
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSucrose modulates insulin amyloid-like fibril formation: effect on the aggregation mechanism and fibril morphology†
Carlotta Marasini *a,
Vito Foderàb and
Bente Vestergaard*a
*a,
Vito Foderàb and
Bente Vestergaard*a
aDepartment of Drug Design and Pharmacology, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark. E-mail: carlotta.marasini@sund.ku.dk; bente.vestergaard@sund.ku.dk
bSection for Biologics, Department of Pharmacy, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark
First published on 7th February 2017
Abstract
Co-solutes, such as sugars, are used in in vitro protein aggregation experiments to mimic crowding and, in general, complex environments. Sugars often increase the stability of the native protein structure by affecting inter- and intramolecular protein–protein interactions. This, in turn, modifies the protein self-assembly pathways. Using a combination of fluorescence spectroscopy, synchrotron radiation circular dichroism and transmission electron microscopy, we study the kinetics of formation and structural properties of human insulin fibrils in the presence of sucrose. The presence of sucrose results in a delay of the onset of fibrillation. Moreover, it leads to a dramatic change in both the morphology and overall amount of fibrils. Our results emphasize that the detailed composition of protein surroundings likely influences not only the fibrillation kinetics but also the balance between different species, potentially determining fibril strains with different biological activities. This aspect is crucial in the etiology of pathologies associated with amyloidosis.
Introduction
The environment in a living cell consists of a multitude of components which co-exist in a soluble state at very high concentrations.1 Proteins, RNA and DNA often occupy 30–40% of the entire volume.1–4 This both reduces the mobility and modifies the extent of the physico–chemical interactions between macromolecules.5 In vitro, it is possible to mimic such a complex environment by adding macromolecular crowding agents (MCA). Typical agents are inert, un-charged and can be of different sizes (e.g. polysaccharides, dextran, sucrose, trimethylamine oxide). Osmolytes can act either as stabilizers or destabilizers depending on whether the osmolyte is excluded or accumulated around the protein backbone, i.e. preferential hydration.6 Sucrose was used in several studies on the folding and stability of, e.g. lysozyme,4 cytochrome C,7 RNase8 and insulin.9 In all these cases the proteins tend to assume a more folded and compact structure in the presence of sucrose.Protein amyloid-like aggregation is related to devastating pathologies as Alzheimer's and Parkinson's diseases. During this process, the protein native state converts into a partially unfolded non-native state and then associates into smaller, soluble oligomers of different sizes. Subsequently, elongated fibrils are formed (i.e. fibrillation). The initial formation of oligomeric species is considered to be a nucleated reaction,10–12 while the formation of the elongated structures is also controlled by auto-catalytic processes known as secondary nucleation processes.13,14 Importantly, monomers, oligomers, and fibrils coexist in an equilibrium that is influenced by the environment.15–17 In vivo, proteins experience a naturally complex environment that affects inter- and intra-protein interactions. This may modify the fibrillation process17,18 via changing both protein conformational stability and the tendency to form complexes. As a consequence, the effect of MCA on the fibrillation reaction is expected to be multifactorial. It acts both at the level of the reaction and the final structures, influencing the complex equilibrium between the different structural species. This can determine the formation of species with different toxic potency and with various biological effects.19,20 As a consequence, disentangling the different effects of MCA on protein self-assembly processes is crucial in the etiology of amyloid-related diseases. Some general trends are evident from the existing literature on in vitro experiments. MCA have different effects on the aggregation process of intrinsically disordered proteins (IDPs)21,22 compared to the one of (natively) folded proteins. In an aggregation process, IDPs convert from a disordered state into a relatively defined and aggregation-prone conformation.23 The latter is favored by the presence of MCA,7,24,25 making the aggregation process more likely. In contrast, MCA prevent a globular/folded protein to adopt a relatively unfolded state. This, in turn, inhibits the self-assembly process. In the case of IDPs, it has been shown that both the onset and the speed of fibrillation are enhanced in a complex environment.26–29 For globular/folded protein there is evidence of either an enhancement15,27 or inhibition of the fibrillation process.19,30–33 And finally, the effect of chaperones on protein aggregation in different environments has been studied.34 The above studies are focused on how and if a complex environment affects the fibrillation kinetics. On the contrary, an analysis of the fibril structural changes induced by MCA is still lacking, with only few evidences reported in the literature.35
In this article, we aim to clarify how sucrose can affect structural and kinetic aspects of the amyloid fibril formation in the case of a natively globular protein. We used human insulin as a model system11,36,37 and we monitored the fibrillation process in the presence of sucrose using microscopy and spectroscopy techniques. Previous studies suggest that sucrose favors a more compact and folded state of native insulin.9,33 However, it is still unclear how and if sucrose influences the insulin fibrillation kinetics and the morphology of the final fibrils. We focus on the overall effect induced by sucrose (from 10 to 40% w/v) on both the fibrillation kinetics and the fibril morphologies. We highlight an effect on the temporal features of the kinetics. Moreover, our data show that the structure of a soluble native-like species co-existing with the mature fibrils is significantly stabilized by the presence of sucrose. This provides a proof of sucrose-induced changes not only on the fibrillation kinetics but also on the structural equilibria between the occurring species.
