
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of new acetylcholinesterase and butyrylcholinesterase inhibitors through structure-based virtual screening†
Yao Chena,
Hongzhi Linb,
Hongyu Yangb,
Renxiang Tana,
Yaoyao Biana,
Tingming Fua,
Wei Lia,
Liang Wua,
Yuqiong Pei*a and
Haopeng Sun *b
*b
aSchool of Pharmacy, Nanjing University of Chinese Medicine, Nanjing, 210023, China. E-mail: peiyuqiong@126.com; Tel: +86-15952007562
bDepartment of Medicinal Chemistry, China Pharmaceutical University, Nanjing, 210009, China. E-mail: sunhaopeng@163.com; Tel: +86-25-85863169
First published on 13th January 2017
Abstract
Small molecule cholinesterase (ChE) inhibitors represent one of the most effective therapeutic strategies for the treatment of Alzheimer's disease (AD). However, only three of these drugs have entered the market. Understanding of the structures of successful ChE inhibitors is still very limited; therefore, it is an urgent task to identify new scaffolds for the design of ChE inhibitors. In the present study, we report an effective computer-aided workflow using a hierarchical structure-based virtual screening to identify new ChE inhibitors. 3D shape-based similarity screening combined with structure-based pharmacophore models was applied to obtain efficiently narrow potential hits from large compound collections, such as Chemdiv. Molecular docking was then used to further restrict the screening results. Five new ChE inhibitors, namely C629-0196, G070-1566, G115-0283, G801-0274, and F048-0694, were identified from the biological validation of 24 potential hits from the virtual screening. These compounds provide interesting templates for the design of new ChE inhibitors. Among the compounds, G801-0274 exhibited the lowest nanomolar range IC50 (0.031 ± 0.006 μM) and the highest selectivity (ratio = 66.13) against BuChE. Another compound, C629-0196, showed a low micromolar range IC50 (1.28 ± 0.83 μM) against AChE; however, it had no activity against BuChE. Most importantly, these two compounds provide good starting points for the discovery of highly selective ChE inhibitors.
1. Introduction
Alzheimer's disease (AD) is the one of the most severe causes of cognitive impairment in humans. It is predicted that AD will affect about 6% of the population aged over 65,1 with more than 70 million patients by 2050.2 AD is characterized by progressive memory impairment, decline in cognitive function, and altered behavioral and psychiatric symptoms.3 Although the pathogenic causes of AD are not yet fully understood, the multifactorial nature of AD, which is derived from a complex array of neurochemical factors, is well accepted by researchers worldwide. The pathogenesis of AD now includes cholinergic dysfunction,4 β-amyloid (Aβ) oligomerization,5 τ-protein hyperphosphorylation and aggregation,6 the cell cycle hypothesis,7 oxidative stress,8 metal dyshomeostasis,9 and mitochondrial dysfunction.10 Understanding of this pathogenesis has resulted in multiple therapeutic strategies, which have further inspired the discovery and development of numerous agents, including both small synthetic compounds and macromolecules.Many compounds are currently at the stage of pre-clinical or clinical study; unfortunately, however, the failure rate of drug discovery in this area is extremely high. Most of the current treatments for AD are based on the cholinergic dysfunction hypothesis, which asserts that dysregulation of the cholinergic system, mainly due to the decline of acetylcholine (ACh) levels, is the ultimate reason for this cognitive disorder. Therefore, recovery of Ach levels has become a determinant for the treatment of AD.11 It is well accepted that two types of cholinesterases (ChEs), namely acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), are responsible for the hydrolysis of ACh within the human brain. Thus, administration of ChE inhibitors will upregulate the concentration of ACh in the brains of AD patients and benefit their treatment.12 Previous studies have shown that AChE is more substrate-specific than BuChE in the human brain; as a result, it is believed that acetylcholinesterase inhibitors (AChEIs) can provide more specific therapeutic effects than BuChE inhibitors (BuChEIs).13 Indeed, although there are now many potential drug targets for AD treatment, AChEIs still serve as the main therapeutic agents applied clinically for AD. Additionally, an increasing number of studies have confirmed the key function of BuChE in late-stage AD patients, whose AChE is progressively lost.14–16 Therefore, the discovery of selective BuChE inhibitors is also attracting the attention of drug developers. To date, three ChE inhibitors have been used in the clinic. Among these, donepezil and galantamine are selective AChE inhibitors, while rivastigmine is a dual AChE–BuChE-inhibiting compound.17
The active site of human AChE is a long gorge with a length of approximately 20 Å. Two key sites, namely the catalytic active site (CAS) at the bottom of the gorge and the peripheral anionic site (PAS) near the entrance of the gorge, are linked by a narrow groove.18,19 CAS is responsible for the hydrolysis of Ach through a catalytic triad consisting of Ser200, Glu327, and His440.20 PAS consists of several aromatic residues, including Tyr70, Tyr121, and Trp279.21 It is considered that PAS exerts significant functions related to both ACh hydrolysis and Aβ aggregation.22 Compounds that can interact with both CAS and PAS are believed to exert multiple therapeutic effects; this strategy has inspired the design of multi-target-directed ligands (MTDLs).23 The shape and arrangement of the active site of BuChE is similar to that of AChE; however, the volume of the catalytic site in BuChE is much larger than that of AChE.24 This difference provides an opportunity for the design of selective BuChE inhibitors.
To date, the chemical scaffolds for approved ChE inhibitors have been very limited; meanwhile, the compounds which have entered the market only enable palliative treatment, rather than curing or preventing neurodegeneration.25 Therefore, obtaining ChE inhibitors with new scaffolds is still an urgent task for drug developers. Herein, we describe our efforts toward discovering new ChE inhibitors aided by computational methods. Donepezil, one of the most active AChEIs, was selected as a molecular template for the model generation. The workflow combines shape-based comparison, structure-based pharmacophore (SBP)-mediated virtual screening, and molecular docking. Aided by this method, we successfully discovered five new compounds that show good inhibitory activities toward ChEs. Especially, compound G801-0274 exhibited a low nanomolar range IC50 and high selectivity against BuChE. Another compound, C629-0196, showed a moderate micromolar range IC50 against AChE; however, it had no activity against BuChE. These compounds provide a good starting point for the discovery of highly selective AChEIs. To the best of our knowledge, these compounds have not been previously reported as ChE inhibitors. Further structure-guided optimizations can lead to new types of ether selective or dual-active ChE inhibitors.
2. Results and discussion
2.1 Structure-based virtual screening of potential hits on ChEs
In this study, we screened the Chemdiv compound collection, which contains 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 293896 molecules. The collection was first prepared by the Omega module in the Openeye suite to perform ligand minimization and generate multiple conformations. The prepared collection was then subjected to a 3D shape-based virtual screening mediated by rapid overlay of chemical structures (ROCS). Donepezil, one of the most potent AChEIs, was used as the template for the generation of the 3D shape-based model. ROCS is an efficient shape comparison application for the evaluation of molecular shape similarity. The principle of ROCS can summarized as follows: molecules will form similar shapes if their volumes overlay well. The program applies a Gaussian function to describe the molecular volume of one compound.26 When evaluating the shape similarity of a compound to the model, ROCS will score the result in the range from 0 to 1. In addition to molecular volume, ROCS uses a color force field, which is a spatial arrangement of chemical features, to describe the structural characters of one molecule. The chemical features include six types: hydrogen bond donors, hydrogen bond acceptors, hydrophobes, anions, cations, and rings. The similarity is also scored ranging from 0 to 1.26 In this study, we used a combo score (volume shape plus color force field) ranging from 0 to 2. The higher the score, the more similar a given compound is to donepezil.
