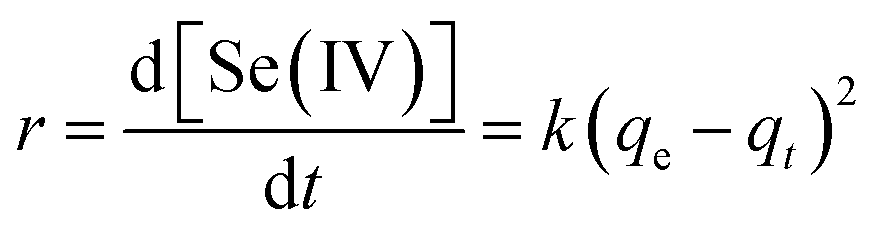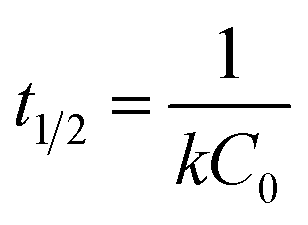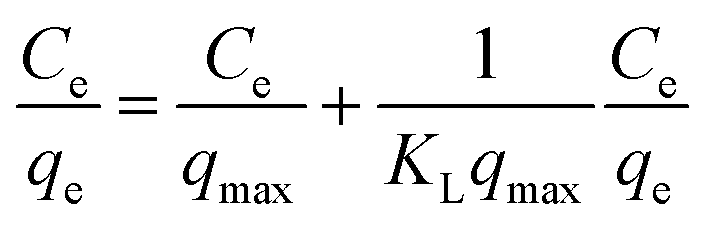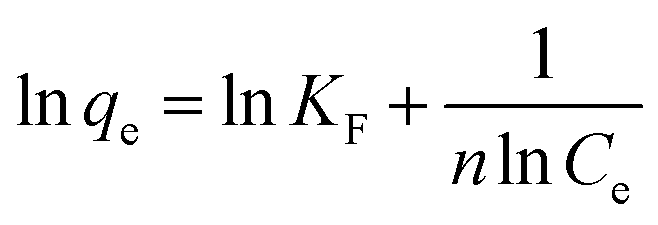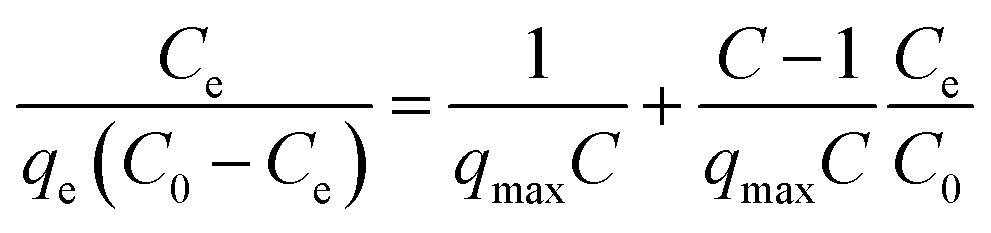DOI:
10.1039/C6RA26225B
(Paper)
RSC Adv., 2017,
7, 13398-13405
Immobilization of selenite from aqueous solution by structural ferrous hydroxide complexes†
Received
3rd November 2016
, Accepted 9th January 2017
First published on 27th February 2017
1. Introduction
Selenium (Se) is a fission-product nuclide in nuclear reactions. It has a half-life of 2.95 × 105 years. Se has chemical and radiant dual-toxicity, and is a high-level radioactive waste in geological disposal systems.1,2 Se is an indispensable element in humans, flora and fauna. Humans and animals are very sensitive to its intake, and inappropriately high intake is harmful and toxic. A daily intake of Se > 400 mg is toxic for humans.3 The average concentration of Se is in the ng L−1 range in water and is 0.4 mg kg−1 in soil.4 Se(IV) originates from the products of agriculture, mining, smelting and coal-burning industries, and can be released from the natural weathering of Se-rich black shales and other geological deposits.5–7 Se is an element with various valence states (−II, −I, 0, +IV, +VI), and Se(−II), Se(−I) and Se(0) (elemental Se) are the dominant species in reducing conditions. Se(IV) mainly exists in weak oxidizing conditions and Se(VI) exists in strong oxidizing conditions. According to Eh-pH phase diagrams,8 the dissolved states of Se(IV) are SeO32−, HSeO3− and H2SeO3 (aq). HSeO3− is the principal species (pH = 2.64–8.36). The dissolved state of Se(VI) is dominantly SeO42−, and HSeO4− is the predominant species (pH < 2). The principal dissolved state of Se(−II) is HSe−, and H2Se (aq) is the main form (pH < 4). As reported by the Environmental Protection Agency (FRL-5649-7), the acute toxicity of Se(IV) is nearly tenfold higher than that of Se(VI) when they simultaneously coexist in surface water with the same concentration in the presence of oxygen.9
The chemical properties of Se solutions are considerably unique, with extremely strong mobility that is not influenced by solubility, and poor absorption by clay or granite. Therefore, reduction from high valence Se(IV) to Se(0) deposited into sediment is the primary reason for the hindrance of Se mobility. Fe(II)-mineral is a type of ubiquitous mineral that is reducible in nature, but there are others, including magnetite, iron pyrite, pyrrhotite and siderite. Retardation of selenite reduction by Fe(II)-mineral has aroused extensive concern.2,9–13 Studies have shown that zero-valent iron and nano zero-valent iron14,15 exhibit a low removal rate on Se because dense iron oxides can form on the surface and lead to low reaction activity. However, ferrous hydroxyl complexes (FHCs) can achieve good performance in this respect. For example, reductive transformation of reactive black 5 by FHCs has shown that the latter can reduce azo dyes by cleavage of chemical bonds and generate aniline.16 Owing to the low removal efficiency, it is hard to introduce these natural Fe(II)-minerals into environmental engineering. It is well-known that Se(IV) in the environment is absorbed on the surface of zero-valent iron with chemisorption, and is gradually reduced to Se(0), Se(−I) or Se(−II). A similar reduction mechanism has been mentioned by immobilization of selenite on Fe3O4 and Fe/Fe3C ultra-small particles.9,17 The mechanism of Fe–Mn hydrate application on Se(IV) involves loading of Se(IV) onto the active site of the amorphous form of Fe(III)–Mn(III) hydrate, which forms an inner-orbital coordination compound (bidentate binuclear). Meanwhile, the active sites on the surface of Fe(III)–Mn(III) hydrate increase with decreasing pH value.18 The green rusts (GRs) formed in the reaction system of OH− and Fe(II) or Fe(III) belong to layered double hydroxides (LDHs). Myneni et al.19 studied the reducing effect of GRs on selenite and estimated that GR(SO42−) could reduce selenate to selenite with Se(0) and a small amount of Se(−II). The mechanism was found to involve physical absorption (ion exchange and ion coexistence) due to the LDH structure of GR. Meanwhile, chemisorptions of Se(IV) and bidentate binuclear iron polyhedrons occur in the pH range 7.0–9.3, which is accompanied by the production of Se(0) and FeSex without Fe2(SeO3)3.
FHCs in the presence of a structural Fe(II) complex possess high reduction activation. Wu et al.16,20 evaluated the disposal of organic pollutants by structural Fe(II). They also discovered that structural Fe(II) had better removal efficiency at lower cost than GRs or siderite.21 However, few studies have focused on Se(VI) removal by structural Fe(II) from aqueous solution, and the mechanism for the removal of Se(IV) by structural Fe(II) has not been studied. Therefore, the objectives of this study were to: (1) investigate the removal efficiency of Se(IV) with various dosages of structural Fe(II) and reaction kinetics, (2) explore the effect of various ([Fe(II)]/[OH−]) ratios by structural Fe(II) on removal of Se(IV), and (3) clarify the envelopes of Se(IV) reduction removal by structural Fe(II).
2. Materials and methods
2.1. Materials
All reagents were of analytical grade and were used without further purification. Ultrapure water (18.2 MΩ cm−1, Millipore, USA) was used for all experiments. The water was deoxygenated by purging with high-purity nitrogen for >30 min. Se(IV) stock solution was prepared with K2SeO3 in an amber bottle with deoxygenated ultrapure water.