Materials and methods
Materials and sample preparation
Thioflavin T (ThT), recombinant human insulin (91077C) and all other chemicals are obtained from Sigma, St. Louis, MO. Stock solutions of 60% sucrose (w/v, 1.75 M) in 20% acetic acid, pH 1.8 are prepared weighting the correct amount of sucrose in a solution of 20% acetic acid (v/v) and used for making diluted solutions at 45%, 30%, and 15% w/v sucrose. The diluted solutions are stored at −18 °C. Before each preparation, the sucrose solutions are thawed, heated and filtered at 80 °C using a 0.22 μm syringe filter (Ministart, 16534 Sartorius). The final concentration of sucrose is further evaluated by monitoring the mass per volume, as well as the refractive index measured by an optical refractometer (data not shown). A protein stock solution is obtained by dissolving the insulin powder at a concentration of 20 mg ml−1 in 20% acetic acid, 0.03 M NaCl, pH 1.8. The solution is filtered using a 0.20 μm spin filter (UFC30GV00, Millipore). The concentration is determined by absorption at 280 nm (extinction coefficient of 1.0675 cm−1) measured by using a Nanodrop UV-vis spectrophotometer (Thermo Scientific). From this stock solution, insulin samples at a final concentration of 5 mg ml−1 insulin, 20% acetic acid, pH 1.8, 10 mM NaCl and 0, 10, 20, 30 and 40% w/v of sucrose are prepared. Here after, % of sucrose always indicates % w/v of sucrose.Fibrillation assays
Fibrillation is thermally induced and monitored by in situ ThT (20 μM).38,39 Fluorescence is detected using a Fluostar Optima plate reader (BMG labtech) in 96-well optical bottom plates (Thermo Scientific). 150 μl is the total volume for each well and samples are incubated at 60 °C or 45 °C without shaking. The measurements are carried out at an excitation wavelength of 450 nm, and the ThT emission is recorded at 480 nm (using 5 nm excitation and emission bandwidth). Fibrils are collected after the ThT fluorescence intensity reaches the plateau value for the imaging analysis.Experiments on sonicated samples (after fibrillation) are performed at 30 °C in the plate reader for 24 hours. The fibrils are sonicated for 1 minute using a micro-sonicator Sonopuls from Bandelin, setting the intensity to 50% with 30 pulses at 0.1 s frequency. 20 μM of ThT are added, and the fluorescence is followed during 24 hours.
The ThT signal is recorded as a function of the incubation time and is normalized by the average of the fluorescence value in the plateau. Temporal profiles are described using two parameters the time at which insulin fibrillation initiated (lag time) and the ThT fluorescence final value (FFV).40,41 The data presented are an average of 20 different curves, for each sucrose concentration, recorded from five different experiments.
Transmission electron microscopy
Grids are prepared as described by Smith and co-workers.40 Briefly, 5 μl of the sample (0.5 mg ml−1) are loaded onto copper 400 mesh grids (Agar Scientific, Stansted, UK) coated with Formvar and carbon film. After 60 s, 10 μl of distilled water is added, and excess water is removed. Subsequently, 10 μl of 2% (v/v) uranyl acetate (Agar Scientific) is placed on the grid and left for 30 s. Finally, 2 × 10 μl distilled water are added and again excess water is removed. The grid is then left to dry. Images are acquired at the Core Facility for Integrated Microscopy (CFIM), at the Department of Health and Medical Sciences, University of Copenhagen. Images are collected using a CM100 transmission electron microscope operating at an acceleration voltage of 80 kV. The program ImageJ is used for performing a statistical analysis of the fibril dimensions. Twenty different images are analyzed to estimate radius and clustering parameters. The statistical ensemble for the imaging analysis consists of a total number of 200 fibrils for each sucrose concentration, made in 3 different sample preparations.Synchrotron radiation circular dichroism (SRCD)