293896 molecules. The collection was first prepared by the Omega module in the Openeye suite to perform ligand minimization and generate multiple conformations. The prepared collection was then subjected to a 3D shape-based virtual screening mediated by rapid overlay of chemical structures (ROCS). Donepezil, one of the most potent AChEIs, was used as the template for the generation of the 3D shape-based model. ROCS is an efficient shape comparison application for the evaluation of molecular shape similarity. The principle of ROCS can summarized as follows: molecules will form similar shapes if their volumes overlay well. The program applies a Gaussian function to describe the molecular volume of one compound.26 When evaluating the shape similarity of a compound to the model, ROCS will score the result in the range from 0 to 1. In addition to molecular volume, ROCS uses a color force field, which is a spatial arrangement of chemical features, to describe the structural characters of one molecule. The chemical features include six types: hydrogen bond donors, hydrogen bond acceptors, hydrophobes, anions, cations, and rings. The similarity is also scored ranging from 0 to 1.26 In this study, we used a combo score (volume shape plus color force field) ranging from 0 to 2. The higher the score, the more similar a given compound is to donepezil.
For the generation of a shape-based model, the accuracy of the conformation from which the model is built plays a significant role. Therefore, the bio-conformation of donepezil bound to human AChE was extracted from the co-crystal structure (PDB ID: 4EY7) and directly used to generate the model (Fig. 1A). The molecular shape of donepezil is displayed in a yellow shadow. The model contained four hydrophobes derived from the aromatic ring of donepezil. The oxygen atoms of the two methoxyl groups and the carbonyl oxygen atom were recognized as three hydrogen-bond acceptors; the protonated nitrogen atom supplied the cation. This model was then subjected to virtual screening of the Chemdiv collection. 12939 compounds with combo scores within the top 1% were retained as primary hits.
 | ||
| Fig. 1 (A) ROCS 3D shape-based model generated from donepezil. (B) Structure-based pharmacophore model generated from the co-crystal structure of donepezil bound to human AChE (PDB ID: 4EY7). Donepezil is shown in light blue stick mode. Key residues of AChE are depicted in yellow thin stick mode. Only polar hydrogen atoms are shown. In the pharmacophore model, the hydrogen bond acceptor, cation, aromatic ring, hydrophobic portion and excluded volumes are colored green, red, orange and gray, respectively. | ||
An SBP model was generated on the basis of the donepezil–AChE co-crystal structure (PDB ID: 4EY7) using the Receptor-Ligand Pharmacophore Generation module in the BioVIA Discovery Studio (DS) suite. The top-ranked model was retained as the final model (Fig. 1B). The carbonyl oxygen atom of donepezil was also recognized as a hydrogen-bond acceptor. The phenyl ring acted as the aromatic ring, while the protonated nitrogen atom located at the center functioned as the cation. The excluded volumes were generated according to the spatial arrangements of the key residues. The volumes could exclude the intermolecular collision of a potential hit; thus, the model is more precise and more consistent with the nature of the active binding site. Primary hits from the last step were subjected to a second round of virtual screening, and 1089 compounds with fit values above 3.0 predicted by the SBP model were screened out.
The binding patterns of these compounds were then predicted using the CDOCKER docking module in DS.27 After visualization of the binding modes, 106 compounds were retained for subsequent manual selection to ensure the structural diversity of the hits. Finally, 24 compounds (Fig. 2) were purchased from the Chemdiv collection, with purity >95.0%, and their ChE inhibitory activities were determined. The detailed screening workflow is shown in Fig. 3.
 | ||
| Fig. 2 The structures of the 24 potential hits from the virtual screening. | ||
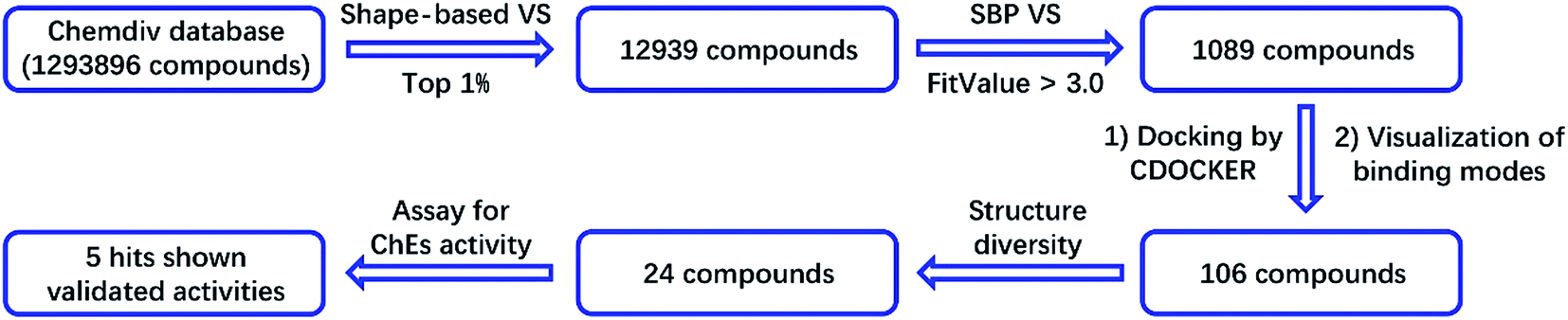 | ||
| Fig. 3 Virtual screening workflow for the identification of new ChE inhibitors. | ||
2.2 ChE inhibitory activities of the five hit compounds
