots of these PSCs; Table 3 summarizes the PV properties of these PSCs, including their values of VOC, JSC, fill factor (FF), and photoconversion efficiency (η). For the PEGA13/PC61BM-based PSCs (PSC I-1–PSC I-3), the value of VOC did not change significantly in response to the content of PC61BM. Moreover, the values of JSC, FF, and η of the PSCs increased upon increasing the PC61BM content. The highest values of JSC and η were observed for the PSC based on a photoactive layer of PEGA13/PC61BM at a weight ratio of 1:2. Higher concentrations of PC61BM in the photoactive layer favored the formation of phase-separated interpenetrating networks with sizable domains, which in turn led to effective charge separation and charge transfer.27 Typically, efficient dissociation of excitons and higher degrees of charge collection to the electrode are favorable for PSCs displaying enhanced values of JSC. Nevertheless, an excessive content of PC61BM does not favor the charge transfer behavior and PV performance of PSCs. The PV performance of PSC I-3 was poorer than that of PSC I-2. This finding implies that the optimal PT-to-PC61BM composition was approximately 1:2 (w/w) for the PEGA13-based PSCs. In addition, the hole mobility of a photoactive layer also plays an important role affecting the PV performance of a PSC. We used the space-charge limited current method to measure the hole mobilities of our PT/PC61BM blend films, with a typical hole-only device structure of ITO/PEODT/PT:PC61BM (or PC71BM)/Au.40 Fig. 6 presents the results plotted as the current density (J) with respect to voltage (V). Table 3 summarizes the hole mobilities of the blend films. The hole mobility of the 1:2 (w/w) PEGA13/PC61BM blend film (2.04 × 10−5 cm2 V−1 s−1) was greater than those of the corresponding 1:1 (1.79 × 10−5 cm2 V−1 s−1) and 1:3 (1.86 × 10−5 cm2 V−1 s−1) blend films. The relatively higher hole mobility of the PEGA13/PC61BM (1:2, w/w) blend film resulted in the better PV performance of PSC I-2. On the other hand, the PV properties of the PEGA11- and PEGA12-based PSCs (PSC II and PSC III, respectively) were much poorer than those of the PEGA13-based PSCs, reflecting the lower average molecular weights and poorer hole mobilities of PEGA11 and PEGA12. The hole mobilities of PEGA11/PC61BM and PEGA12/PC61BM were lower than that of PEGA13/PC61BM (Table 3).
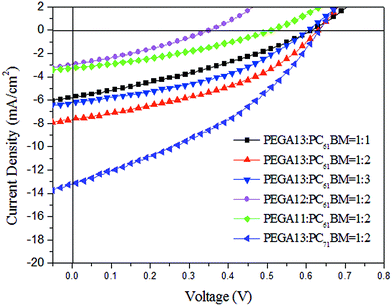 |
| | Fig. 5 Current density–potential characteristics of illuminated (AM 1.5G, 100 mW cm−2) PSCs incorporating (PEGA13, PEGA12, PEGA11)/PC61BM and PEGA13/PC71BM blends. | |
Table 3 Photovoltaic performance of PSCs incorporating films of (PEGA13, PEGA12, PEGA11)/PC61BM and PEGA13/PC71BM blends