Structural Fe(II) was synthesized by precipitation from Fe(II) sulfate salts with sodium hydroxide (NaOH). Briefly, FeSO4·7H2O (0.1 M) was dissolved in 80 mL deoxygenated ultrapure water in a 100 mL flask under magnetic stirring. Then, 100 mL of NaOH (2.5 M) solution, corresponding to [Fe(II)]/[OH−] ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1, 1:2, 1:3, and 1:4, respectively, was added to the solution under the same magnetic-stirring and anaerobic conditions. Finally, structural Fe(II) was generated, with ratios referred to as FHC(2:1), FHC(1:1), FHC(1:2), FHC(1:3), and FHC(1:4). All preparation work was performed in an anoxic atmosphere. Note that structural Fe(II) was freshly prepared to avoid possible oxidation.
:1, 1:1, 1:2, 1:3, and 1:4, respectively, was added to the solution under the same magnetic-stirring and anaerobic conditions. Finally, structural Fe(II) was generated, with ratios referred to as FHC(2:1), FHC(1:1), FHC(1:2), FHC(1:3), and FHC(1:4). All preparation work was performed in an anoxic atmosphere. Note that structural Fe(II) was freshly prepared to avoid possible oxidation.
2.2. Batch experiments on selenite removal and equilibrium
Adsorption kinetics experiments were performed in glass bottles (100 mL) containing 80 mL of selenite solution (30.0 mg L−1) with specific levels of adsorbent (228.0 mg L−1). Individual bottles were sacrificed after specific time intervals (5, 10, 20, 30, and 60 min) and immediately filtered using a 0.45 μm membrane.
For the sake of simplicity of computation, equilibrium experiments were performed by adding 200.0 mg L−1 adsorbent into a series of 100 mL bottles filled with 80 mL selenite solutions at different concentrations (20.0, 40.0, 50.0, 70.0, 100.0 and 150.0 mg L−1); bottles were sealed and magnetically stirred at room temperature under the protection of a N2 atmosphere for 1 h to ensure equilibrium.
2.3. Sequential extractions
Se speciation, from reacting various [Fe(II)]/[OH−] ratios with Se(IV) sediments, was determined by a sequential extraction scheme according to previously published methods.22,23 A five-stage sequential extraction procedure was used in sediment with the following extraction: (1) water-soluble Se (ultrapure water); (2) exchangeable Se (0.1 M NaH2PO4, pH = 7); (3) elemental state Se (1.0 M Na2SO3, pH = 7), water bath ultrasonication for 3 min; (4) oxide-containing Fe state (15% CH3COOH and digestion);24 (5) Se compound states (selenide) (0.5 g KClO3, concentrated hydrochloric acid). Extractions were performed in 50 mL Falcon tubes, and each extraction step used 20 mL of extractant, with shaking or ultrasound, centrifugation (3500 rpm) for 15 min and filtration.
2.4. Analytical methods and characterization
Aqueous Se(IV) concentration was determined using ICP (720ES; Agilent, USA). pH was measured using pHSJ-3F (Shanghai Precision and Scientific Instruments, China). After filtration, solid products were rinsed with ultrapure water and immediately freeze-dried. Morphology of structural Fe(II) solid phase was obtained using a field emission scanning electron microscope (S-4800; Hitachi, Japan) equipped with an energy dispersive X-ray spectrometer at 200 kV. XRD patterns were collected using a D8 Advance Powder X-ray diffractometer (Bruker, USA) and a Cu Ka (λ = 1.5406 Å) radiation source (40 kV, 40 mA); the diffraction angle (2θ) was recorded from 10° to 80° with a scanning speed of 1° min−1 and a step size of 0.02°. Related oxidation states on the surface of Fe(III)-contained solid products were analyzed by X-ray photoelectron spectroscopy (XPS) using a PHI-5000 Versaprobe spectrometer equipped with a rotating Al anode generating Al Kα X-ray radiation at 1486.6 eV.
3. Results and discussion
3.1. Batch experiments on selenite removal and equilibrium
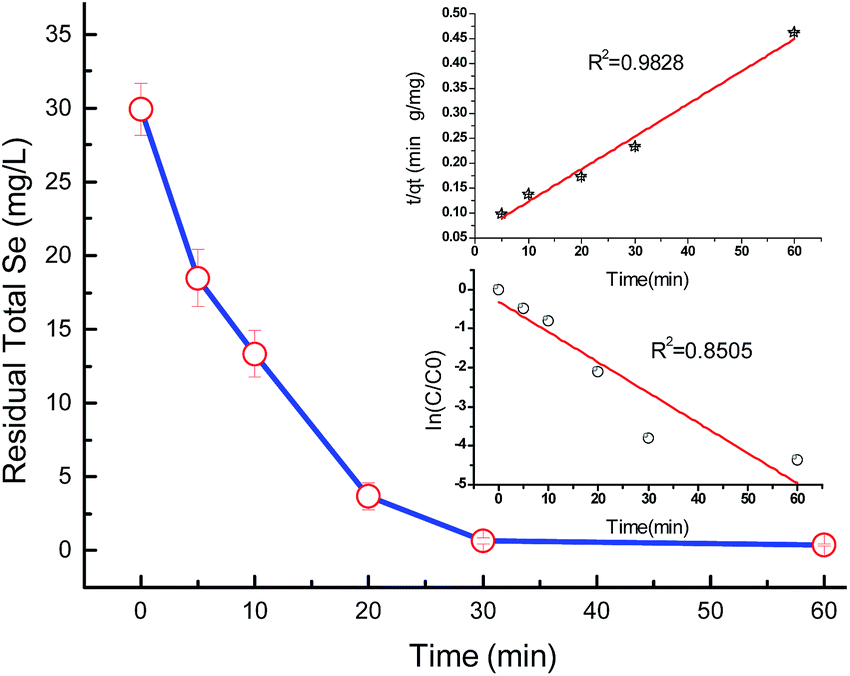
Selenite removal studies have important roles in adsorption studies because they can predict the equilibrium time and removal rate of selenite from aqueous solution. Fig. 1 presents the selenite removal in aqueous solution as a function of contact time. With an initial selenite solution at 30 mg L−1, the selenite removal efficiency was >99% within 30 min and remained constant over 60 min, which is in agreement with the study published by Myneni et al., who used Fe(II)/Fe(III) oxide (GRs) to remove selenite.19 The aqueous Se(IV) concentration dropped quickly, >48% in the first 0.01 h (36 s). As reported by Han et al., Se(IV) can be completely removed by FeS (1.0 g L−1) within 30 min.25 During the course of the experiment, the milky white suspension gradually became atrovirens and, 1 h later, reddish-brown substances formed. A possible explanation for this phenomenon can be that structural Fe(II) was oxidized to Fe(II)/Fe(III) and/or Fe(III) hydroxides in the reaction. Another possibility is that the red Se(0) formed, which was also observed by Breynaert et al. in which red Se(0) particles formed on the FeSe surface after FeS reacted with Se(IV) for 3 days.2 Se(IV) removal by structural Fe(II) hydroxides could be described by a generalized pseudo-first order kinetic model and generalized pseudo-second order kinetic model. Meanwhile, the correlation coefficient (R2) of the generalized pseudo-first order kinetic model was 0.8505, whereas the generalized pseudo-second order kinetic model was 0.9828 (Fig. 1 inset). Se(IV) removal by structural Fe(II) was more coincident with the pseudo-second order kinetic model. In the following section, the generalized pseudo-second order kinetic model was further studied, with an expression shown as eqn (1) and the corresponding half-life (t1/2) could be calculated using eqn (2).| |
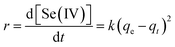 | (1) |
| |
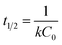 | (2) |
where [Se(IV)] shows Se(IV) concentration (mg L−1) at time t (min), k is the pseudo-second-order reaction rate constant and t1/2 stands for the half-life of Se(IV) removal. Calculation revealed the reaction rate constant (k) and t1/2 to be 1.05 × 10−3 mg g−1 min−1 and 31.7, respectively.