The inhibitory activities of the 24 hits against AChE from Electrophorus electricus (eeAChE) and BuChE from equine serum (eqBuChE) were investigated, following Ellman's method.28 Donepezil and tacrine were applied as positive controls for AChE and BuChE, respectively. The hits were first screened for their inhibitory activities on AChE and BuChE at a concentration of 10 μM. This preliminary assay resulted in five hits (C629-0196, G070-1566, G115-0283, G801-0274, and F048-0694) which exhibited over 50.0% inhibitory effects on AChE and/or BuChE (Table 1). They were subsequently assayed for dose-dependent inhibitory activity (Fig. 4), and their IC50 values were calculated (Table 1). For AChE, the five hits showed IC50 values of 1.28 ± 0.83, 2.10 ± 0.40, 6.61 ± 2.73, 2.05 ± 0.11, and 3.78 ± 0.32 μM, respectively. The AChE inhibitory curves (Fig. 4A) showed dose-dependent activities for all the compounds. However, C629-0196 did not achieve complete inhibition at the highest concentration (100 μM), leading to a large S.E.M. value. This was due to the poor solubility of the compound at concentrations above 100 μM. Interestingly, C629-0196 did not show any inhibition of BuChE (only 5.63% IR at 10 μM), indicating that it may serve as a good template for the design of a selective AChE inhibitor. For BuChE, dose-dependent curves of the 4 compounds, G070-1566, G115-0283, G801-0274, and F048-0694, were well fitted (Fig. 4B), resulting in IC50 values of 0.78 ± 0.14, 5.11 ± 4.21, 0.031 ± 0.006, and 2.03 ± 0.35 μM, respectively. Surprisingly, G801-0274 exhibited a remarkable nanomolar-range potency and very high selectivity for BuChE (AChE IC50/BuChE IC50 ratio = 66.13). Recent studies have demonstrated the significant role of BuChE, especially in the AChE-deficient brains of AD patients;14–16 there is currently an urgent call for the discovery of selective BuChE inhibitors. This compound will no doubt provide a very good starting point for the discovery of highly selective BuChE inhibitors.| Chemdiv ID | AChEa | BuChEb | Ratioe | ||
|---|---|---|---|---|---|
| % IRc | IC50d (μM) | % IRc | IC50d (μM) | ||
| a AChE (EC 3.1.1.7) from electric eel.b BuChE (EC 3.1.1.8) from horse serum.c % inhibitory rate (IR) at a concentration of 10 μM.d Concentration of the compound required for 50% inactivation of ChEs; data are shown as mean ± S.E.M. of three experiments.e Ratio = AChE IC50/BuChE IC50.f This compound was not soluble under the test conditions. | |||||
| C749-0087 | 28.60 | nd | 12.90 | nd | — |
| 8003-0197 | 6.28 | nd | 16.13 | nd | — |
| C611-0434f | nd | nd | nd | nd | — |
| C629-0196 | 54.12 | 1.28 ± 0.83 | 5.63 | nd | — |
| C437-0121 | 38.59 | nd | 5.63 | nd | — |
| 4964-1479 | 5.65 | nd | 3.42 | nd | — |
| 5905-2743 | 8.94 | nd | −2.73 | nd | — |
| C590-0093 | 10.35 | nd | 4.11 | nd | — |
| K284-5464 | 2.59 | nd | 4.79 | nd | — |
| 6048-0476 | 4.41 | nd | 11.64 | nd | — |
| G070-1566 | 82.94 | 2.10 ± 0.40 | 86.30 | 0.78 ± 0.14 | 2.69 |
| G421-0271 | 44.72 | nd | 16.20 | nd | |
| G900-0252 | 29.73 | nd | 17.53 | nd | |
| D305-0628 | 7.67 | nd | −0.64 | nd | |
| F687-0560 | 16.84 | nd | 7.14 | nd | |
| D434-0535 | 4.59 | nd | 4.76 | nd | |
| G115-0283 | 53.80 | 6.61 ± 2.73 | 47.89 | 5.11 ± 4.21 | 1.29 |
| G801-0274 | 81.61 | 2.05 ± 0.11 | 98.70 | 0.031 ± 0.006 | 66.13 |
| F048-0694 | 90.28 | 3.78 ± 0.32 | 76.62 | 2.03 ± 0.35 | 1.86 |
| C599-1080 | 4.13 | nd | 8.84 | nd | — |
| C590-0150 | 8.38 | nd | −2.04 | nd | — |
| S211-1317 | 29.28 | nd | 4.08 | nd | — |
| S211-1044 | 26.81 | nd | 13.38 | nd | — |
| P867-0190 | 13.86 | nd | 7.75 | nd | — |
| Donepezil | — | 0.008 ± 0.001 | — | nd | — |
| Tacrine | — | 0.07 ± 0.01 | — | 0.03 ± 0.002 | 2.33 |
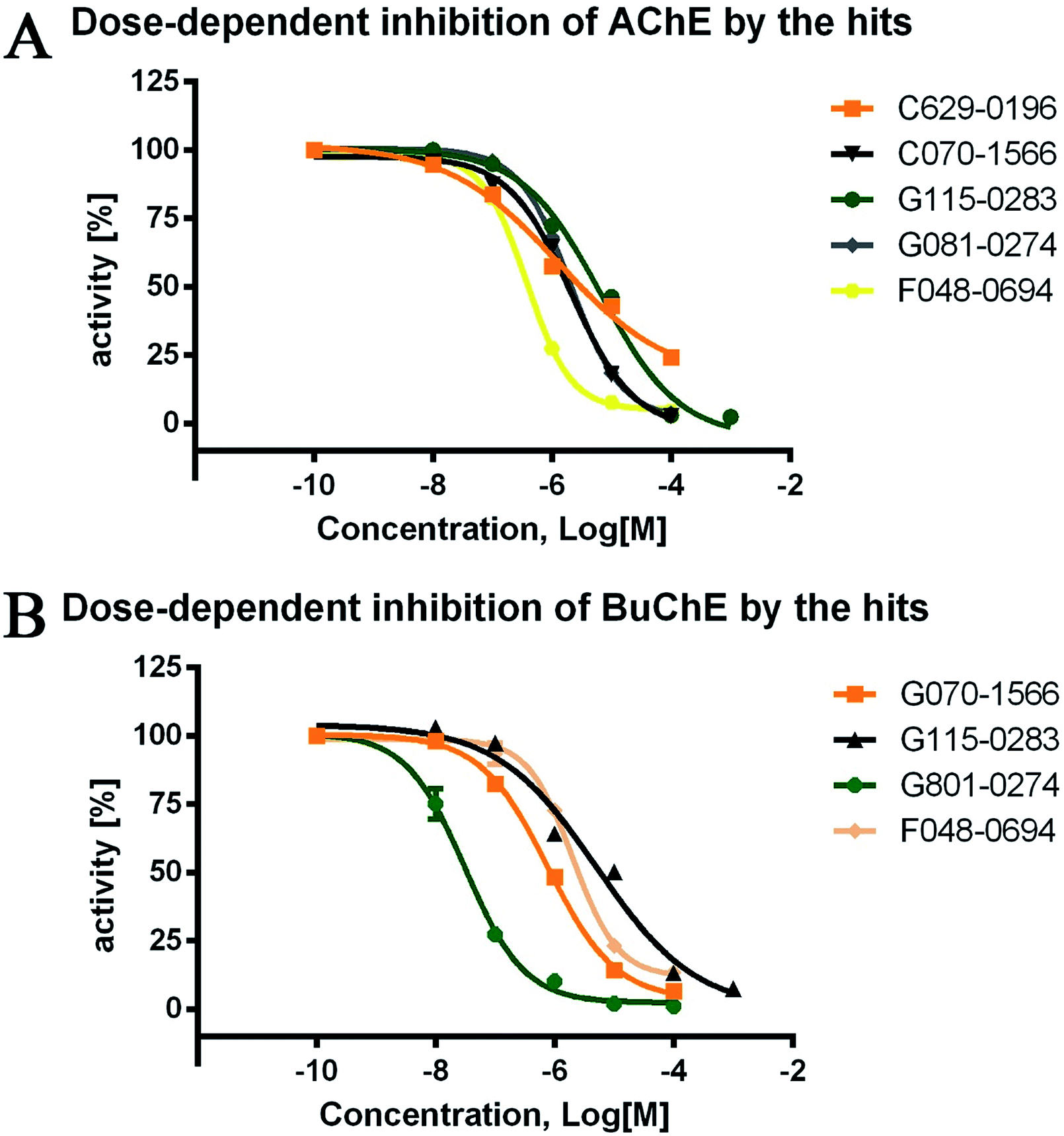 | ||
| Fig. 4 Dose-dependent inhibitory curves of the five hits against AChE and BuChE. The initial concentration was set as 100 μM (except for G115-0283, which was set as 1 mM) and then diluted 10 times to obtain the three other concentrations. The lowest concentration of the test compounds was set as 0.1 nM. Donepezil and tacrine were used as the positive controls for AChE and BuChE, respectively. | ||
To remove potential false positive compounds, the 24 hits were then projected to the Pan Assay Interference Compounds (PAINS) filter; this was performed through an online server (http://www.cbligand.org/PAINS/, created and maintained by Prof. Xiang-Qun (Sean) Xie's laboratory, School of Pharmacy, University of Pittsburgh). The results showed that among the 24 potential hits, 22 compounds passed the PAINS filter, including all 5 validated active hits. Detailed results from the PAINS filter are summarized in the ESI (Table S1†).