The measurements are performed at the Aarhus University, CD beam line on ASTRID2. Samples are measured at an insulin concentration of approximately 5 mg ml−1 using a cuvette with a path length of 16 μm. The short path length enables a relatively high protein concentration, thereby allowing for direct comparison between CD spectra and ThT fluorescence observations. Ellipticity is monitored at wavelengths from 170 nm to 280 nm. The ellipticity at each wavelength is measured 15 times and averaged automatically. Several spectra are recorded over around 20 minutes for checking the stability of the signal over time. The spectra are then averaged and normalized to the concentration including only curves where the protein structure is stable. All spectra are smoothed using the Savitzky–Golay algorithm.42Results and discussion
Effect of sucrose on the human insulin fibrillation kinetics
Using the same approach as previously described,38,39 in situ ThT fluorescence is measured for monitoring the amyloid aggregate formation at 60 °C of 5 mg ml−1 human insulin at five different sucrose concentrations: 0%, 10%, 20%, 30% and 40% (black, green, red, pink and blue respectively in Fig. 1a). In all cases, a clear sigmoidal trend is observed. FFV is the fluorescence value at the plateau (approx. 5 hours) and, at the insulin concentration investigated here, it can be related to the overall amount of fibrils formed.37 Additionally, we determined the lag time calculated as the time at which the fluorescence ThT signal has increased to 10% of the FFV.41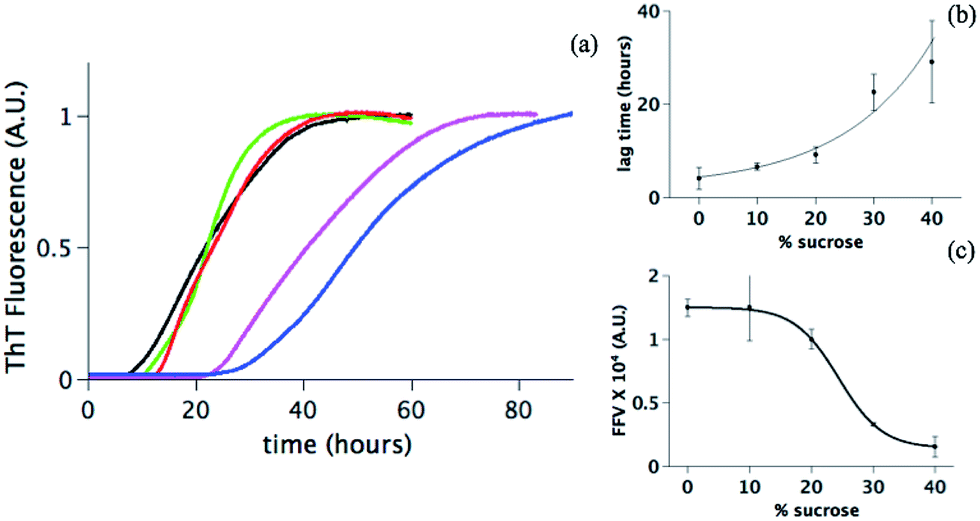 | ||
| Fig. 1 (a) Fibrillation of human insulin in 20% acetic acid, pH 1.8, 10 mM NaCl at different concentrations of sucrose: 0% (black line), 10% (green line), 20% (red line), 30% (pink line) and 40% (blue line). Each curve is an average (with standard deviation as an error) of at least 5 experiments where 20 different wells are recorded for each sucrose concentration. Two parameters are quantified (see Materials and methods) from the curves in panel a as a function of the amount of sucrose: (b) the lag time and (c) the final fibrils value (FVV) with a full line as a guide for the eyes. In both panel (b) and (c) each point is the average (with a standard deviation) of the value obtained from six independent experiments. | ||
Data in Fig. 1 suggest that sucrose changes the fibrillation kinetics. Upon increasing the sucrose concentration, the lag time increases, indicating that sucrose delays the onset of the fibril formation (Fig. 1b). Longer lag times could reflect a partial suppression of primary nucleation.43,44 This step is dependent on at least two factors: (1) the distribution of conformations, i.e. folded or (partially) unfolded vs. nucleation-prone, and (2) the diffusion of these molecules in solution determining the rate of interactions between molecules. Sucrose may modify both these factors.
The FFV values change significantly in the presence of sucrose (Fig. 1c). This can be due to (i) a reduced sensitivity of the ThT assay due to the presence of sucrose or (ii) a direct effect of sucrose on the fibril formation. In the first scenario, one possibility can be that sucrose interferes with the ThT signal. Sucrose molecules could indeed either organize themselves around or directly interact with the protein fibrils, thereby excluding the ThT molecules from the fibril accessible surfaces and hence reducing the fluorescence values.45,46 Alternatively, the drop of the FFV can be due to an increased tendency of the fibrils to cluster at increasing sucrose concentrations.47 The surface of fibrils is less exposed in a cluster; this reduces the possibility for ThT to bind to the cross-β structure and decreases the fluorescence signal.47–49 A final explanation can be that the presence of sucrose effectively reduces the quantity of formed fibrils. Our further investigations enable a distinction between these scenarios.
Sucrose affects the fibril morphology and tendency of clustering