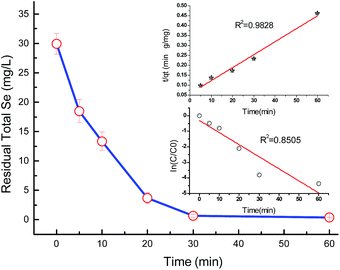 |
| | Fig. 1 Adsorption kinetics of Se(IV). Error bars represent standard error of the mean. Initial [Se(IV)] = 30.0 mg L−1 and FHC(1:2) dosage = 228.0 mg L−1. | |
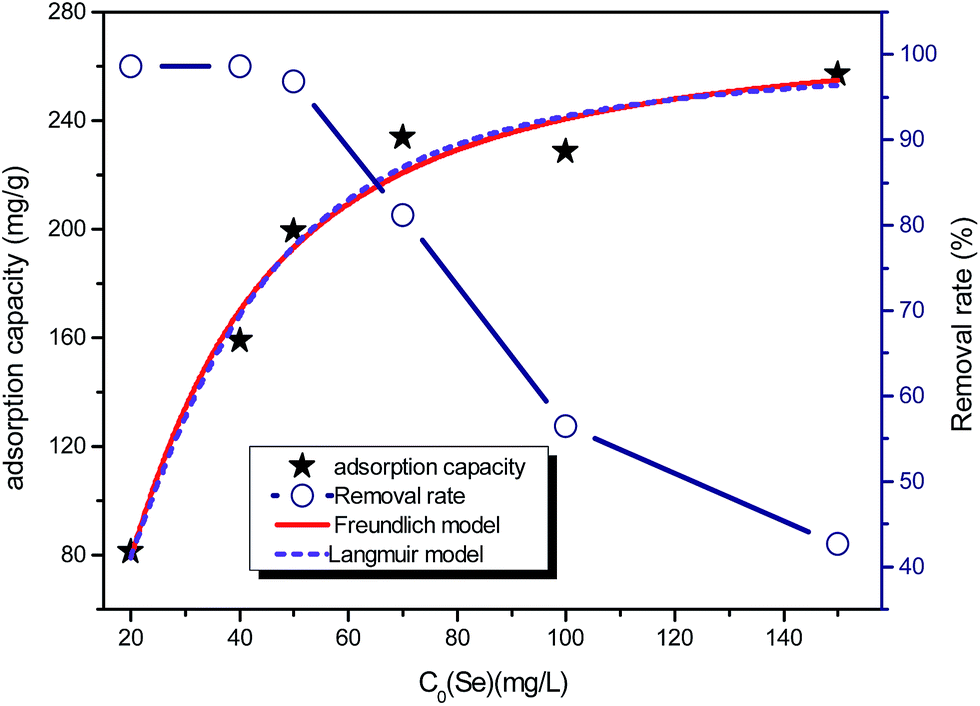
Fig. 2 shows the selenite removal rate and adsorption capacity equilibrium concentration. The results were fitted with the Langmuir model (3), Freundlich model (4) and BET isotherm (5), respectively:
| |
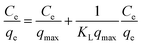 | (3) |
| |
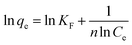 | (4) |
| |
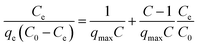 | (5) |
where
Ce is the equilibrium concentration of selenite (mg L
−1),
C0 is the assumed initial Se concentration,
qe is the equilibrium adsorption capacity (mg g
−1),
qmax is the maximum adsorption capacity (mg g
−1),
KL is the adsorption constant (L mg
−1), and
KF and 1/
n are Freundlich isotherm constants related to adsorption capacity and intensity of adsorption, respectively. Models (BET, Langmuir, Freundlich) were fitted to data, and the values of model parameters are summarized in
Table 1. The BET isotherm was the best-fit model, whose
R2 was 0.9902, whereas there was little difference between the Freundlich isotherm (
R2 = 0.9609) and Langmuir isotherm (
R2 = 0.9650). Se(
IV) removal was well described by the BET isotherm, so it can be inferred that selenite primarily experienced adsorption on the surface of structural Fe(
II). Afterwards, Se(
IV) could be reduced to Se(0), Se(−
II) and precipitated as FeSe or FeSe
x. The maximum adsorption capacity was 256.41 mg g
−1, suggesting that structural Fe(
II) was more potential and effective than other related adsorbents.
25–28
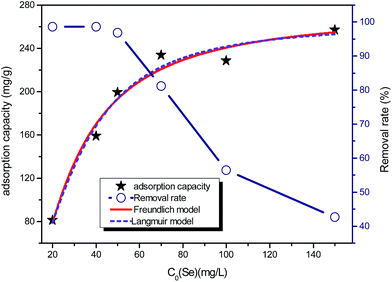 |
| | Fig. 2 Se(IV) removal (mg g−1) capacity and percentage removal rate. FHC(1:2) dosage = 200.0 mg L−1, initial [Se(IV)] concentrations = (20.0, 40.0, 50.0, 70.0, 100.0 and 150.0 mg L−1), and reaction time = 1 h. | |
Table 1 Summary of model parameters for Se(IV) removal by structural Fe(II)
| Langmuir isotherm |
Freundlich isotherm |
BET isotherm |
| qmax (mg g−1) |
KL (L mg−1) |
R2 |
n |
KF |
R2 |
R2 |
| 256.41 |
0.9793 |
0.965 |
6.67 |
144.89 |
0.9609 |
0.9902 |
Although the BET model was developed to describe multilayer gas sorption, it could also represent continuous accumulation of adsorbates on the solid surface until a maximum level of sorption. The BET model was preferable for use over a solid solution system because of its simplicity and ability to ascertain if continuous adsorption (e.g., polymeric cluster and surface precipitates) occurs. The constant KF of the Freundlich model was defined as adsorption capacity,29 and the unit equilibrium concentration of structural Fe(II) for selenite removal was 144.89, which was higher than that for nZVI.30 Moreover, the value 1/n in the Freundlich model for selenite was >1, suggesting that this isotherm was nonlinear, which can be attributed to adsorption-site heterogeneity, electrostatic attraction and other sorbent–sorbate interactions. Fig. 2 shows that the selenite removal rate and adsorption capacity had a significant negative correlation. Chemical precipitation may have an important role at a higher initial selenite concentration; therefore, the selenite-removal mechanism at high initial selenite concentrations (100–1000 mg L−1) may include adsorption and co-precipitation processes. However, Chubar et al. reported that surface precipitation was not part of the processes at the interface between Fe–Mn mixed adsorbent and aqueous selenite.31 Therefore, the mechanism of selenite removal by structural Fe(II) required further study, as shown in subsequent sections.
