
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of hybrid multi-shell hollow structured CeO2–MnOx materials for selective catalytic reduction of NO with NH3†
Kaili Maab,
Weixin Zouab,
Lei Zhangc,
Lulu Liab,
Shuohan Yuab,
Changjin Tang*ab,
Fei Gaob and
Lin Dong *ab
*ab
aKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: tangcj@nju.edu.cn
bJiangsu Key Laboratory of Vehicle Emissions Control, Center of Modern Analysis, Nanjing University, Nanjing 210093, P. R. China. E-mail: donglin@nju.edu.cn
cSchool of Environmental and Chemical Engineering, Chongqing Three Gorges University, Wanzhou 404000, P. R. China
First published on 18th January 2017
Abstract
Hollow structured CeO2–MnOx hybrid materials with up to three shells were prepared successfully by using carbon spheres as the hard template. It was found that the shells could be well controlled by simply adjusting the calcination rate. Elemental mapping results showed Mn and Ce species exhibited a homogeneous spatial distribution and the existence of Mn could improve the diffusion of CeO2 into the carbon spheres during the construction of the multi-shell structure, suggesting the intensive cooperative interaction between Mn and Ce. When triple-shell CeO2–MnOx hollow spheres were used as a catalyst for selective catalytic reduction of NO with NH3, superior low-temperature catalytic performance was exhibited, compared with traditional CeO2–MnOx nanoparticles, single-shell and double-shell hollow spheres. Combined with XRD, H2-TPR and XPS characterization, it was indicated that the synergistic effects and surface active species enhanced by the special multi-shell CeO2–MnOx hollow structures could account for the excellent performances. The results of the present study shed light on the creation of complex and hybrid hollow structured materials for superior performance in catalysis fields, like NO reduction for environmental protection.
1 Introduction
Ceria, as a typical rare earth oxide, owns considerable intrinsic properties such as abundant oxygen vacancy defects, high oxygen storage capacity and alterable valence states,1 which result in its extensive applications in the field of diesel soot oxidation,2 CO oxidation,3 photocatalysis,4,5 NO reduction,6,7 and so on. Nevertheless, pure ceria is rarely used in catalysis due to its inherent poor activity. It is recognized that the combination of CeO2 and transition as well as noble metals, for instance, Cu,8 Mn,6 Nb,9 Pt10 and Au,11 is beneficial to its properties. Besides, manganese hybrid oxides, with variable valences and resulting intrinsic environmentally benign features as well as excellent catalytic activity, have been considered as promising catalysts for redox reactions.12–14 Thus, much attention has been paid to CeO2–MnOx composites due to their admirable performance in catalysis, for example, the low-temperature deNOx reaction.6,15,16 The advantageous interactions between Ce and Mn species bring about the excellent redox ability, amphoteric property and thermal stability, which are helpful for the catalytic application of CeO2–MnOx composites.17Generally, compared with the irregular particles, catalysts with well-designed structure are supposed to exhibit amazing functions on account of the size, shape, interface effects, etc.18 Thus, considerable researches have been focused on preparing CeO2–MnOx composites with specific structures. Chen et al. prepared the Ce–Mn binary oxide hollow cubes by a novel method, which obtained excellent CO oxidation activity and remarkable adsorption capacity of Congo red.19 Putla and the co-workers designed a heteronanostructure of CeO2 nanocubes (100) with dispersed MnOx nanoparticles for diesel soot oxidation and continuous-flow benzylamine oxidation.2 Besides, Wei et al. reported a kind of MnO2 doped CeO2 catalyst with tailored 3-D channels, which possessed large surface area for chemical absorption, as well as proper channels for diffusion.20 Hence, the controlled fabrication of well-structured CeO2–MnOx composites is of great significance to catalysis field.
Recently, hollow structured materials with features of low density, high permeability and confined inner cavity have captured tremendous attention.5,21–24 Especially, the fabrication of multi-shell hollow spheres with special isolated chambers and controllable shell structures attracted considerable attention.25,26 According to the literatures, multi-shell CeO2 hollow spheres have been successfully synthesized.4,11,27 Nevertheless, to the best of our knowledge, hybrid multi-shell ceria-based hollow spheres with diverse compositions have rarely been prepared, and the relationship of their structure and catalytic performance is not reported yet.
Therefore, in this study, by using carbon spheres as a hard template, we reported for the first time the fabrication of CeO2–MnOx hollow spheres with adjustable shells via simple modification of calcination rate for the first time. The obtained samples were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET), X-ray photoelectron spectroscopy (XPS), X-ray fluorescence analysis (XRF), H2-temperature programmed reduction (H2-TPR) and tested in the reaction of NO reduction by NH3 (NH3-SCR). Results showed that Mn and Ce were well distributed on the hybrid hollow spheres, and there existed strong cooperative interaction between Mn and Ce in formation of the multi-shells. In comparison with traditional CeO2–MnOx nanoparticles, the multi-shell hollow sphere catalysts exhibited superior low-temperature performance in NH3-SCR, particularly for the triple-shell sample.