To further investigate the binding manner of the hits to the ChEs, F048-0694 and G801-0274 were selected to perform kinetic studies with AChE and BuChE, respectively. Lineweaver–Burk reciprocal plots were applied as described previously29 to elucidate the kinetic properties and inhibitory modes of the two compounds. Generally, Lineweaver–Burk plots can be described by reciprocal rates versus reciprocal substrate concentrations for different inhibitor concentrations resulting from the substrate–velocity curves for ChEs. The detailed values of Km and Vmax for the two compounds at different concentrations are listed in Table 2. For F048-0694 (Fig. 5A), both slopes (decreased Vmax), and the intercepts (higher Km) varied with increasing concentration (0.2, 0.4, 0.6, and 1.0 μM), suggesting mixed inhibition of AChE by this compound. The substrate–velocity curve (Fig. 5B) showed that F048-0694 reduced the enzymatic velocity of the AChE-substrate catalytic reaction in a dose-dependent manner. For G801-0274 (Fig. 5C and D), data from different concentrations (10, 30, 50, and 70 nM) also exhibited mixed inhibition of BuChE and a dose-dependent decrease of the enzymatic velocity of the BuChE-substrate catalytic reaction, similar to that of F048-0694. These results indicated that the two compounds may simultaneously bind to CAS and PAS when interacting with the targets.
| Concentration (μM) | Vmax (μM min−1) | Km (μM) | R squared |
|---|---|---|---|
| F048-0694 against AChE | |||
| 0 | 3.66 ± 0.34 | 319.70 ± 53.13 | 0.98 |
| 0.2 | 2.38 ± 0.26 | 265.20 ± 55.72 | 0.96 |
| 0.4 | 1.94 ± 0.20 | 254.50 ± 50.42 | 0.96 |
| 0.6 | 2.24 ± 0.09 | 319.30 ± 60.48 | 0.97 |
| 1.0 | 0.95 ± 0.07 | 223.00 ± 30.95 | 0.96 |
|
|||
| G801-0274 against BuChE | |||
| 0 | 0.36 ± 0.008 | 85.12 ± 5.12 | 0.99 |
| 10 | 0.30 ± 0.014 | 128.00 ± 14.51 | 0.98 |
| 30 | 0.28 ± 0.019 | 178.50 ± 27.04 | 0.97 |
| 50 | 0.17 ± 0.007 | 89.02 ± 10.97 | 0.97 |
| 70 | 0.14 ± 0.005 | 95.78 ± 10.23 | 0.98 |
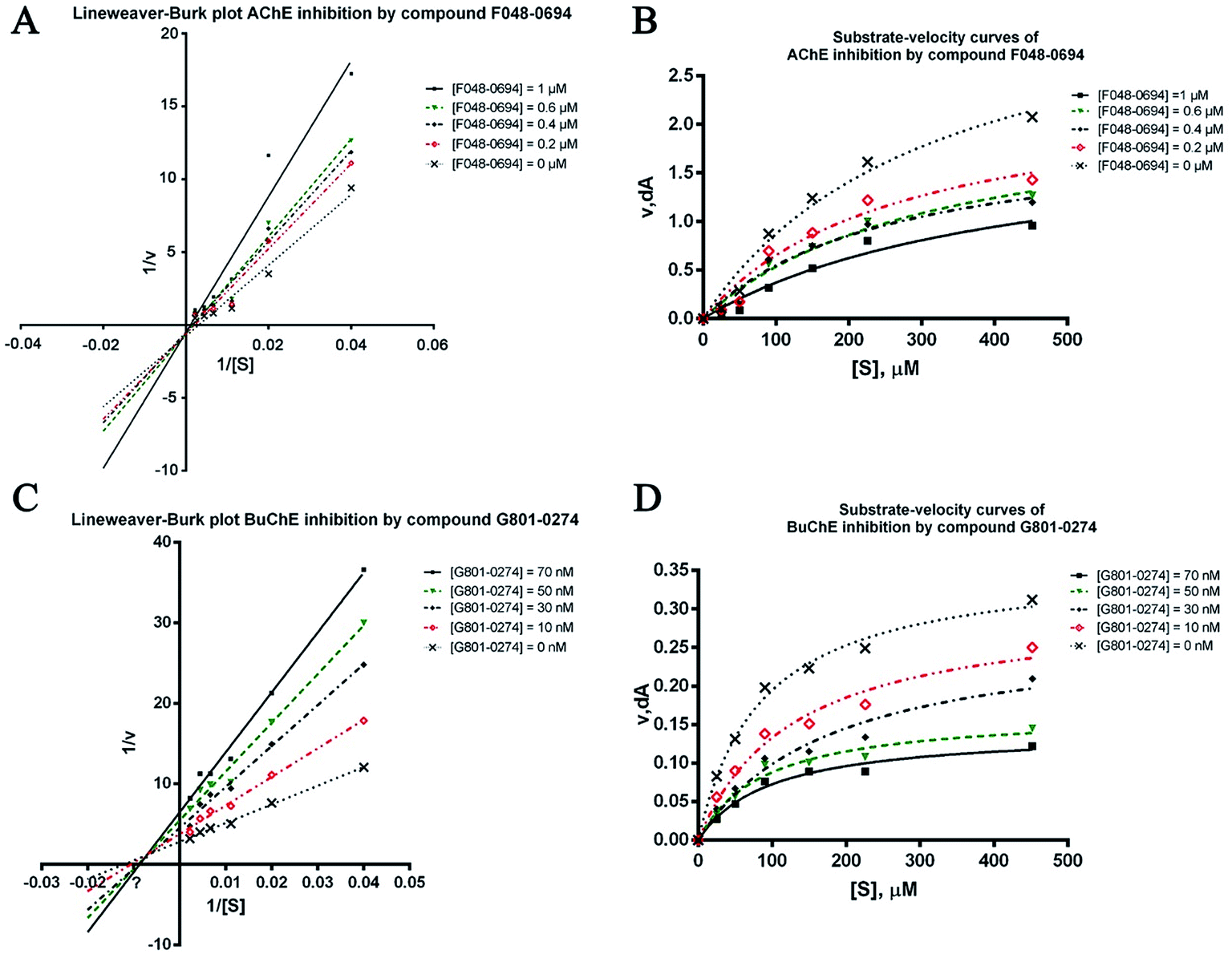 | ||
| Fig. 5 Lineweaver–Burk plots resulting from subvelocity curves of the AChE and BuChE activities with different substrate concentrations (25 to 450 μM) in the absence and presence of F048-0694 and G801-0274 at the indicated concentrations. | ||
2.3 Binding mode elucidation of the five hit compounds
Detailed binding patterns of the five hits to AChE were investigated by the CDOCKER molecular docking module implemented in BioVIA Discovery Studio 2016. Generally, all the hits were inserted well into the binding groove of AChE by simultaneously interacting with CAS and PAS, indicating a mixed binding pattern, which was consistent with the results of the kinetic studies. For C629-0196 (Fig. 6A), the tricyclic ring was bound to the CAS of AChE, forming strong π–π stacking contact with the aromatic side chain of Trp84 and Tyr121. The piperidine amide linker was located in the narrow groove between CAS and PAS. The terminal phenyl group bound to PAS through interactions with Trp279 and Tyr 334. For G070-1566 (Fig. 6B), the phenyl ring became located in the CAS by interacting with Trp84. Hydrophobic contacts could also be observed between this phenyl ring with the side chains of residues in the CAS. The sulphanilamide of G070-1566 formed a hydrogen bond with the hydroxyl group of the side chain of Tyr121. This interaction could improve the binding affinity and stabilize the binding conformation to provide the interaction in the PAS, which was achieved by the pyrrole ring. The binding mode of G115-0283 (Fig. 6C) was similar to that of G070-1566. The sulphanilamide also formed a hydrogen bond with Tyr121, and the benzyl group became bound to the PAS by interacting with Trp279. The bicyclic ring was inserted into the CAS. It is noteworthy that the lactam moiety of the seven-membered ring formed two hydrogen bonds with His440, which is a critical residue in the Ser200–Glu327–His440 catalytic traid of AChE. Therefore, these interactions can hinder the approach of acetylcholine to the CAS, thus enhancing enzyme inhibition. For G801-0274 (Fig. 6D), the 1,3-dimethyl-1,3-dihydro-2H-benzo[d]imidazol-2-one moiety acted as the binder for CAS by interacting with the side chains of Trp84 and Phe330. The sulphanilamide also formed a hydrogen bond with Tyr121, enhancing the binding affinity. The benzyl group was located at the PAS and formed hydrophobic contact with Trp279. For F048-0694 (Fig. 6E), the benzyl ring was inserted deeply into the CAS and made contact with Trp84 through π–π interactions. Additionally, the bicyclic ring occupied the rest of the CAS, forming cation–π interactions with Asp72 through the thiazole ring. Another π–π contact to Tyr334 was also observed through this moiety. The 2,3-dihydrobenzo[b][1,4]dioxine moiety became bound to the PAS and made contact with Trp279 through π–π interactions.