3.2. Effect of FHC dosage
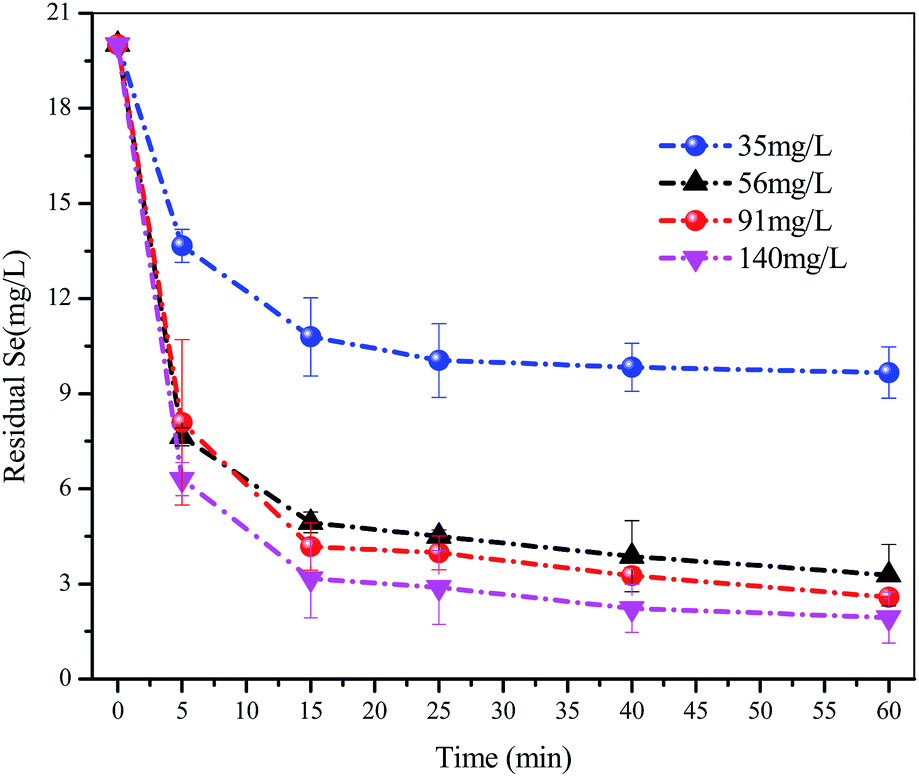
As shown in Fig. 3, the residual concentration of Se(IV) continuously decreased with increasing FHC(1:2) dosage. When the dosage was >56.0 mg L−1, the removal rate slowed and residual concentration of Se(IV) was 4.70 mg L−1. After a 1 h reaction, when the dosage of FHC(1:2) was 35.0 mg L−1, the removal rate of Se(IV) was 51.7%. When the dosage of FHC(1:2) was 91.0 mg L−1, the removal rate of Se(IV) was ≈90%. However, when the dosage of FHC(1:2) was >91.0 mg L−1, the increase in the Se(IV) removal rate was not obvious. Agglomeration may be the more possible reason according to the scanning electron micrograph shown in Fig. 6.
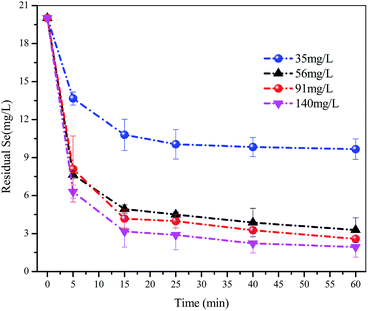 |
| | Fig. 3 The selenite solutions (80 mL and 20 mg L−1) and different adsorbent dosages (35.0, 56.0, 91.0 and 140.0 mg L−1) were mixed in separate batch bottles with a reaction time = 1 h. | |
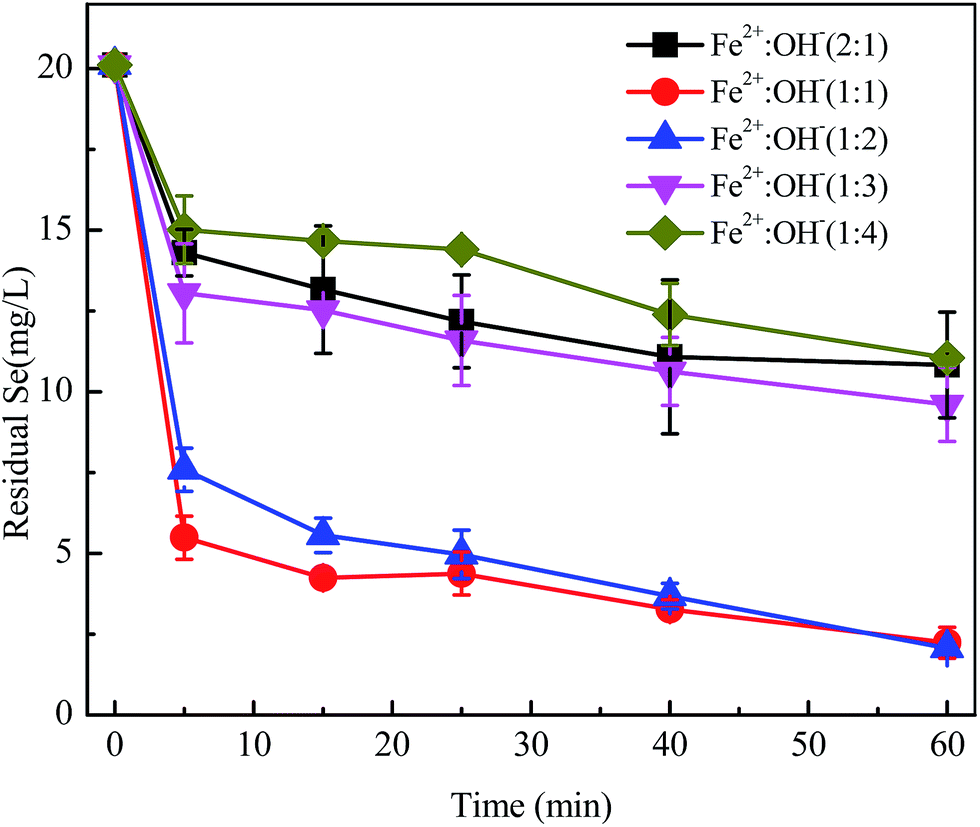
3.3. Effect of the [Fe(II)]/[OH−] ratio
Min et al.32 discovered that GRs have an LDH structure with a high anion exchange capacity according to their study on the sorption of iodine and Se onto GRs. Structural Fe(II) with poly-hydroxyl conjoined with ferrous ions is similar to GRs and is bound up with mole ratios of [Fe2+] to [OH−]. The effect of various mole ratios of [Fe2+] to [OH−] was studied for the reactive properties of FHC structures. As shown in Fig. 4, the removal efficiencies of SeO32− with FHC(1:1) and FHC(1:2) were higher than those of FHC(1:3) and FHC(1:4). The solution pH value before and after the reaction is labeled in Table 2. FHC with various ratios of [Fe2+] to [OH−] caused changes in the solution pH. FHC(1:3) or FHC(1:4) could increase the solution pH to ≥11.5 and hindered the removal of SeO32−. Possible causes may be the competition relationship between OH− and Se(IV). Also, at higher pH, the positive charge on Fe(II) hydroxides decreased and lower attraction to negative ions led to a decrease in removal rate. At lower pH, a greater positive charge could accumulate on FHC(2:1) but, with a decrease in pH, the layered structure of FHC could be undermined and structural Fe(II) transformed into free ferrous, leading to lower selenite removal. For example, research on reductive transformation of reactive black 5 by FHCs showed that structural Fe(II) became free ferrous at pH 2 and reductive removal ability significantly decreased.16 These results were consistent with the experiments by Hana et al.25 Another reason may be that redundant OH− is unfavorable for FHC structural stability. The third reason might be that the mixed pH affected the forms of Fe(II) present. Judging from Fig. S1 (ESI†) and Table 2, Fe2+ was the main form present for FHC(2:1) and FHC(1:1), FeOH+ was the preponderant form for FHC(1:2), and Fe(OH)3− was the major form for FHC(1:3) and FHC(1:4). Therefore, FHC(2:1), FHC(1:1) and FHC(1:2) were pH-dependent positive charge surfaces and FHC(1:3) and FHC(1:4) were pH-dependent negative charge surfaces. A positive charge is better for selenite adsorption and then reduction, whereas electrostatic repulsion of a negative charge antagonizes selenite sequestration. There was a special phenomenon in the solution pH value before and after the reaction. When FHC(2:1) and FHC(1:1) were used as adsorbents, the pH mainly decreased. When FHC(1:2)–FHC(1:4) were used, the pH was increased or changed slightly. This phenomenon is possible due to a deviation in pH determination. The authentic interpretation of Se(IV) removed by structural Fe(II) should be obtained in further investigations at the molecular level using spectroscopic methods such as XPS.