A morphological investigation of the samples can highlight potential macroscopic structural differences and the overall extent of fibrillation. The Transmission Electron Microscopy (TEM) images in Fig. 2 show that fibrils are formed in all samples. However, as highlighted in Fig. 2 (colored squares), different morphologies can be observed as a function of the sucrose concentration.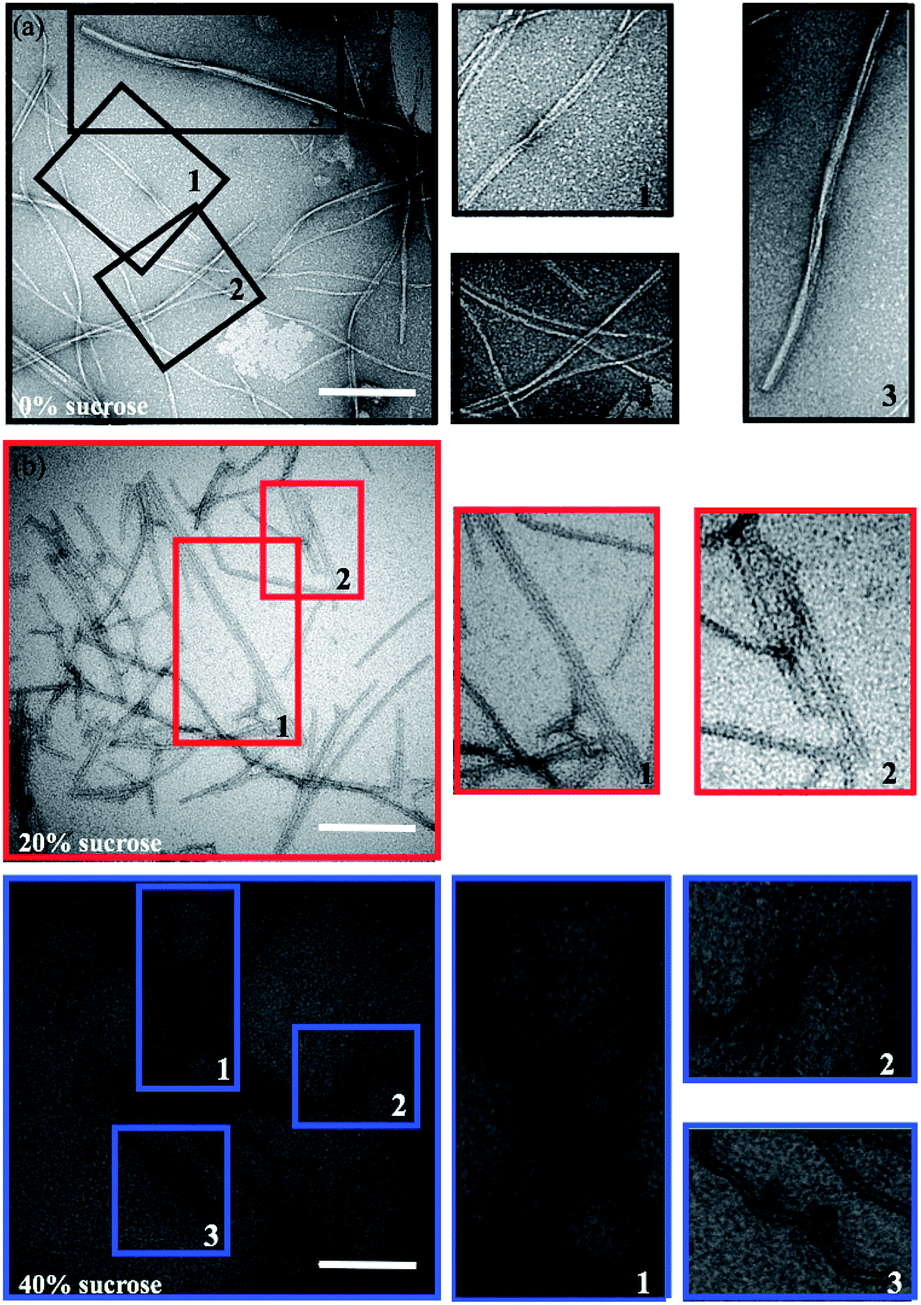 | ||
| Fig. 2 Representative TEM (a–c) images of insulin fibrils formed in presence of different amounts of sucrose: (a) 0% (black), (b) 20% (red) and (c) 40% (blue). Scale bars are 200 nm. The colored boxes in panels (a–c) indicate the areas acquired at higher resolution (right column). Magnifications highlight the different morphologies: insulin fibrils with a twisted helices morphology, straight parallel wires, and ribbon-like macroscopic structure. | ||
In the absence of sucrose, the fibrils have the typical helical twist morphology as observed for most amyloid fibrils (Fig. 2a).10,50 In the presence of 20% sucrose, fibrils become longer and linear and align in pairs (Fig. 2b). This is in accordance with a previous report.35 If the concentration of sucrose is 40%, fibrils are isolated, rather than clustered, with a pronounced length up to 10 μm (Fig. 2c). These fibrils are also less linear than the fibrils formed in 20% sucrose (additional examples of these different morphologies are shown in Fig. S1 in the ESI†). We estimated the section diameters of 200 fibrils for each sample using the program ImageJ (Fig. 3a). A decrease of the fibril section diameter is obtained already at 20% sucrose (Fig. 3a), and then is maintained at 40% sucrose (Table 1). As shown in Table 1 the number of isolated fibrils increases upon increasing the amount of sucrose. These data reject the hypothesis that the decreasing of the FFV at 40% sucrose may be due to a quenching of the fluorescence signal determined by fibril clustering.47
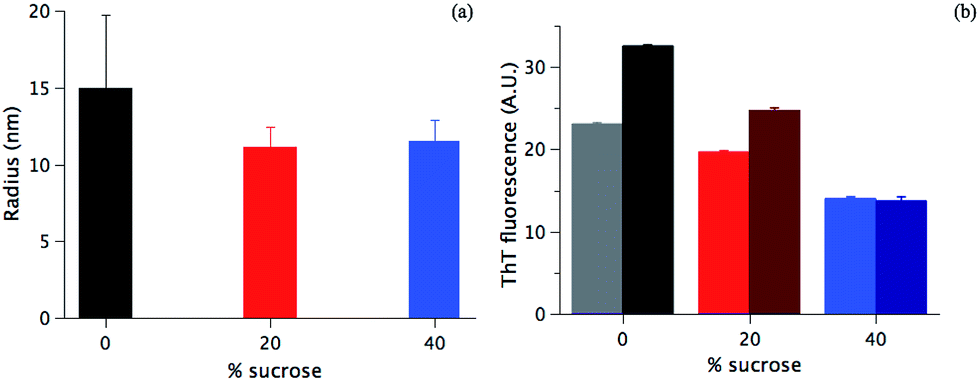 | ||
| Fig. 3 (a) Diameters of the fibril section formed in the presence of 0% (black column), 20% (red colour) and 40% (blue colour) sucrose (see Materials and methods), (b) ThT fluorescence signal of fibrils formed in a different amount of sucrose, the colour code as in (a) before (light column) and after gentle sonication (dark column). | ||
| Sample | Single fibrils | Clusters (2 fibrils) | Clusters (>2 fibrils) |
|---|---|---|---|
| 0% sucrose | 88% | 10% | 2% |
| 20% sucrose | 96% | 4% | 0% |
| 40% sucrose | 98% | 2% | 0% |
To verify the suggestion from the TEM data in Fig. 2a–c and Table 1, fibrils were sonicated (see Materials and methods), and the ThT fluorescence values were detected before and after the treatment. If there is a significant fibril cluster formation, the mechanical disruption would likely lead to a more efficient ThT staining of the single fibrils, i.e. an increase of ThT accessibility to the fibrils and ThT fluorescence signal.47,51 On the contrary, if sucrose is not affecting the fibril clustering, the sonication will have a minor effect, if any. As shown in Fig. 3b, at 40% sucrose no significant difference in the fluorescence intensity before and after the sonication is detected (light and dark blue column, respectively). On the contrary, in the absence of sucrose and at 20% sucrose, an increase of 45% and 28% of the signal is observed after sonication, respectively. Data in Fig. 3b corroborate the image analysis performed (Table 1) and shows that clustering mainly takes place in the absence of sucrose. This allows us to conclude that (1) sucrose is not increasing the fibril clustering and (2) likely does not affect the ThT staining. For both of the above hypotheses, an increase of the signal after the sonication would indeed have been detected, and such an increase would be expected to be increasingly significant at higher sucrose concentrations. This is not observed in our case.