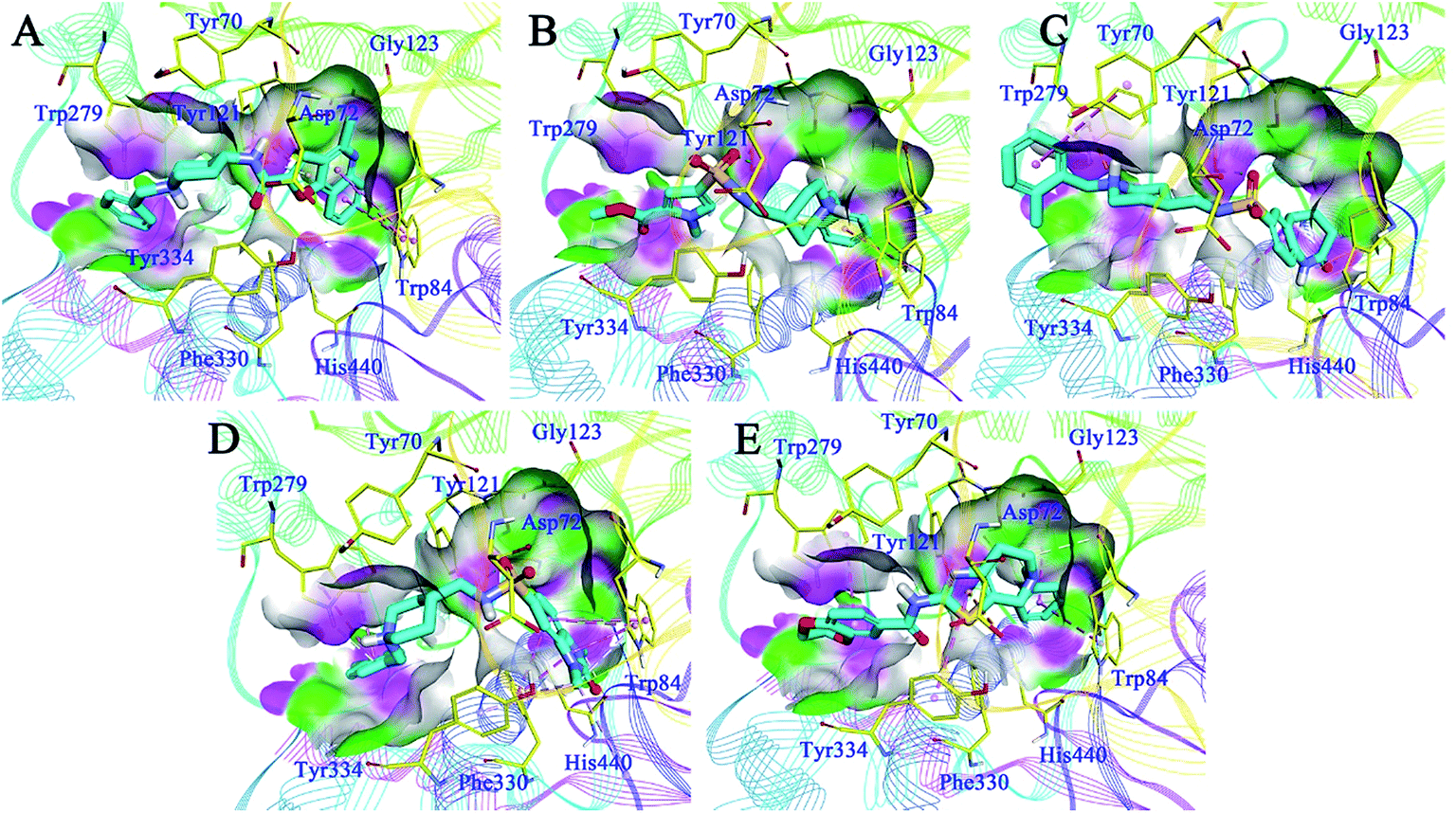 | ||
| Fig. 6 Binding mode predictions for C629-0196 (A), G070-1566 (B), G115-0283 (C), G801-0274 (D), and F048-0694 (E) with AChE (PDB ID: 2CKM). The compounds are shown in light blue stick mode; key residues are shown in yellow stick mode. Only polar hydrogen atoms are shown. Hydrophobic contacts and π–π stacking are depicted with purple dotted lines; H-bonds are represented with green dotted lines. | ||
The binding patterns of the four active compounds with BuChE were also analysed by molecular docking. Generally, these compounds bound to BuChE in similar U-shaped conformations, except for F048-0694; this can be attributed to its more rigid scaffold compared to the other three compounds. The conformations were much different from these line-shaped conformations when bound to AChE. In detail, for G070-1566 (Fig. 7A), the benzyl group was inserted into the bottom of the acyl pocket and formed hydrophobic contacts with Trp82, which is a key residue in the CAS of BuChE. Aided by the U-shaped binding conformation, the pyrrole ring became located at the middle of the CAS; thus, it occupied a much larger pocket compared to AChE. The ester moiety provided two hydrogen bonds with Gly116 and Ser198, which further stabilized the conformation. The piperidine sulphanilamide linker of G070-1566 was located at the open mouth of the CAS. The protonated nitrogen atom on the piperidine ring formed multiple cation–π interactions with the side chain of Tyr332, while the sulphanilamide moiety formed hydrogen bonds with Val288; this further improved the polar intermolecular recognition and resulted in sub-micromolar potency. For G115-0283 (Fig. 7B), the bicyclic ring was located at the bottom of the CAS; the oxygen atom of the lactam moiety functioned similar to that of the ester group of G070-1566, forming three hydrogen bonds with Gly116 and Ser198. The sulphanilamide group was involved in the hydrogen bond contact with Trp82. The protonated nitrogen atom on the piperidine ring formed a cation–π interaction with the side chain of Tyr332. The terminal benzyl group was located at the open mouth of the pocket. Remarkably, we observed polar conditions in this region; therefore, introducing polar substitutions into this benzyl ring may form rational polar contacts and thus improve the inhibitory potency. For G801-0274 (Fig. 7C), the 1,3-dimethyl-1,3-dihydro-2H-benzo[d]imidazol-2-one moiety inserted deeply into the bottom of the acyl pocket and formed strong hydrophobic interactions with the aromatic side chain of Trp82. The imidazolidin-2-one ring formed hydrogen bond networks with Gly116; these can provide strong intermolecular recognitions between the compound and BuChE. The methyl group on this ring interacted with Phe329 through hydrophobic contact, which further improved its binding affinity. The terminal benzyl group pointed to the outside of the pocket. These interactions resulted in very strong binding at the bottom of the pocket, thus preventing binding of the substrate and leading to high inhibitory potency. F048-0694 (Fig. 7D) became bound to BuChE in a line-shaped conformation, as mentioned above. The benzyl group was inserted into the bottom of the CAS and interacted with Trp82 through π–π stacking. The thiazole ring formed a π–π stacking interaction with Tyr332. The 1,4-dioxane moiety was located at the open edge of the pocket. The oxygen atoms formed polar contacts with the backbone of Val288, supporting the above hypothesis that the introduction of polar groups at this region can provide additional intermolecular recognition.