| PSC |
Photoactive layer |
PT/PC61BM (or PC71BM) (w/w) |
VOC (V) |
JSC (mA cm−2) |
FF |
η (%) |
Mobility (10−5 cm2 V−1 s−1) |
| PSC I-1 |
PEGA13/PC61BM |
1:1 |
0.61 |
5.8 |
0.32 |
1.1 |
1.79 |
| PSC I-2 |
PEGA13/PC61BM |
1:2 |
0.63 |
7.7 |
0.41 |
2.0 |
2.04 |
| PSC I-3 |
PEGA13/PC61BM |
1:3 |
0.60 |
6.7 |
0.40 |
1.6 |
1.86 |
| PSC II |
PEGA12/PC61BM |
1:2 |
0.35 |
2.9 |
0.32 |
0.3 |
1.54 |
| PSC III |
PEGA11/PC61BM |
1:2 |
0.51 |
3.2 |
0.33 |
0.5 |
1.36 |
| PSC IV |
PEGA13/PC71BM |
1:2 |
0.67 |
9.0 |
0.42 |
2.5 |
3.17 |
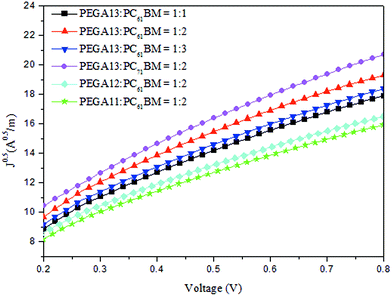 |
| | Fig. 6 Plots of (J)0.5 vs. V of hole-only devices incorporating PT/PC61BM and PEGA13/PC71BM blends. | |
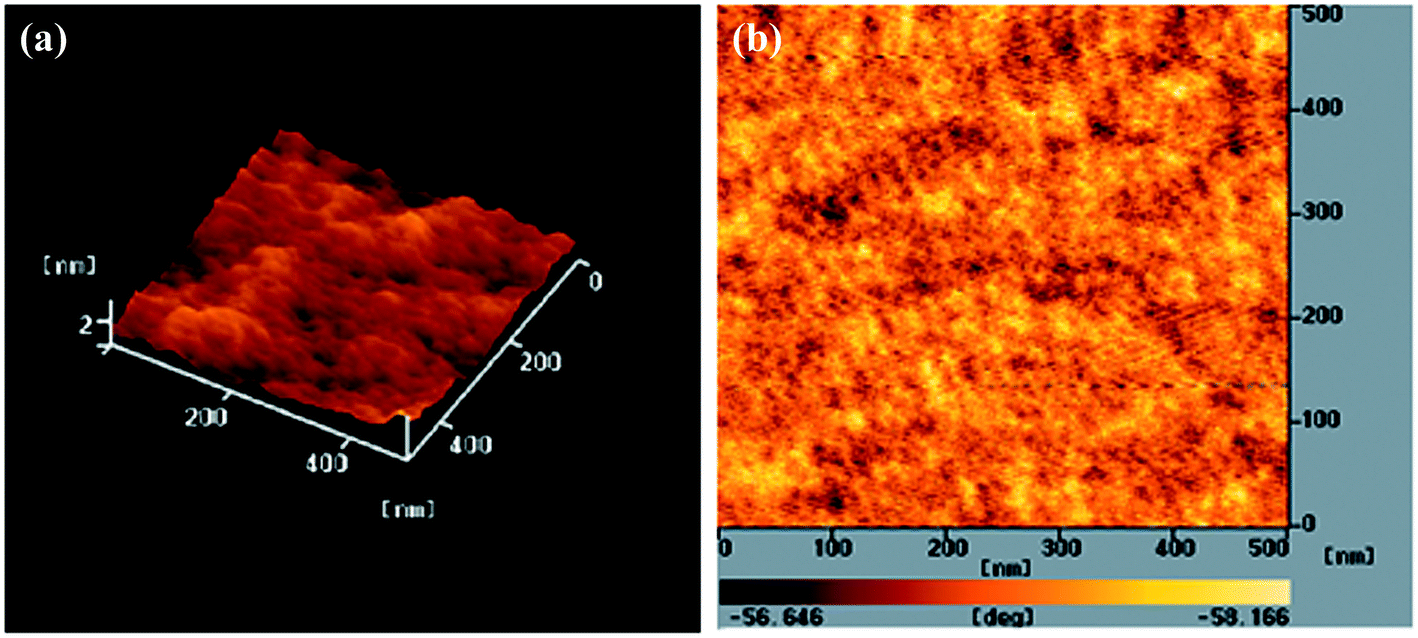
To further improve the PV performance of the PEGA13-based PSCs, we introduced PC71BM as the electron-acceptor in the photoactive layers of the PSCs. Fig. 5 displays the photocurrent density–voltage plots of the PSCs based on the PEGA13/PC71BM blend film (PSC IV). Table 3 summarizes the PV properties of these PSCs. Because the optimal composition of the PEGA13/PC61BM blend was 1:2 (w/w), we studied the PSC IV based on a PEGA13/PC71BM blend also having a composition of 1:2 (w/w). The AFM images in Fig. 7 indicate that phase-separated interpenetrating networks with sizable domains were formed in the photoactive layer for the PEGA13/PC71BM blend film. In addition, we used TEM to further characterize the distribution of PC71BM in the PT/PC71BM films. Fig. 8 presents the TEM image of the PEGA13/PC71BM blend film (1:2, w/w). The dark areas in the image represent PC71BM domains, because the electron scattering density of PC71BM is greater than that of the conjugated polymer. The formation of phase-separated interpenetrating networks with sizable domains in the photoactive layer was confirmed. The hole mobility of the 1:2 (w/w) PEGA13/PC71BM blend film was approximately 3.17 × 10−5 cm2 V−1 s−1 (Table 3). As displayed in Fig. 5, we observed much higher values of JSC and PCE for the PSCs based on the PEGA13/PC71BM blend films, as compared with those based on PT/PC61BM blends, presumably because of the stronger and broader absorption of PC71BM in the visible region and the greater hole mobility of the PEGA13/PC71BM blend film.27 Apart from that, PV performance of the PEGA13/PC71BM based PSC was much better than that of the r-TPATh-PT/PC71BM based PSC.24 As compared to the r-TPATh-PT, the incorporation of the electron-deficient DG-TDPP units onto the polymer backbone resulted in a broader absorption band for PEGA13. Therefore, better PV performance was observed for the PEGA13 based PSC.