The effect of sucrose on the equilibrium between different species during fibrillation
The evidence presented in Fig. 1, 2 and 3a could in principle be due to sucrose-induced changes to the initial structural state prior to the aggregation. Changes in the structural state will influence inter-protein interactions and hence the aggregation pathway.52,53 However, under the experimental conditions investigated here, the initial human insulin secondary structure is not affected by the presence of sucrose (Fig. S2a in ESI†). SRCD spectra show the same secondary structural content (85% of α-helices, 10% of random coil and 5% of β-turns) for the samples at 0%, 20%, and 40% sucrose. This initially suggests that the expected preferential hydration effect6,54 cannot explain the observed changes, since the structural state of the starting material is unaltered.From the data in Fig. 1 and 2, we can argue that at 40% sucrose concentration, we have a significant reduction in fibril formation, even if a potential role of sucrose-layers in altering the detection of fibrils cannot be completely ruled out. We used SRCD to have an indication of the residual monomer concentration at the endpoint of aggregation. Fig. S2b in ESI† shows the different SRCD spectra of the final state of the fibrillation in the presence of various amounts of sucrose. At the end of the kinetics, the protein is expected to be fully fibrillated53,55–57 and should hence show a CD signal typical for β-strands with a minimum at 216 nm and a maximum around 190 nm (as shown in Fig. S2b† in the absence of sucrose or with 10% sucrose). Seemingly, 40% sucrose lowers the fibril content, revealing an increasing occurrence of α-helical secondary structure. The presence of these structural elements either reflects significant structural changes in the fibril structure, or, alternatively, a shift in the equilibrium towards a monomeric state that co-exists with the fibrils in solution, i.e. a decrease of the fraction of protein in the fibril form. To distinguish between these options, we centrifuged the fibril samples, and the supernatant is characterized by SRCD (Fig. 4). If the sample is fully fibrillated, centrifugation should leave a very low concentration of the monomeric species and small oligomers in the supernatant, resulting in a low SRCD signal. For concentration of sucrose > 20%, the supernatant shows an increasingly intense signal upon increasing the sucrose concentration. Moreover, the SRCD signal from the soluble fraction sample at 40% sucrose is not statistically different from the initial structural state before fibrillation (Fig. S2a†). This suggests that a minor fraction of insulin molecules is converted into fibrils, while a larger fraction remains in a native-like soluble state. These results, thus, do highlight that even if the secondary structures of insulin at different sucrose concentrations are undistinguishable, the preferential hydration (modulated by the sucrose content) plays a significant role in stabilizing a native-like soluble state58 of the molecules that co-exist with the fibril state. Hence, in presence of sucrose the equilibrium between different states (native-like vs. fibrillation-prone)58 is likely to be altered, rather than the intrinsic structure of the soluble starting state.
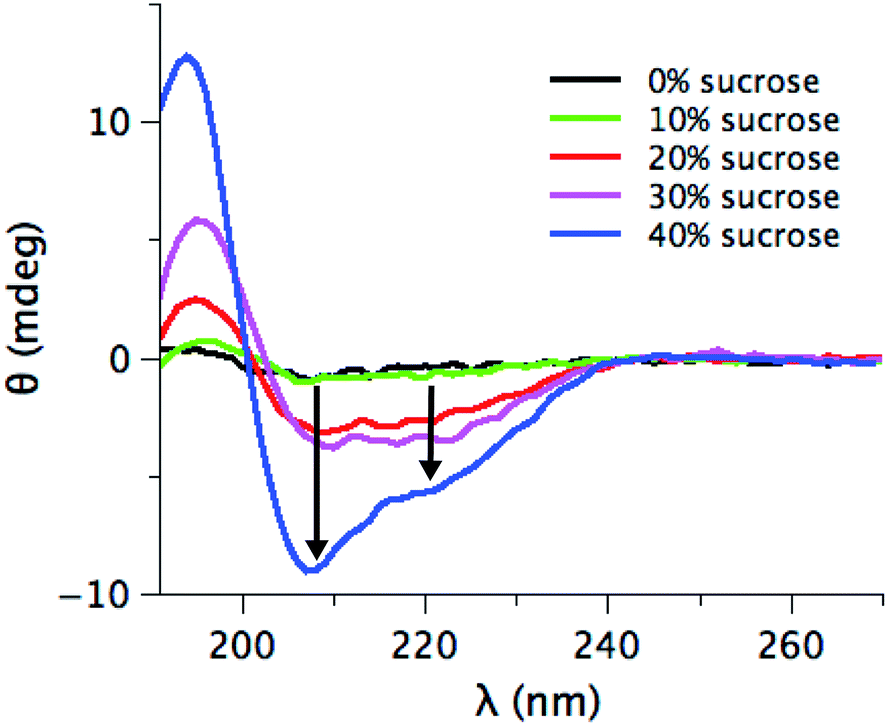 | ||
| Fig. 4 SRCD spectra of the supernatant obtained by centrifugation the final product of the fibrillation with different percentages of sucrose: 0% (black), 10% (green), 20% (red), 30% (pink) and 40% (blue). The two black arrows marked the typical minima for alpha-helix secondary structures. | ||
Data in Fig. 4 thus univocally suggest that the FFV decrease observed in Fig. 1c is mainly due to a shift of the protein ensemble towards a less fibrillation-prone insulin conformation, determining a dramatic reduction in the fraction of protein molecules converted into fibrils.