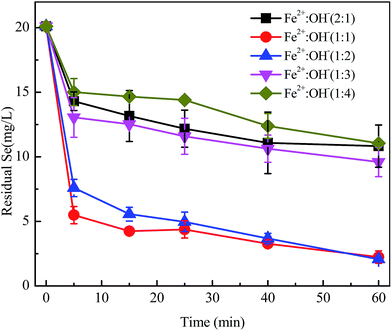 |
| | Fig. 4 Various [Fe(II)]/[OH−] ratios were mixed in batch bottles, initial [Se(IV)] = 20.0 mg L−1, FHC dosage = 114.0 mg L−1, and reaction time = 1 h. | |
Table 2 pH value of aqueous solution before and after the reaction
| [Fe(II)]/[OH−] |
FHC(2:1) |
FHC(1:1) |
FHC(1:2) |
FHC(1:3) |
FHC(1:4) |
| Initial pH |
7.65 |
7.82 |
10.20 |
11.58 |
11.85 |
| Reaction pH |
6.75 |
6.93 |
10.58 |
11.68 |
11.90 |
| Residual Se concentration (mg L−1) |
10.82 |
2.24 |
2.06 |
9.61 |
11.05 |
3.4. Solid-phase analyses
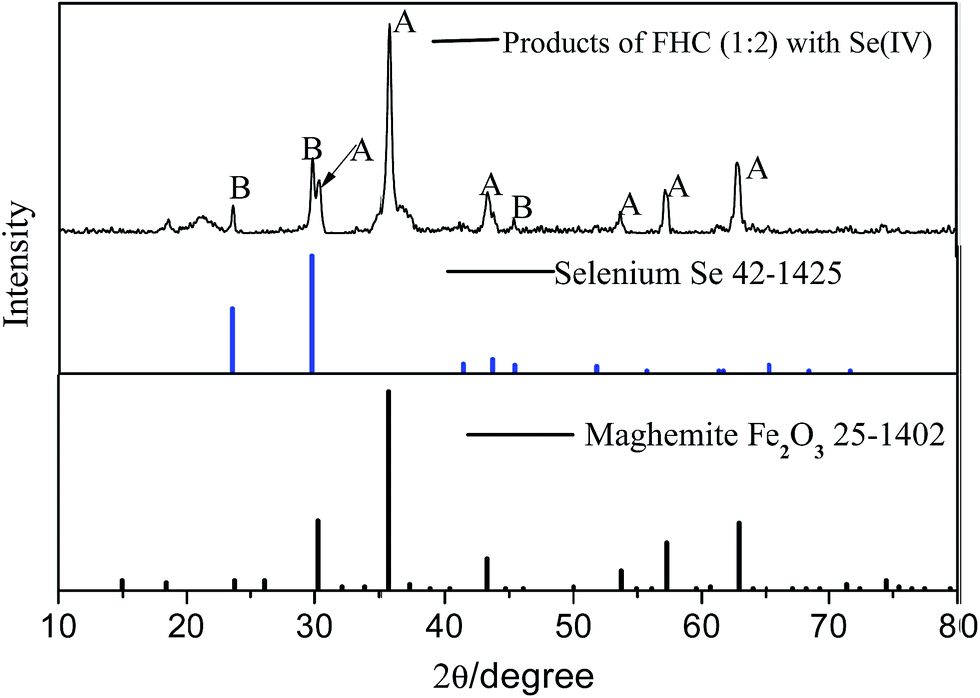
XRD diffraction patterns showed products of FHC(1:2) with Se(IV) (Fig. 5). Maghemite (PDF: 25-1402) is consistent with the products of GRs with Se(IV).19 The intermediate products of GRs were formed in the reaction, which was followed by the adsorption of Se(IV) onto them in a subsequent process. In addition, characteristic strong reflections at 2θ = 23.5°, 29.7° and 43.6° were detected, which were taken to be for Se (PDF: 42-1425). The result showed Se(IV) was reduced to crystalline Se(0) in the reaction.
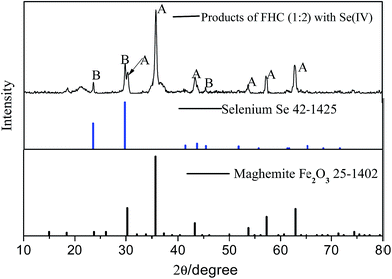 |
| | Fig. 5 XRD analyses of reacted FHC(1:2) with Se(IV). | |
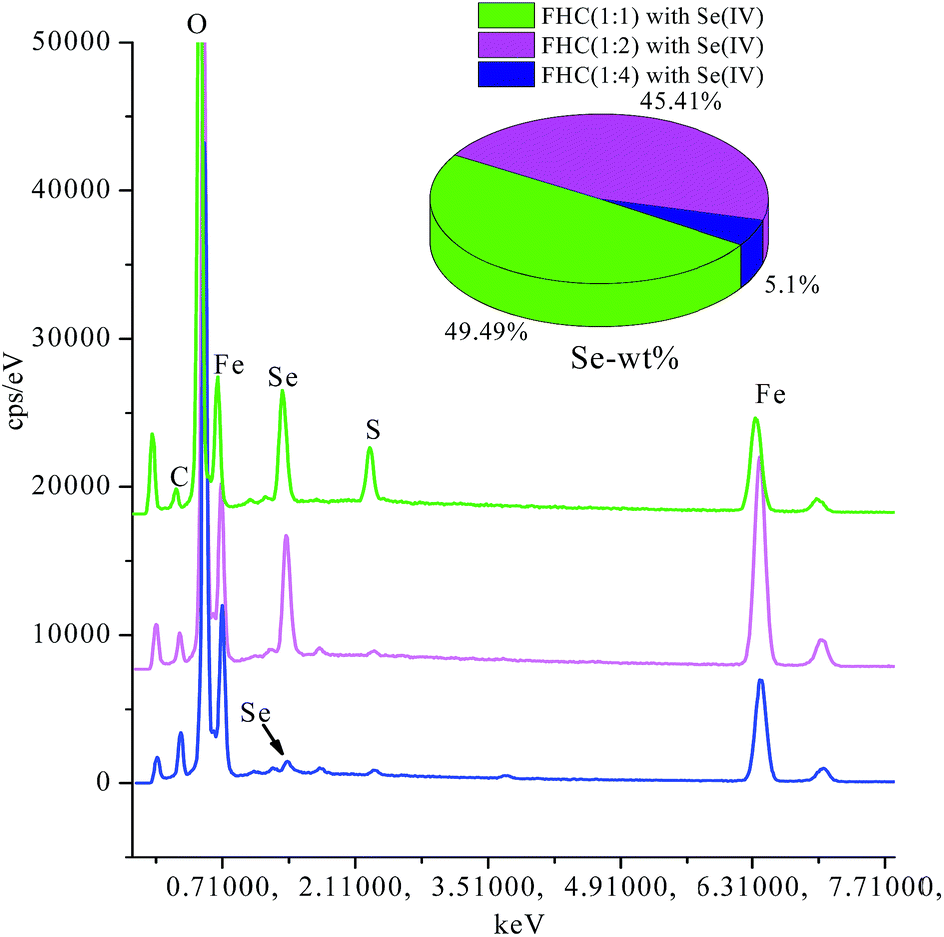
Fig. 6 displays the scanning electron microscopy (SEM) images of fresh FHC (A) and reacted FHC (B and C). Many intervals were observed in the image of fresh FHC, and it was obvious that the reacted FHC formed an agglomeration, and FHC(1:4) granules apparently grew larger, which was a possible reason for the lower removal rate of Se(IV). According to energy dispersive X-ray spectrometry (EDS) (Fig. 7 ), the level of solid Se on FHC(1:1) or FHC(1:2) was obviously higher than that for FHC(1:4) in the final products, which could also demonstrate that the removal capacity of the former was higher than that for the latter.