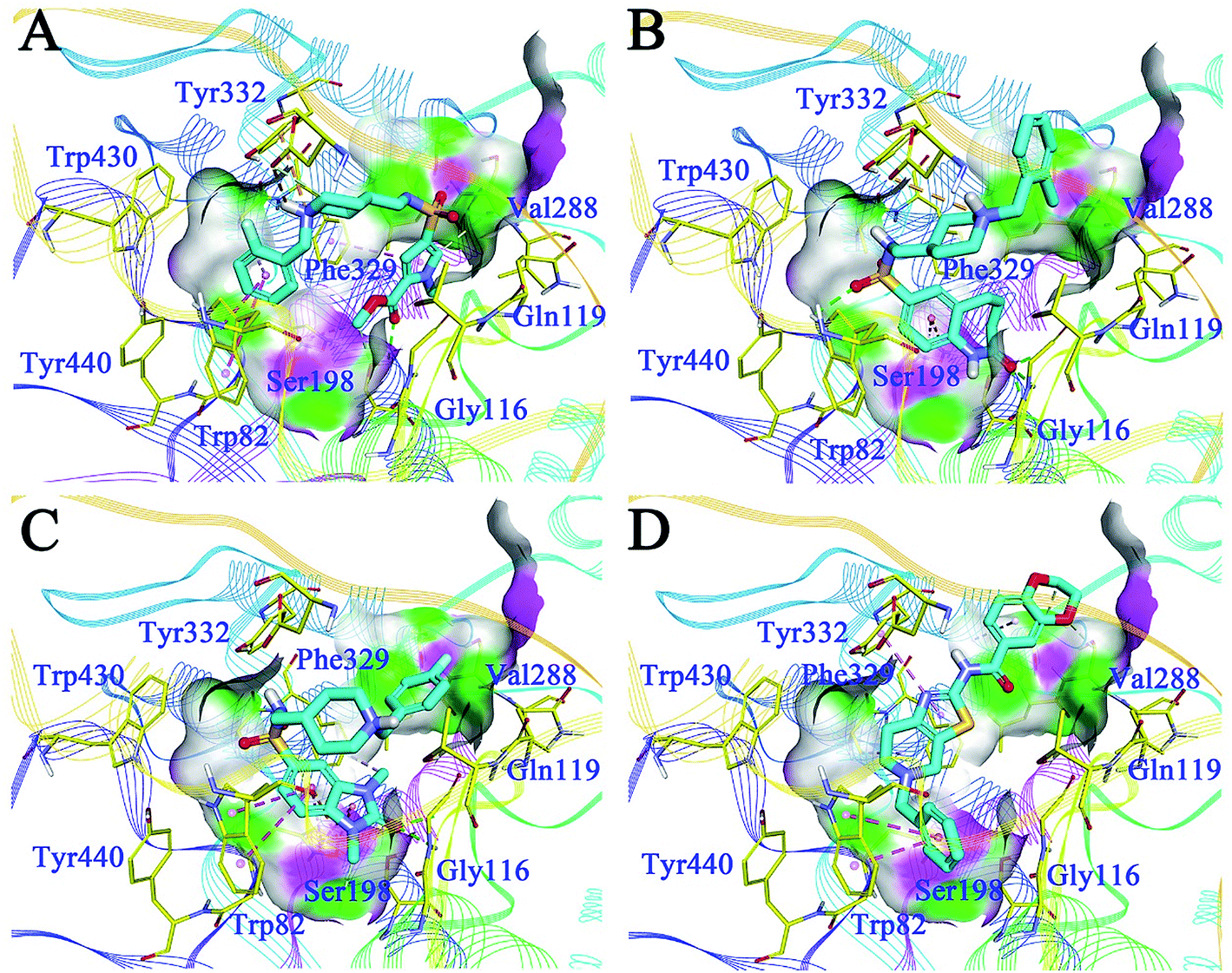 | ||
| Fig. 7 Binding mode predictions for G070-1566 (A), G115-0283 (B), G801-0274 (C), and F048-0694 (D) with BuChE (PDB ID: 4TPK). Compounds were shown in light blue stick mode; key residues were shown in yellow stick mode. Only polar hydrogen atoms were shown. Hydrophobic contacts and π–π stacking are depicted with purple dotted lines; H-bonds are represented by green dotted lines. | ||
As compound G801-0274 exhibited the highest selectivity for BuChE among all the hits, we next analysed the reason for this phenomenon. We superimposed the binding structure of this compound on AChE and BuChE (Fig. 8A). The carbon atoms of G801-0274 and the key residues of AChE are colored white, while the corresponding atoms in BuChE are colored blue. Compared to the conformation in AChE, G801-0274 exhibited a more contracted pose in BuChE in order to occupy the larger active site. However, if G801-0274 uses the same conformation to bind with AChE, there are two intermolecular collisions; one involves the sulphanilamide moiety, and the other takes place at the phenyl ring (the two positions are highlighted with dotted lines). Considering the larger binding site of BuChE, introducing bulky groups at these positions may enhance the target selectivity. Additionally, there is no doubt that the U-shaped conformation of G801-0274 will cost additional energy and lead to higher internal energy during the induced-fit binding process; introduction of bulky groups may help to restrict the flexible conformation when the compound binds to BuChE. Finally, introducing hydrogen bond donating or accepting groups at the imidazolidin-2-one moiety may enhance the polar intermolecular recognition and thus improve the activity. The detailed design strategy is summarized in Fig. 8B.