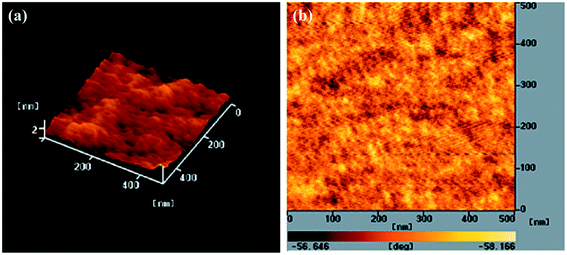 |
| | Fig. 7 AFM (tapping mode) (a) topographic and (b) phase images of the PEGA13/PC71BM (1:2, w/w) blend film after annealing at 110 °C for 10 min. | |
 |
| | Fig. 8 TEM image of the PEGA13/PC71BM (1:2, w/w) blend film after annealing at 110 °C for 10 min. | |
4. Conclusion
We have synthesized a series of PTs featuring di(ethylene glycol)-substituted 2,5-thienyl diketopyrrolopyrrole as conjugated units in the polymer backbones and tTPA-containing moieties as pendant units. Incorporating the electron-deficient DG-TDPP moieties into the polymer backbone and appending the tTPA units promoted charge balance and efficient conjugation within the extended conjugated frameworks of the polymers. The absorption ability and hole mobility of the PTs both improved upon optimization of the molar ratio of the tTPA-based electron-donating pendant units and the electron-withdrawing DG-TDPP units in the polymer chains. The PV performance of the PEGA13-based PSCs was better than those of the PEGA12- and PEGA11-based PSCs.
Acknowledgements
We thank the Ministry of Science and Technology (MOST) of Taiwan for financial support.
References
- M. Helgesen, R. Sondergaard and F. C. Krebs, J. Mater. Chem., 2010, 20, 36 RSC.
- J. D. Chen, C. Cui, Y. Q. Li, L. Zhou, Q. D. Ou, C. Li, Y. F. Li and J. X. Tang, Adv. Mater., 2015, 27, 1035 CrossRef CAS PubMed.
- Y. W. Su, W. H. Lin, Y. J. Hsu and K. H. Wei, Small, 2014, 10, 4427 CrossRef CAS PubMed.
- Y. W. Su, S. C. Lan and K. H. Wei, Mater. Today, 2012, 15, 554 CrossRef CAS.
- M. H. Chen, J. Hou, Z. Hong, G. Yang, S. Sista, L. M. Chen and Y. Yang, Adv. Mater., 2009, 21, 4238 CrossRef CAS.
- B. Xu, Z. Zheng, K. Zhao and J. Hou, Adv. Mater., 2016, 28, 434 CrossRef CAS PubMed.
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. C. Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H. Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702 CrossRef CAS PubMed.
- J. Wang, M. Xiao, W. Chen, M. Qiu, Z. Du, W. Zhu, S. Wen, N. Wang and R. Yang, Macromolecules, 2014, 47, 7823 CrossRef CAS.
- L. Wang, S. Shi, D. Ma, S. Chen, C. Gao, M. Wang, K. Shi, Y. Li, X. Li and H. Wang, Macromolecules, 2015, 48, 287 CrossRef CAS.
- Y. R. Cheon, Y. J. Kim, J. J. Ha, M. J. Kim, C. E. Park and Y. H. Kim, Macromolecules, 2014, 47, 8570 CrossRef CAS.
- A. J. Heeger, Adv. Mater., 2014, 26, 10 CrossRef CAS PubMed.
- M. C. Scharber, D. Wuhlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. L. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- C. Cui, W. Y. Wong and Y. F. Li, Energy Environ. Sci., 2014, 7, 2276 CAS.
- Y. Liang, Z. Xu, J. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Energy Mater., 2010, 22, E135 CrossRef CAS PubMed.
- F. Liu, Y. Gu, X. Shen, S. Ferdous, H. W. Wang and T. P. Russell, Prog. Polym. Sci., 2013, 38, 1990 CrossRef CAS.
- R. H. Lee and L. Y. Lee, Colloid Polym. Sci., 2011, 289, 1215 CAS.