Earlier studies have shown that lysozyme,4 cytochrome C,7 RNase8 and insulin9 assume a more folded and compact structure in the presence of sucrose. Indeed, our results are also in agreement with the presence of a folded and native-like structure. An explanation for the observed phenomena may be the fact that sucrose is influencing both the protein–protein interactions (PPIs) and the diffusion of the molecules in solution. Data in Fig. S2a† show that sucrose does not directly alter the secondary structure of the insulin molecules. This result means that changes of PPIs are mainly related to solvent effects rather than specific changes in the structurally defined protein–protein interactions.59–61 Sucrose can shield the protein by transient interactions (i.e. an enthalpic effect), thereby decreasing the tendency of association,62,63 i.e. an increase in the repulsion barrier.64–67 In addition to this, in the presence of sucrose, the viscosity of the solution increases, strongly affecting the diffusion of the protein molecules in solution. This results in a decrease in the number of attempts for a single molecule to cross the repulsion barrier and associate to another molecule,68–70 affecting the lag time of the process. The combination of these two effects determines a shift of all the equilibria towards the monomeric state, delaying the aggregation process and the efficiency of the conversion into the aggregated state.
Conclusions
We presented an experimental study on insulin amyloid fibril formation in the presence of sucrose. Our data prove that sucrose delays insulin aggregation kinetics and the extent of fibrillation is strongly reduced upon increasing the sucrose concentration. This is mainly due to a shift of the equilibrium towards a less fibrillation-prone monomeric soluble state at increasing sucrose concentrations, which can be explained by preferential hydration.58 Moreover, sucrose influences fibril morphology and clustering, favoring the formation of isolated and long fibrils.Our findings highlight the role of sucrose not only as a stabilizer of the native state, as previously suggested in the literature, but also as a key factor for the formation of morphologically distinct amyloid fibrils. This is particularly relevant in the etiology of amyloidosis. Morphologically different fibrils can underlie different biological activities, such as different toxicity to neuronal cells or deposition patterns in diseases.71–76 As a consequence, the results reported here point toward a key role of MCA for in vivo protein aggregation.
Acknowledgements
CM and BV acknowledge funding from the Copenhagen University Programme of Excellence CoNEXT. Great collegial support during SRCD beamtime is acknowledged from Thomas S. Pedersen and Fatima Herranz-Trillo. We acknowledge the beamtime and the highly competent support by the staff at the SRCD beamline at ASTRID2. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen for access to performing the Transmission Electron Microscopy experiments. We acknowledge Valeria Vetri from Dipartimento di Fisica e Chimica & Aten Center, University of Palermo and Marco van der Weert for the preliminary measurements. VF acknowledges support from the FP7 Marie-Curie Actions Intra European Fellowship (IEF) for Career Development 2012–2014, project nr. 299385 “FibCat” (University of Copenhagen).Notes and references
- R. J. Ellis and A. P. Minton, Nature, 2003, 425, 27–28 CrossRef CAS PubMed
.