 |
| | Fig. 6 SEM images of fresh FHC(1:1) and reacted FHC(1:1) and FHC(1:4). | |
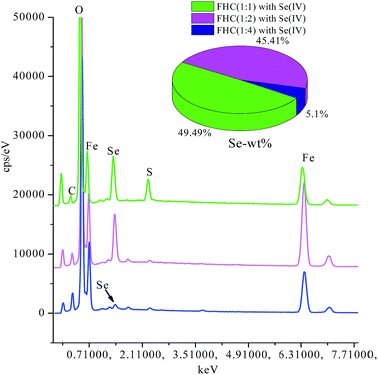 |
| | Fig. 7 EDS analyses of FHC with Se(IV). | |
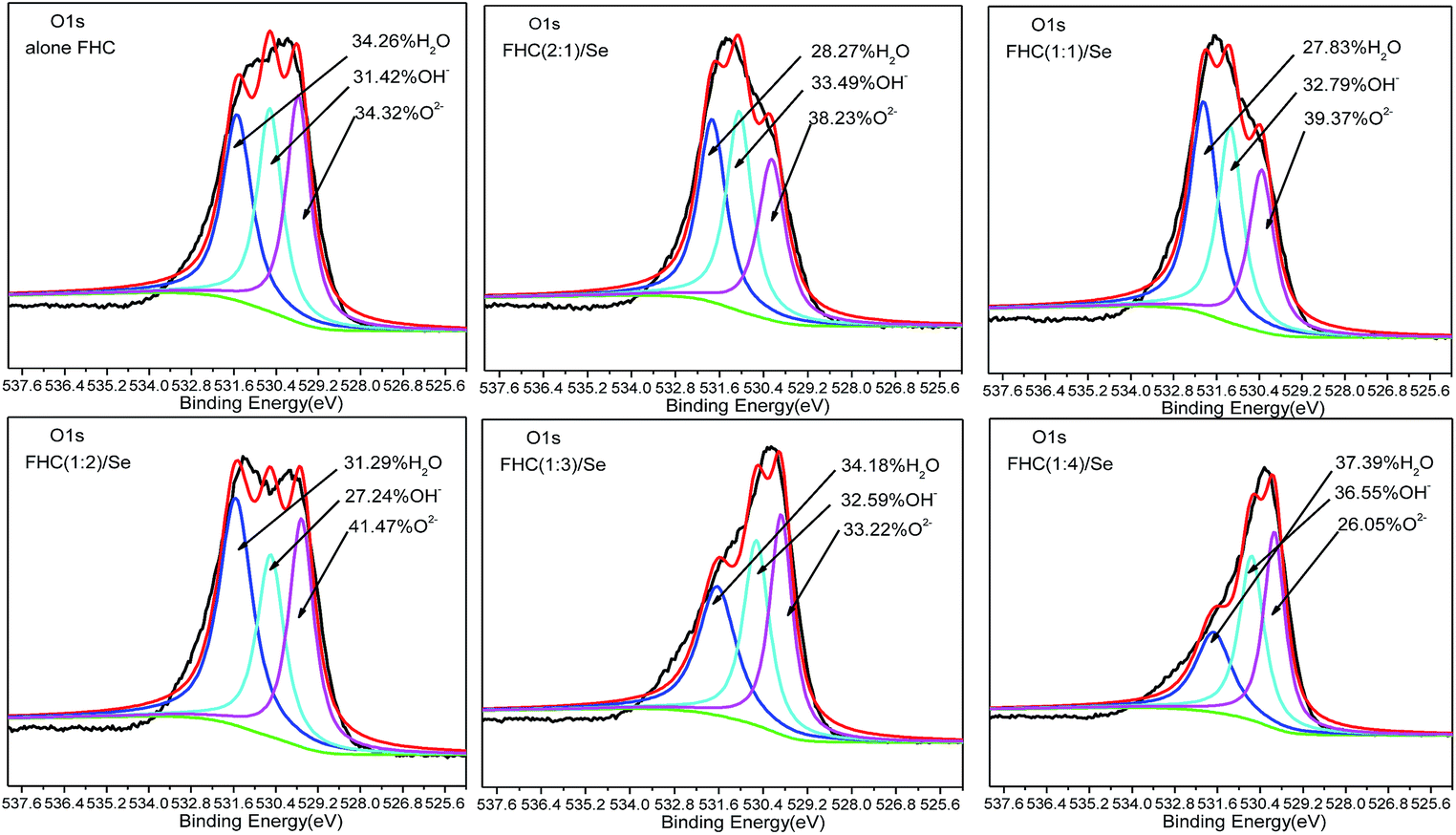
Structural Fe(II) exists as a poly-hydroxyl conjoined with Fe(II), whose structure is similar to GRs, and the removal mechanism for this heavy metal is also similar.33 Some studies have demonstrated ion exchange during processing, making it necessary for future study on structural Fe(II) in solid products. Fig. 8 shows the O1s XPS spectra of FHC(2:1), FHC(1:1), FHC(1:2), FHC(1:3) and FHC(1:4) products that were reacted with Se(IV). The O1s spectra were fitted with three components at 529.0–529.7 eV, 530.2–530.7 eV, and 531.1–531.7 eV, which corresponded to the oxygen ion (O2−), structural hydroxide (OH−), and adsorbed water molecule (H2O), respectively.34 The structural hydroxide originated from hydroxyl ions in solution or dissociation of absorbed water molecules and, when present, atmospheric O2 could originate from the combination of a proton (H+) and O2−, which was produced by the reduction of O2.35 Although all experiments were conducted in an anaerobic chamber, the residual O2 on the solution surface could never be completely expelled, and the solid samples were exposed to air when transferred for XPS analyses. Fig. 8 shows that the peak area of O2−, H2O and OH− changed with the ratios of ([Fe(II)]/[OH−]). Due to the effect of the [Fe(II)]/[OH−] ratio, the peak area of structural hydroxide [OH−] was moderate and the removal rate was better. When the [Fe(II)]/[OH−] ratio was 1:4, however, the affinity of residual OH− was superior to Se(IV) and hindered the removal of Se(IV).
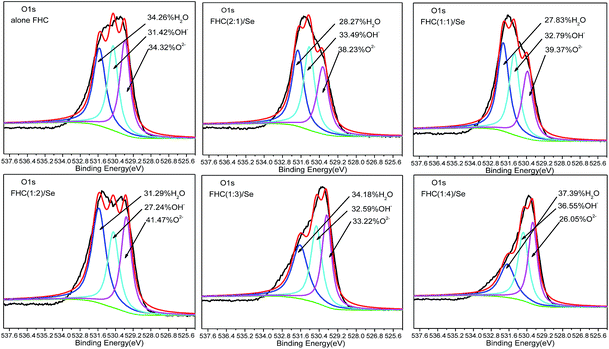 |
| | Fig. 8 O1s XPS spectra of FHC of different [Fe(II)]/[OH−] ratios on the removal of Se(IV). Initial [Se(IV)] = 20.0 mg L−1, FHC dosage = 114.0 mg L−1, and reaction time = 1 h. | |
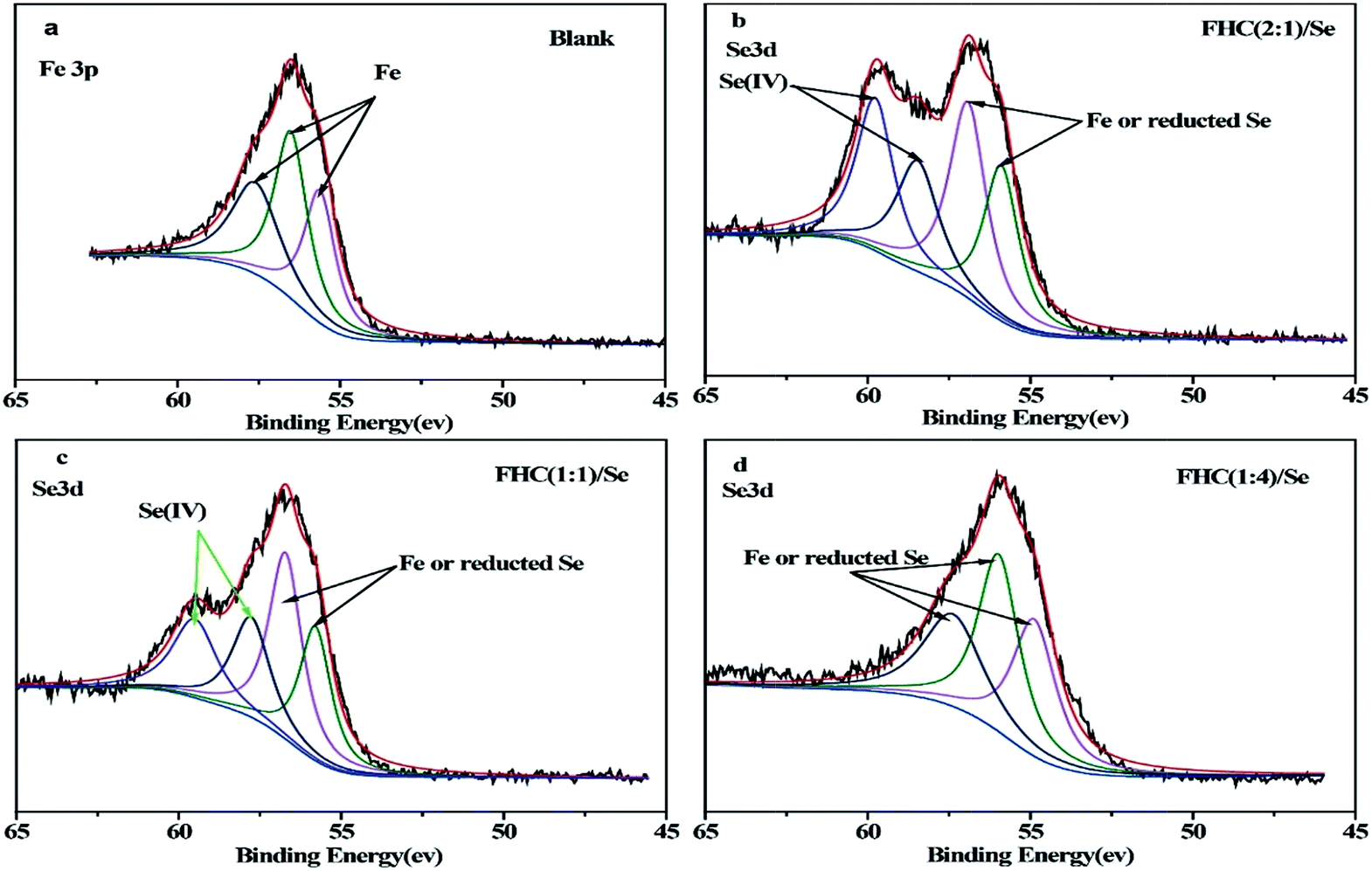
There are some limitations in the analyses of XPS spectra that contain Fe and Se due to the interference between Fe3p and Se3d. Fortunately, because there was no overlap in the binding energy between Se(IV) 3d and Fe3p, the changes in Se(IV) in the solid stage could be observed. Based on the National Institute of Standards and Technology binding energy database, Fe3p spectra of structural Fe(II) are at 55.9 eV, Se3d spectra of Se(IV) are at 59.1 eV, and spectra peaks of Se(0) are at 54.6–55.9 eV.36 Obviously, it was difficult to analyze the existence of Fe and Se(0) on the solid surface from XPS spectra. According to Fig. 9a, Fe3p spectra of structural Fe(II) without Se were present at 55.6–57.6 eV, which overlapped with the Se3d peaks 54.8–57.8 eV in Fig. 9b–d. As shown in Fig. 9b–d, the peak intensity was obviously stronger in Fig. 9b and the contents of Se(IV) in the solid state were ranked in the following order: b > c > d. Therefore, Se(IV) removal by structural Fe(II) changed with various [Fe(II)]/[OH−] ratios.