- G. Li, C. Kang, X. Gong, J. Zhang, C. Li, Y. Chen, H. Dong, W. Hu, F. Li and Z. Bo, Macromolecules, 2014, 47, 4645 CrossRef CAS.
- C. Duan, K. S. Chen, F. Huang, H. L. Yip, S. Liu, J. Zhang, A. K. Y. Jen and Y. Cao, Chem. Mater., 2010, 22, 6444 CrossRef CAS.
- Y. Li, H. Xia, B. Xu, S. Wen and W. Tian, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3970 CrossRef CAS.
- Y. T. Chang, S. L. Hsu, M. H. Su and K. H. Wei, Adv. Mater., 2009, 21, 2093 CrossRef CAS.
- Z. G. Zhang, S. Y. Zhang, J. Min, C. H. Chui, J. Zhang, M. J. Zhang and Y. F. Li, Macromolecules, 2012, 45, 113 CrossRef CAS.
- J. H. Tsai, W. Y. Lee, W. C. Chen, C. Y. Yu, G. W. Hwang and C. Ting, Chem. Mater., 2010, 22, 3290 CrossRef CAS.
- Z. Gu, P. Shen, S. W. Tsang, Y. Tao, B. Zhao, P. Tang, Y. Nie, Y. Fang and S. Tan, Chem. Commun., 2011, 47, 9381 RSC.
- H. J. Wang, L. H. Chan, C. P. Chen, S. L. Lin, R. H. Lee and R. J. Jeng, Polymer, 2011, 52, 326 CrossRef CAS.
- N. Chakravarthi, K. Gunasekar, C. S. Kim, D. H. Kim, M. Song, Y. G. Park, J. Y. Lee, Y. Shin, I. N. Kang and S. H. Jin, Macromolecules, 2015, 48, 2454 CrossRef CAS.
- S. Mei, F. Wu, Y. Huang, B. Zhao and S. Tan, Eur. Polym. J., 2015, 67, 31 CrossRef CAS.
- H. J. Wang, J. Y. Tzeng, C. W. Chou, C. Y. Huang, R. H. Lee and R. J. Jeng, Polym. Chem., 2013, 4, 506 RSC.
- S. Li, J. Yuan, P. Deng, W. Ma and Q. Zhang, Dyes Pigm., 2014, 106, 121 CrossRef CAS.
- D. F. Zeigler, K. A. Mazzio and C. K. Luscombe, Macromolecules, 2014, 47, 5019 CrossRef CAS.
- P. Zhou, D. Dang, J. Fan, W. Xiong, C. Yang, H. Tan, Y. Wang, Y. Liu and W. Zhu, Dyes Pigm., 2015, 112, 99 CrossRef CAS.
- Y. R. Liu, L. H. Chan and H. Y. Tang, J. Polym. Sci., Polym. Chem. Ed., 2015, 53, 2878 CrossRef CAS.
- K. Kranthiraja, K. Gunasekar, W. Cho, M. Song, Y. G. Park, J. Y. Lee, Y. Shin, I. N. Kang, A. Kim, H. Kim, B. Kim and S. H. Jin, Macromolecules, 2014, 47, 7060 CrossRef CAS.
- D. Dang, P. Zhou, Q. Peng, K. He, H. Jiang, P. Yang, H. Tan, Y. Wang, Y. Liu, G. Lei and W. Zhu, Dyes Pigm., 2014, 109, 6 CrossRef CAS.
- H. Liang, X. Zhang, R. Peng, X. Ouyang, Z. Liu, S. Chen and Z. Ge, Dyes Pigm., 2015, 112, 145 CrossRef CAS.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Sol. Energy Mater. Sol. Cells, 2011, 95, 1168 CrossRef CAS.
- S. Y. Shiau, C. H. Chang, W. J. Chen, H. J. Wang, R. J. Jeng and R. H. Lee, Dyes Pigm., 2015, 115, 35 CrossRef CAS.
- M. A. Naik, N. Venkatramaiah, C. Kanimozhi and S. Patil, J. Phys. Chem. C, 2012, 116, 26128 CAS.
- R. H. Lee and L. W. Liu, Dyes Pigm., 2010, 84, 190 CrossRef CAS.
- O. Kwon, S. Barlow, S. A. Odom, L. Beverina, N. J. Thompson, E. Zojer, J. L. Brédas and S. R. Marder, J. Phys. Chem. A, 2005, 109, 9346 CrossRef CAS PubMed.
- C. Y. Huang, W. H. Lee and R. H. Lee, RSC Adv., 2014, 4, 48150 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26245g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b
b