- S. B. Zimmerman and A. P. Minton, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 27–65 CrossRef CAS PubMed
- G. Rivas, F. Ferrone and J. Herzfeld, EMBO Rep., 2004, 5, 23–27 CrossRef CAS PubMed
- B. van den Berg, R. Wain, C. M. Dobson and R. J. Ellis, EMBO J., 2000, 19, 3870–3875 CrossRef CAS PubMed
- P. Ball, Chem. Rev., 2008, 108(1), 74–108 CrossRef CAS PubMed
- S. N. Timasheff, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(15), 9721–9726 CrossRef CAS PubMed
- K. Sasahara, P. McPhie and A. P. Minton, J. Mol. Biol., 2003, 32, 1227–1237 CrossRef
- Y. Qu and D. W. Bolen, Biophys. Chem., 2002, 101–102, 155–165 CrossRef CAS PubMed
- Y. S. Kim, L. S. Jones, A. Dong, B. S. Kendrick, B. S. Chang, M. C. Manning, T. W. Randolph and J. F. Carpenter, Protein Sci., 2003, 12, 1252–1261 CrossRef CAS PubMed
- F. A. Aprile, G. Meisl, A. K. Buell, P. Flagmeier, C. M. Dobson, M. Vendruscolo and T. P. J. Knowles, Biophys. J., 2014, 106, 268 CrossRef
- B. Vestergaard, M. Groenning, M. Roessle, J. S. Kastrup, M. van de Weert, J. M. Flink, S. Frokjaer, M. Gajhede and D. I. Svergun, PLoS Biol., 2007, 5, e134 Search PubMed
- C. C. Lee, A. Nayak, A. Sethuraman, G. Belfort and G. J. McRae, Biophys. J., 2007, 92, 3448–3458 CrossRef CAS PubMed
- F. A. Ferrone, J. Hofrichter and W. A. Eaton, J. Mol. Biol., 1985, 183, 611–631 CrossRef CAS PubMed
- A. Cacciuto, S. Auer and D. Frenkel, Nature, 2004, 428, 404–406 CrossRef CAS PubMed
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS PubMed
- B. Morel, L. Varela, A. I. Azuaga and F. Conejero-Lara, Biophys. J., 2010, 99, 3801–3810 CrossRef CAS PubMed
- R. L. Morris, K. Eden, R. Yarwood, L. Jourdain, R. J. Allen and C. D. Macphee, Nat. Commun., 2013, 4, 1891 CrossRef PubMed
- A. Nayak, C. C. Lee, G. J. McRae and G. Belfort, Biotechnol. Prog., 2009, 25, 1508–1514 CrossRef CAS PubMed
- M. Verma, A. Vats and V. Taneja, Ann. Indian Acad. Neurol., 2015, 18, 138–145 CrossRef PubMed
- L. Mucke and D. J. Selkoe, Cold Spring Harbor Perspect. Med., 2012, 7, a006338 Search PubMed
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS PubMed
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS PubMed
- Z. A. Levine, L. Larini, N. E. LaPointe, S. C. Feinstein and J. E. Shea, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2758–2763 CrossRef CAS PubMed
- J. Aden and P. Wittung-Stafshede, Biochemistry, 2014, 53, 2271–2277 CrossRef PubMed
- H. X. Zhou, FEBS Lett., 2013, 587, 1053–1061 CrossRef CAS PubMed
- V. N. Uversky, K. S. Bower, J. Li and A. L. Fink, FEBS Lett., 2002, 515, 99–103 CrossRef CAS PubMed
- L. A. Munishkina, E. M. Cooper, V. N. Uversky and A. L. Fink, J. Mol. Recognit., 2004, 5, 456–464 CrossRef PubMed
- D. M. Hatters, A. P. Minton and G. J. Howlett, J. Biol. Chem., 2002, 277, 7824–7830 CrossRef CAS PubMed
- Q. Ma, J. Y. Hu, J. Chen and Y. Liang, Int. J. Mol. Sci., 2013, 14, 21339–21352 CrossRef CAS PubMed
- J. Seeliger, A. Werkmuller and R. Winter, PLoS One, 2013, 8, e69652 CAS
- T. S. Choi, J. W. Lee, K. S. Jin and H. I. Kim, Biophys. J., 2014, 107, 1939–1949 CrossRef CAS PubMed
- S. Choudhary, N. Kishore and R. V. Hosur, Sci. Rep., 2015, 5, 17599 CrossRef CAS PubMed
- N. Estrela, H. G. Franquelim, C. Lopes, E. Tavares, J. A. Macedo, G. Christiansen, D. E. Otzen and E. P. Melo, Proteins: Struct., Funct., Bioinf., 2015, 83, 2039–2051 CrossRef CAS PubMed
- M. Gao, K. Estel, J. Seeliger, R. P. Friedrich, S. Dogan, E. E. Wanker, R. Winter and S. Ebbinghaus, Phys. Chem. Chem. Phys., 2015, 17, 8338–8348 RSC
- T. Hoppe and A. P. Minton, Biophys. J., 2015, 108, 957–966 CrossRef CAS PubMed
- F. Librizzi, V. Fodera, V. Vetri, C. Lo Presti and M. Leone, Eur. Biophys. J., 2007, 36, 711–715 CrossRef CAS PubMed
- V. Fodera, F. Librizzi, M. Groenning, M. van de Weert and M. Leone, J. Phys. Chem. B, 2008, 112, 3853–3858 CrossRef CAS PubMed
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed
- S. G. Bolder, L. M. Sagis, P. Venema and E. van der Linden, Langmuir, 2007, 23, 4144–4147 CrossRef CAS PubMed
- M. I. Smith, V. Fodera, J. S. Sharp, C. J. Robertsand and A. M. Donald, Colloids Surf B Biointerfaces, 2012, 89, 216–222 CrossRef CAS PubMed
- R. S. Pagano, M. Lopez Medus, G. E. Gomez, P. M. Couto, M. S. Labanda, L. Landolfo, C. D'Alessio and J. J. Caramelo, Biophys. J., 2014, 107, 711–720 CrossRef CAS PubMed