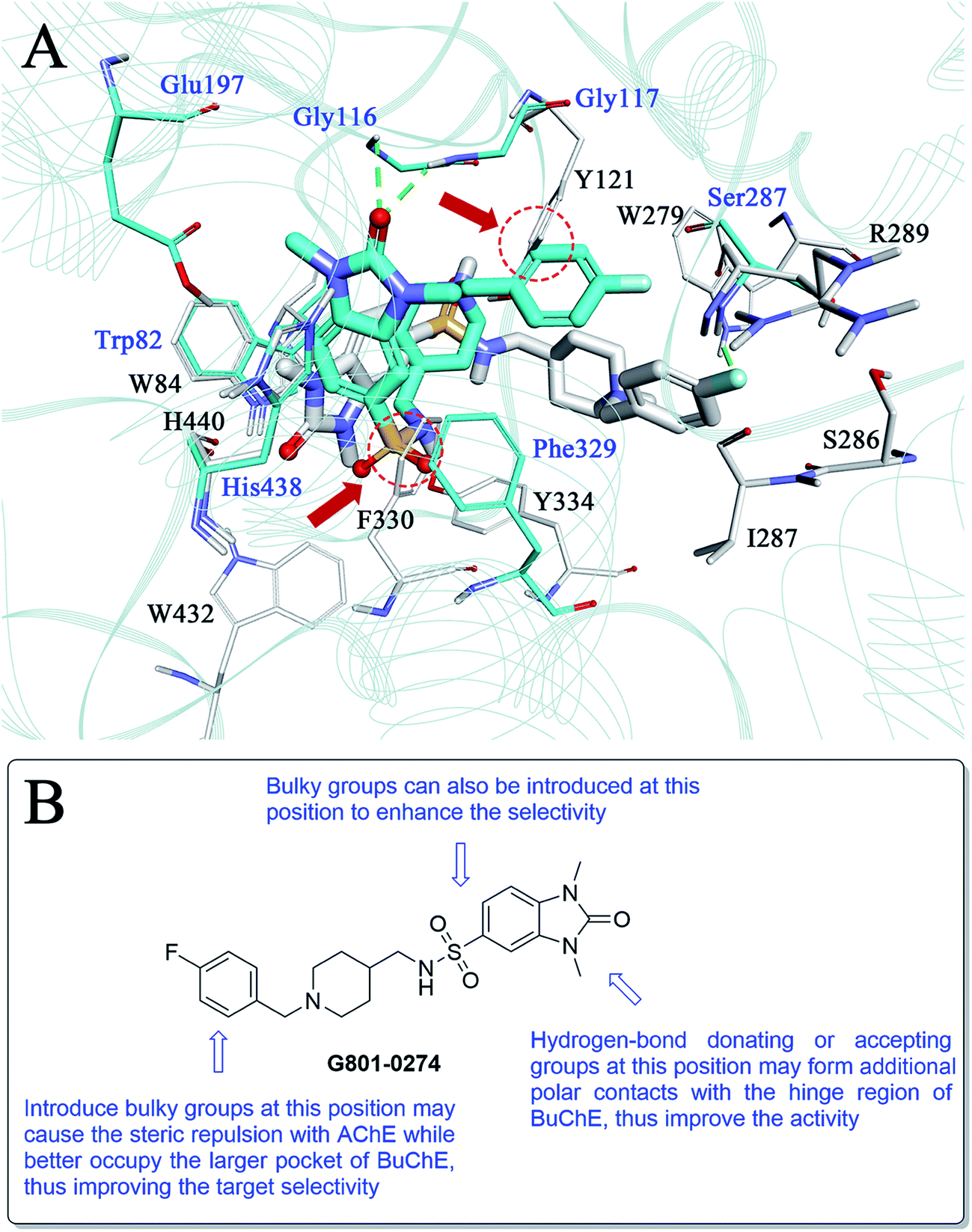 | ||
| Fig. 8 (A) Binding mode differences between G801-0274–AChE and G801-0274–BuChE. The carbon atoms of G801-0274 and the key residues of AChE are colored white, while the corresponding atoms in BuChE are colored blue. (B) Detailed modification strategy of G801-0274 to improve the target selectivity. | ||
3. Conclusion
In the present study, we demonstrate the effectiveness of using a hierarchical structure-based virtual screening workflow to identify new ChE inhibitors. Specifically, we applied 3D shape-based similarity screening combined with the SBP model to efficiently narrow down 1293896 compounds to 1089 compounds, which were further reduced to 106 compounds using molecular docking. The final 24 hits were subjected to biological validation with ChEs, leading to five active hits. To the best of our knowledge, these compounds have not been previously reported to be ChE inhibitors; thus, they provide interesting templates for the design of new inhibitors. Among them, G801-0274 exhibited a low nanomolar range IC50 (0.031 ± 0.006 μM) and high selectivity (ratio = 66.13) against BuChE. Another compound, C629-0196, showed a moderate micromolar range IC50 (1.28 ± 0.83 μM) against AChE; however, it had no activity against BuChE. These compounds provide good starting points for the discovery of highly selective ChE inhibitors. Additionally, we also determined the ClogP values of the 5 active hits (Table S2†). Although lower than that of donepezil, the ClogP values of the hits still remained in an acceptable range (all of them were above 3); C629-0196 shows a comparable value (ClogP = 4.21) to donepezil. Therefore, these hits possess fundamentally lipophilic character which can be improved by further structural optimization.
4. Experimental section
4.1 Creation of the ROCS overlay and SBP model
Shape-based overlays used in further virtual screening were generated using the ROCS module (version 3.1.2) of the Openeye suite. Donepezil was used as a template molecule. The binding conformation of donepezil in complex with human AChE (PDB ID: 4EY7) was extracted and applied directly in the generation of the model. The Chemdiv compound collection was handled by the Omega module (version 2.4.6) to minimize the ligands and generate multiple conformations. For the superimposition of the molecules, a smooth Gaussian function was used to represent the molecular volume. Subsequently, the overlay of the molecules was corrected by simple matching of chemical functionalities.The SBP model was generated using the Receptor–Ligand Pharmacophore Generation module in DS. The co-crystal structure of donepezil in complex with human AChE (PDB ID: 4EY7) was used to generate the model. The structure was first prepared using the Prepare Protein module in DS and was then subjected to model generation. The minimum and maximum features of the model were set to 4 and 6, respectively. A maximum of ten models were generated.
4.2 Virtual screening of the Chemdiv database
For the ROCS model, virtual screenings were performed based on the query overlay of donepezil. The parameters for the ROCS run were set as follows: rankby = combo and besthits = 1. Other parameters were set as default. For the SBP model, the hits from the ROCS screening were first subjected to the generation of multiple conformations using the Build 3D Database module. Align Ligand was set to True, and Search Method was set to Best. Other parameters were set as default.4.3 Molecular docking
The docking studies were performed with the CDOCKER module implemented in DS. The principle of CDOCKER can be briefly summarized as follows: CDOCKER generates ligand “seeds” to populate the binding pocket. Each seed is then subjected to high temperature molecular dynamics (MD) using a modified version of the CHARMm force field. The structure after the MD run was then fully minimized under the forcefield. The solutions were then clustered according to position and conformation and were ranked by energy. The co-crystal structures of AChE bound with bis(7)-tacrine (PDB ID: 2CKM) and BuChE with a small molecule inhibitor, naphthamide (PDB ID: 4TPK) were used for molecular docking. The binding sites were defined according to the native ligand. The heating steps, cooling steps, and cooling temperatures were set to 5000, 5000, and 310, respectively. Other parameters were maintained as the defaults.Finally, 24 hits were purchased from the Chemdiv database, with purity >95% (liquid chromatography-mass spectrometry, LC-MS).