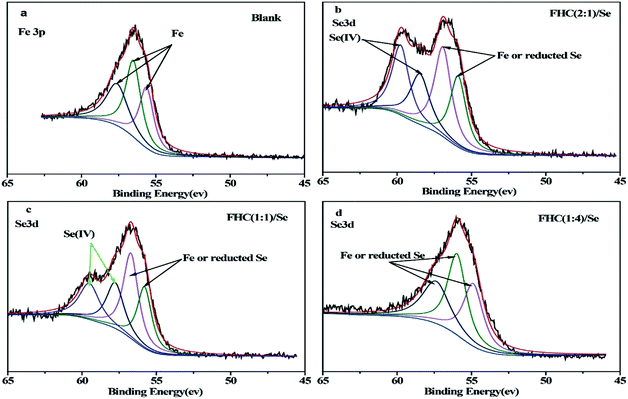 |
| | Fig. 9 Se 3d XPS spectra of FHC of different [Fe(II)]/[OH−] ratios on the removal of Se(IV). Initial [Se(IV)] = 20.0 mg L−1, FHC dosage = 114.0 mg L−1, reaction time = 1 h. | |
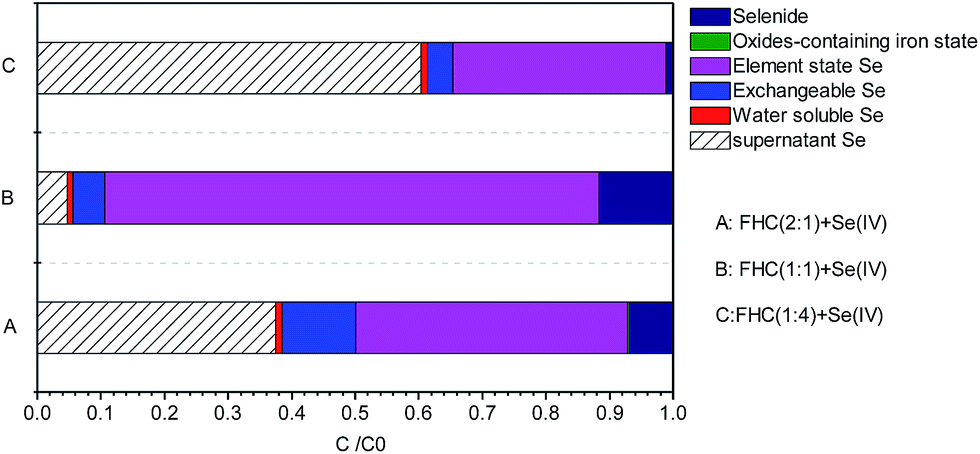
With use of a sequential extraction procedure, Se in sediments was classified into the following five groups: water-soluble, exchangeable, elemental Se, oxides-containing Fe state, and selenide. Fig. 10A–C, respectively, show FHC(2:1), FHC(1:1) as well as FHC(1:4) reacted with Se(IV). The percentage content of water-soluble and oxide-containing Fe-state Se might be neglected. Se(0) was the main reduction product and accounted for 77% of the five groups when FHC(1:1) was reacted with Se(IV). The presence of exchangeable Se indicated that FHC had the function of flocculation adsorption. The percentage content of exchangeable Se on FHC(2:1) was obviously higher than that for FHC(1:4) or FHC(1:1). This conclusion was consistent with XPS spectra analyses of Se. The results of sequential extraction treatment indicated that reduction immobilization was the principal approach for selenite removal.
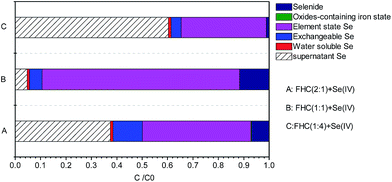 |
| | Fig. 10 Various [Fe(II)]/[OH−] ratios with Se(IV) were sequentially extracted; initial [Se(IV)] = 20.0 mg L−1 and FHC dosage = 114.0 mg L−1. | |
The bidentate inner-sphere surface complexes formed on the structural Fe(II) surface was consistent with previous studies.37,38 Shrimpton et al. studied Se(VI) reduction by zerovalent Fe and found Se(IV) could be reduced to Se(0). Under high pH conditions, the main precipitation products were iron selenides. However, at low pH, neither FeSe2 nor FeSe was observed in that study.39 As the reaction time progressed, structural Fe(II) could be gradually oxidized to GRs. Afterwards, Se(IV) was mainly reduced to Se(0) and then participated in interlayer ion exchange, which could contribute to the removal capacity of structural Fe(II) being better than GRs. The reducibility of structural Fe(II) was obviously stronger than that of GRs, which was different from many other removal mechanisms.9,18,40–42 Se(IV) was either absorbed on the surface of structural Fe(II) mineral and reduced to Se(0) or ion-exchange between Se(IV) and other anions occurred.