- A. Savitzky, Anal. Chem., 1964, 8, 1627–1639 CrossRef
- S. K. Shoffner and S. Schnell, Phys. Chem. Chem. Phys., 2016, 18, 21259–21268 RSC
- K. Eden, R. Morris, J. Gillam, C. E. MacPhee and R. J. Allen, Biophys. J., 2015, 108, 632–643 CrossRef CAS PubMed
- V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, V. N. Uversky, I. M. Kuznetsova and K. K. Turoverov, J. Phys. Chem. B, 2008, 112, 15893–15902 CrossRef CAS PubMed
- S. A. Hudson, H. Ecroyd, T. W. Kee and J. A. Carver, FEBS J., 2009, 276, 5960–5972 CrossRef CAS PubMed
- V. Fodera, M. van de Weert and B. Vestergaard, Soft Matter, 2010, 6, 4413–4419 RSC
- M. Groenning, M. Norrman, J. M. Flink, M. van de Weert, J. T. Bukrinsky, G. Schluckebier and S. FrokJaer, J. Struct. Biol., 2007, 159, 483–497 CrossRef CAS PubMed
- M. Groenning, L. Olsen, M. van de Weert, J. M. Flink, S. Frokjaer and F. S. Jorgensen, J. Struct. Biol., 2007, 158, 358–369 CrossRef CAS PubMed
- E. Chatani, Y. H. Lee, H. Yagi, Y. Yoshimura, H. Naiki and Y. Goto, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11119–11124 CrossRef CAS PubMed
- R. Jain and K. L. Sebastian, J. Phys. Chem. B, 2016, 120, 3988–3992 CrossRef CAS PubMed
- M. Muzaffar and A. Ahmad, PLoS One, 2011, 6, e27906 CAS
- M. Bouchard, J. Zurdo, E. J. Nettleton, C. M. Dobson and C. V. Robinson, Protein Sci., 2000, 9, 1960–1967 CrossRef CAS PubMed
- T. O. Street, D. W. Bolen and G. D. Rose, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13997–14002 CrossRef CAS PubMed
- M. I. Ivanova, S. A. Sievers, M. R. Sawaya, J. S. Wall and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2009, 45, 18990–18995 CrossRef PubMed
- M. I. Smith, J. S. Sharp and C. J. Roberts, Biophys. J., 2008, 7, 3400–3406 CrossRef PubMed
- U. Langhorst, J. Backmann, R. Loris and J. Steyaert, Biochemistry, 2000, 39, 6586–6593 CrossRef CAS PubMed
- J. C. Lee and S. N. Timasheff, J. Biol. Chem., 1981, 256, 7193–7201 CAS
- S. Grobelny, M. Erlkamp, J. Moller, M. Tolan and R. Winter, J. Chem. Phys., 2014, 141, 22D506 CrossRef CAS PubMed
- J. Janin and C. Chothia, J. Mol. Biol., 1976, 100, 197–211 CrossRef CAS PubMed
- B. Macdonald, S. McCarley, S. Noeen and A. E. van Giessen, J. Phys. Chem. B, 2015, 119, 2956–2967 CrossRef CAS PubMed
- S. Haldar, S. Mitra and K. Chattopadhyay, J. Biol. Chem., 2010, 285, 25314–25323 CrossRef CAS PubMed
- E. P. Melo, N. Estrela, C. Lopes, A. C. Matias, E. Tavares and V. Ochoa-Mendes, Curr. Protein Pept. Sci., 2010, 8, 744–751 CrossRef
- V. Fodera, A. Zaccone, M. Lattuadaand and A. M. Donald, Phys. Rev. Lett., 2013, 111, 108105 CrossRef PubMed
- E. J. Deeds, O. Ashenberg, J. Gerardin and E. I. Shakhnovich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14952–14957 CrossRef CAS PubMed
- A. Bhattacharya, Y. C. Kim and J. Mittal, Biophys. Rev., 2013, 5, 99–108 CrossRef CAS
- P. L. Kastritis and A. M. Bonvin, J. R. Soc., Interface, 2013, 10, 20120835 CrossRef PubMed
- H. S. Chung, S. Piana-Agostinetti, D. E. Shaw and W. A. Eaton, Science, 2015, 349, 1504–1510 CrossRef CAS PubMed
- F. J. de Meyer, M. Venturoli and B. Smit, Biophys. J., 2008, 95, 1851–1865 CrossRef PubMed
- A. N. Gupta, K. Neupane, N. Rezajooei, L. M. Cortez, V. L. Sim and M. T. Woodside, Nat. Commun., 2016, 7, 12058 CrossRef CAS PubMed
- R. A. Crowther and M. Goedert, J. Struct. Biol., 2000, 130, 271–279 CrossRef CAS PubMed
- A. T. Petkova, R. D. Leapman, Z. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed
- S. B. Malinchik, H. Inouye, K. E. Szumowski and D. A. Kirschner, Biophys. J., 1998, 74, 537–545 CrossRef CAS PubMed
- J. L. Jimenez, G. Tennent, M. Pepys and H. R. Saibil, J. Mol. Biol., 2001, 311, 241–247 CrossRef CAS PubMed
- B. Seilheimer, B. Bohrmann, L. Bondolfi, F. Muller, D. Stuber and H. Dobeli, J. Struct. Biol., 1997, 119, 59–71 CrossRef CAS PubMed
- M. Meyer-Luehmann, J. Coomaraswamy, T. Bolmont, S. Kaeser, C. Schaefer, E. Kilger, A. Neunschwander, D. Abramowski, P. Frey, A. L. Jaton, J. M. Vigouret, P. Paganetti, D. M. Walsh, P. M. Mathews, J. Ghiso, M. Staufenbiel, L. C. Walker and M. Jucker, Science, 2006, 313, 1781–1784 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data and technical details of the experiments. See DOI: 10.1039/c6ra25872g |
| This journal is © The Royal Society of Chemistry 2017 |