4.4 In vitro cholinesterase inhibition assay
The assay followed the method of Ellman et al., using a Shimadzu 160 spectrophotometer. AChE (EC 3.1.1.7, Type VI-S, from Electric Eel, C3389) and BuChE (EC 3.1.1.8, from equine serum, C0663), 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB, D218200), acetylthiocholine iodide (ATC, A5751), and butyrylthiocholine iodide (BTC, B3253) were purchased from Sigma-Aldrich (St. Louis, MO, USA). AChE/BuChE stock solution was prepared by adjusting 500 units of the enzyme and 1 mL of gelatin solution (1% in water) to 100 mL with water. The solution was further diluted before use to give 2.5 units per mL. ATC/BTC iodide solution (0.075 M) was prepared in deionized water. DTNB solution (0.01 M) was prepared in water containing 0.15% (w/v) sodium bicarbonate. For buffer preparation, potassium dihydrogen phosphate (1.36 g, 10 mmol) was dissolved in 100 mL of water. The pH of the solution was adjusted to 8.0 ± 0.1 with KOH. Stock solutions of the test compounds were dissolved in ethanol to give final concentrations of 10−4 M when diluted to the final volume of 3.32 mL. For each compound, a dilution series of at least five different concentrations (normally 10−4 to 10−9 M) were prepared.For measurement, a cuvette containing 3.0 mL of phosphate buffer, 100 μL of AChE or BuChE, and 100 μL of the test compound solution was allowed to stand for 5 min before 100 μL of DTNB were added. After the addition of 20 μL of ATC or BTC, the reaction was initiated and the solution was mixed immediately. Two minutes after substrate addition, the absorption was determined at 25 °C at 412 nm. For the reference value, 100 μL of water replaced the test compound solution. For determining the blank value, 100 μL of additional water replaced the enzyme solution. The measurement for each concentration was performed in triplicate. The inhibition curve was fitted by plotting the percentage enzyme activity (100% for the reference) versus the logarithm of the test compound concentration. The IC50 values were calculated by GraphPad Prism 5, and the data were shown as mean ± SEM.
4.5 Kinetic study
Kinetic studies were performed in the same manner as the determination of ChE inhibition; the substrate (ATC/BTC) was used in concentrations of 25, 50, 90, 150, 226, and 452 μM. For F048-0694, the concentrations were set to 0.2, 0.4, 0.6, and 1.0 μM. For G801-0274, the concentrations were set to 10, 30, 50, and 70 nM. The enzymatic reaction was extended to 4 min before the determination of the absorption. The Vmax and Km values of the Michaelis–Menten kinetics were calculated by nonlinear regression from substrate–velocity curves using GraphPad Prism 5. Linear regression was used to fit the Lineweaver–Burk plots.Acknowledgements
We are grateful for the support from grants 81402851 and 81573281 of the National Natural Science Foundation of China and grant BK20140957 of the Natural Science Foundation of Jiangsu Province. We are also grateful for the support from the Fundamental Research Funds for the Central Universities (2015ZD009), Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP-PPZY2015A070) and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- V. Tumiatti, A. Minarini, M. L. Bolognesi, A. Milelli, M. Rosini and C. Melchiorre, Curr. Med. Chem., 2010, 17, 1825–1838 CrossRef CAS PubMed.
- H. Tang, H. T. Zhao, S. M. Zhong, Z. Y. Wang, Z. F. Chen and H. Liang, Bioorg. Med. Chem. Lett., 2012, 22, 2257–2261 CrossRef CAS PubMed.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741–766 CAS.
- P. T. Francis, A. M. Palmer, M. Snape and G. K. Wilcock, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 137–147 CrossRef CAS.
- E. Karran, M. Mercken and B. De Strooper, Nat. Rev. Drug Discovery, 2011, 10, 698–U1600 CrossRef CAS PubMed.
- C. Ballatore, V. M. Y. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2007, 8, 663–672 CrossRef CAS PubMed.
- K. M. Webber, A. K. Raina, M. W. Marlatt, X. Zhu, M. I. Prat, L. Morelli, G. Casadesus, G. Perry and M. A. Smith, Mech. Ageing Dev., 2005, 126, 1019–1025 CrossRef CAS PubMed.
- B. Su, X. Wang, A. Nunomura, P. I. Moreira, H. G. Lee, G. Perry, M. A. Smith and X. Zhu, Curr. Alzheimer Res., 2008, 5, 525–532 CrossRef CAS PubMed.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- L. Devi and H. K. Anandatheerthavarada, Biochim. Biophys. Acta, 2010, 1802, 11–19 CrossRef CAS PubMed.
- U. Holzgrabe, P. Kapkova, V. Alptuzun, J. Scheiber and E. Kugelmann, Expert Opin. Ther. Targets, 2007, 11, 161–179 CrossRef CAS PubMed.
- E. Giacobini, R. Spiegel, A. Enz, A. E. Veroff and N. R. Cutler, J. Neural Transm., 2002, 109, 1053–1065 CrossRef CAS PubMed.
- M. Pohanka, Biomed. Pap., 2011, 155, 219–229 CrossRef CAS PubMed.
- J. Hartmann, C. Kiewert, E. G. Duysen, O. Lockridge, N. H. Greig and J. Klein, J. Neurochem., 2007, 100, 1421–1429 CrossRef CAS PubMed.
- N. H. Greig, T. Utsuki, D. K. Ingram, Y. Wang, G. Pepeu, C. Scali, Q. S. Yu, J. Mamczarz, H. W. Holloway, T. Giordano, D. Chen, K. Furukawa, K. Sambamurti, A. Brossi and D. K. Lahiri, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17213–17218 CrossRef CAS PubMed.
- I. Manoharan, R. Boopathy, S. Darvesh and O. Lockridge, Clin. Chim. Acta, 2007, 378, 128–135 CrossRef CAS PubMed.
- S. N. Dighe, G. S. Deora, E. De la Mora, F. Nachon, S. Chan, M. O. Parat, X. Brazzolotto and B. P. Ross, J. Med. Chem., 2016, 59, 7683–7689 CrossRef CAS PubMed.
- Y. Bourne, P. Taylor, Z. Radic and P. Marchot, EMBO J., 2003, 22, 1–12 CrossRef CAS PubMed.
- J. L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker and I. Silman, Science, 1991, 253, 872–879 CrossRef CAS PubMed.
- M. L. Raves, M. Harel, Y. P. Pang, I. Silman, A. P. Kozikowski and J. L. Sussman, Nat. Struct. Biol., 1997, 4, 57–63 CrossRef CAS PubMed.
- M. Bartolini, C. Bertucci, V. Cavrini and V. Andrisano, Biochem. Pharmacol., 2003, 65, 407–416 CrossRef CAS PubMed.
- N. C. Inestrosa, J. P. Sagal and M. Colombres, Subcell. Biochem., 2005, 38, 299–317 CAS.
- M. Bajda, N. Guzior, M. Ignasik and B. Malawska, Curr. Med. Chem., 2011, 18, 4949–4975 CrossRef CAS PubMed.
- A. Saxena, A. M. Redman, X. Jiang, O. Lockridge and B. P. Doctor, Biochemistry, 1997, 36, 14642–14651 CrossRef CAS PubMed.
- Y. Chen, Z. L. Liu, T. M. Fu, W. Li, X. L. Xu and H. P. Sun, Bioorg. Med. Chem. Lett., 2015, 25, 3442–3446 CrossRef CAS PubMed.
- J. Kirchmair, S. Distinto, P. Markt, D. Schuster, G. M. Spitzer, K. R. Liedl and G. Wolber, J. Chem. Inf. Model., 2009, 49, 678–692 CrossRef CAS PubMed.
- G. Wu, D. H. Robertson, C. L. Brooks 3rd and M. Vieth, J. Comput. Chem., 2003, 24, 1549–1562 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres Jr and R. M. Feather-Stone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- Y. Chen, J. Sun, L. Fang, M. Liu, S. Peng, H. Liao, J. Lehmann and Y. Zhang, J. Med. Chem., 2012, 55, 4309–4321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The SMILES format of all the hits from the virtual screening, the results from PAINS filtering, the scores of the active hits from molecular docking, and the calculated ClogP values of the active hits. See DOI: 10.1039/c6ra25887e. |
| This journal is © The Royal Society of Chemistry 2017 |