4. Conclusions
FHCs were synthesized and employed to remove selenite. The Se(IV) removal isotherm was adequately described by the BET isotherm, and the calculated maximum removal capacity of Se(IV) was 256.41 mg g−1. The corresponding fitting result was more coincident with the pseudo-second order kinetic model and the reaction rate constant (k) was 1.05 × 10−3 mg g−1 min−1. When the FHC(1:2) dosage was 91.0 mg L−1, the removal rate was ≈90%. The amount of solid Se on FHC(1:1) or FHC(1:2) was obviously more than FHC(1:4) in the final products, which could also demonstrate that the removal capacity of the former was higher than that of the latter. XRD diffraction patterns showed that structural Fe(II) was translated into maghemite and Se(IV) was reduced to crystalline Se(0) by FHC(1:2). XPS spectra indicated that the mechanism of Se(IV) removal by FHC(1:1) involved the combined effects of adsorption and reduction, and Se(IV) removal by FHC(1:4) principally involved reduction. Se(IV) removal by structural Fe(II) changed with various ratios of [Fe(II)]/[OH−]. With use of a sequential extraction procedure, Se in sediments was classified into five groups: water-soluble, exchangeable, elemental Se, oxide-containing Fe state, and selenide. Crystalline Se(0) was the main reduction product. The higher uptake of selenite indicated that structural Fe(II) was a lower-cost, effective material for selenite removal.
Acknowledgements
This study was financially supported by the Natural Science Foundation of China (Grant No. 51278356, 41572211).
Notes and references
- F. Chen, P. C. Burns and R. C. Ewing, J. Nucl. Mater., 1999, 275, 81–94 CrossRef CAS.
- E. Breynaert, C. Bruggeman and A. Maes, Environ. Sci. Technol., 2008, 42, 3595–3601 CrossRef CAS PubMed.
- D. L. Hatfield, Selenium: its molecular biology and role in human health, Springer, 2006 Search PubMed.
- J. A. Plant, D. G. Kinniburgh, P. L. Smedley, F. M. Fordyce and B. A. Klinck, Treatise Geochem., 2003, 9, 17–66 Search PubMed.
- A. D. Lemly, Ecotoxicol. Environ. Saf., 2004, 59, 44–56 CrossRef CAS PubMed.
- L. H. E. Winkel, C. A. Johnson, M. Lenz, T. Grundl, O. X. Leupin, M. Amini and L. Charlet, Environ. Sci. Technol., 2012, 46, 571–579 CrossRef CAS PubMed.
- H. Wen and J. Carignan, Geochim. Cosmochim. Acta, 2011, 75, 1411–1427 CrossRef CAS.
- J. S. Cleverley and E. N. Bastrakov, Comput. Geosci., 2005, 31, 756–767 CrossRef CAS.
- R. L. D. A. Loyo, S. I. Nikitenko, A. C. Scheinost and M. Simonoff, Environ. Sci. Technol., 2008, 42, 2451–2456 CrossRef PubMed.
- A. C. Scheinost and L. Charlet, Environ. Sci. Technol., 2008, 42, 1984–1989 CrossRef CAS PubMed.
- A. C. Scheinost, R. Kirsch, D. Banerjee, A. Fernandez-Martinez, H. Zaenker, H. Funke and L. Charlet, J. Contam. Hydrol., 2008, 102, 228–245 CrossRef CAS PubMed.
- A. Naveau, F. Monteil-Rivera, E. Guillon and J. Dumonceau, Environ. Sci. Technol., 2007, 41, 5376–5382 CrossRef CAS PubMed.
- T. Missana, U. Alonso, A. C. Scheinost, N. Granizo and M. García-Gutiérrez, J. Cont. Educ. Nurs., 2009, 73, 6205–6217 CAS.
- L. P. Liang, W. Yang, X. H. Guan, J. Li, Z. Xu, J. Wu, Y. Huang and X. Zhang, Water Res., 2013, 47, 5846–5855 CrossRef CAS PubMed.
- L. Ling, B. Pan and W. X. Zhang, Water Res., 2015, 71, 274–281 CrossRef CAS PubMed.
- W. U. Deli, Y. Feng and M. A. Luming, J. Huazhong Univ. Sci. Technol., Med. Sci., 2011, 39, 1657–1662 Search PubMed.
- S. Chakraborty, F. Bardelli and L. Charlet, Environ. Sci. Technol., 2010, 44, 1288–1294 CrossRef CAS PubMed.
- N. Chubar, V. Gerda and M. Szlachta, Environ. Sci. Technol., 2014, 48, 13376–13383 CrossRef CAS PubMed.
- S. C. B. Myneni and T. K. Tokunaga, Science, 1997, 278, 1106–1109 CrossRef CAS.
- D. L. Wu, Q. M. Wang, Y. Feng and L. M. Ma, Adv. Mater. Res., 2012, 518–523, 1737–1743 CrossRef CAS.
- B. B. Shao, Y. Y. Guan and D. L. Wu, SICHUAN Environment, 2016, 5, 13–19 Search PubMed.
- S. W. Wu and Q Chi, Soils, 2004, 36, 91–92 Search PubMed.
- B. A. Manning and R. G. Burau, Environ. Sci. Technol., 1995, 29, 2639–2646 CrossRef CAS PubMed.
- M. A. Wilson, R. Burt, W. C. Lynn and L. C. Klameth, Commun. Soil Sci. Plant Anal., 1997, 28, 407–426 CrossRef CAS.
- S. H. Dong, B. Batchelor and A. Abdel-Wahab, J. Hazard. Mater., 2011, 186, 451–457 CrossRef PubMed.
- L. Liang, X. Jiang, W. Yang, Y. Huang, X. Guan and L. Li, Desalin. Water Treat., 2015, 53, 1–9 CrossRef.
- M. Szlachta, V. Gerda and N. Chubar, J. Colloid Interface Sci., 2012, 365, 213–221 CrossRef CAS PubMed.
- Y. T. Chan, H. K. Wen, T. Y. Chen and K. W. Ming, Water Res., 2009, 43, 4412–4420 CrossRef CAS PubMed.
- Z. P. Wen, C. Dai, Y. Zhu and Y. L. Zhang, RSC Adv., 2015, 5, 4058–4068 RSC.
- Z. Wen, Y. Zhang and C. Dai, Colloids Surf., A, 2014, 457, 433–440 CrossRef CAS.
- N. Chubar and V. Gerda, Environ. Sci. Technol., 2014, 48, 13376–13383 CrossRef CAS PubMed.
- J. H. Min, M. H. Baik, J. K. Lee and J. T. Jeong, China Nuclear Technology Report, 2013, 1, 56–72 Search PubMed.
- B. Verbinnen, C. Block, P. Lievens, A. V. Brecht and C. Vandecasteele, Waste Biomass Valorization, 2013, 4, 635–645 CrossRef CAS.
- S. W. Knipe, J. R. Mycroft, A. R. Pratt, H. W. Nesbitt and G. M. Bancroff, Geochim. Cosmochim. Acta, 1995, 59, 1079–1090 CrossRef CAS.
- H. W. Nesbitt and I. J. Muir, Geochim. Cosmochim. Acta, 1994, 58, 4667–4679 CrossRef CAS.
- J. R. Rumble, D. M. Bickham and C. J. Powell, Surf. Interface Anal., 1992, 19, 241–246 CrossRef CAS.
- H. Wijnja and C. P. Schulthess, J. Colloid Interface Sci., 2000, 229, 286–297 CrossRef CAS PubMed.
- A. Fernández-Martínez and L. Charlet, Rev. Environ. Sci. Bio/Technol., 2009, 8, 81–110 CrossRef.
- H. K. Shrimpton, D. W. Blowes and C. J. Ptacek, Environ. Sci. Technol., 2015, 49, 11688–11696 CrossRef CAS PubMed.
- J. T. Olegario, N. Yee, M. Miller, J. Sczepaniak and B. Manning, J. Nanopart. Res., 2010, 12, 2057–2068 CrossRef CAS.
- L. Liang, X. Guan, Z. Shi, J. Li, Y. Wu and P. G. Tratnyek, Environ. Sci. Technol., 2014, 48, 6326–6334 CrossRef CAS PubMed.
- A. E. Schellenger and P. Laresecasanova, Environ. Sci. Technol., 2013, 47, 6254–6262 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26225b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a