2 Experimental section
2.1 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nMn:nNH4Ac = 0.371:0.629:11.86) were dissolved together in 20 mL absolute ethanol to form a clear solution, into which 0.2 g carbon spheres were ultrasonically dispersed. Then the dark brown suspension was transferred into a 50 mL Teflon-lined stainless steel autoclave and maintained at 180 °C for 4 h in oven. After it was cooled down, the brown solid product could be collected via centrifugation at 10000 rpm for 5 min, followed by being washed with distilled water and absolute ethanol several times, respectively. The precursor was dried at 80 °C and then calcined in air at 600 °C for 3 h with different defined heating rates (2 °C min−1, 5 °C min−1 and 10 °C min−1) to get the anticipated MSCMHSs. Individual cerium and manganese hollow spheres could be synthesized without another raw material. Additionally, traditional CeO2–MnOx nanoparticles (NP) with identical molar ratio were also prepared through the same method without carbon spheres (heating rate being of 10 °C min−1) for comparison purposes.
:nMn:nNH4Ac = 0.371:0.629:11.86) were dissolved together in 20 mL absolute ethanol to form a clear solution, into which 0.2 g carbon spheres were ultrasonically dispersed. Then the dark brown suspension was transferred into a 50 mL Teflon-lined stainless steel autoclave and maintained at 180 °C for 4 h in oven. After it was cooled down, the brown solid product could be collected via centrifugation at 10000 rpm for 5 min, followed by being washed with distilled water and absolute ethanol several times, respectively. The precursor was dried at 80 °C and then calcined in air at 600 °C for 3 h with different defined heating rates (2 °C min−1, 5 °C min−1 and 10 °C min−1) to get the anticipated MSCMHSs. Individual cerium and manganese hollow spheres could be synthesized without another raw material. Additionally, traditional CeO2–MnOx nanoparticles (NP) with identical molar ratio were also prepared through the same method without carbon spheres (heating rate being of 10 °C min−1) for comparison purposes.2.2 Characterizations
Scanning electron microscopy (SEM) images were captured on Philips XL30 electron microscope operated at beam energy of 10.0 kV.Transmission electron microscopy (TEM) images were taken on a JEM-2100 instrument with an acceleration voltage of 200 kV. High-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) were determined on a JEM-2100F transmission electron microscope with an accelerating voltage of 200 kV. High-angle annular dark field (HAADF)-STEM characterizations were carried out using a JEOL 2100F microscope equipped with an Oxford energy dispersive X-ray (EDX) analysis system with an acceleration voltage of 200 kV.
Fourier translation infrared spectrum (FT-IR) was collected on a NICOLET iS10 FT-IR spectrometer (Thermo Electron Corporation, USA) from 600 to 4000 cm−1. The number of scans was 32 at a resolution of 4 cm−1.
Powder X-ray diffraction (XRD) patterns were recorded on a Shimadzu XRD-6000 power X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å). The intensity data were collected over a 2θ range of 10–80° with a 0.05° step size.
Textural characteristics of the samples were determined by a nitrogen adsorption operation at −196.15 °C on a Micrometrics ASAP-2020 adsorption analyzer by the Brunauer–Emmett–Teller (BET) method. Ahead of each analysis, a preprocessing of the samples was performed under vacuum at 300 °C for 3 h. The surface areas were calculated by the BET equation and the average pore diameter along with pore size distribution were determined by using the equivalent cylindrical model analysis of BJH.
X-ray fluorescence analysis (XRF) was implemented on an ARL-9800 apparatus to clear the bulk composition of the samples. The X-ray tube was operated at 60 kV and 20 mA.
X-ray photoelectron spectroscopy (XPS) analysis was performed on a PHI 5000 Versa Probe system, using monochromatic Al K (1486.6 eV) radiation operating at an accelerating power of 15 kW. Prior to the measurement, the samples were outgassed at room temperature in a UHV chamber (<5 × 10−7 Pa). All binding energies (BE) of the samples were calibrated with the adventitious C 1s peak at 284.6 eV to compensate the charging effect. The experimental error was within ±0.1 eV.
H2-Temperature programmed reduction (H2-TPR) was carried out with H2–Ar mixture (7% H2 by volume) as reducing agent. Typically, 50 mg sample was used in a quartz U-tube reactor for each measurement. After the sample was pretreated in a N2 stream at 200 °C for 0.5 h, reduction measurement of the sample by H2 began from room temperature to 800 °C with a rate of 10 °C min−1.
2.3 Catalytic activity test
The catalytic performance of the samples for NH3-SCR reaction was determined under the reaction conditions as follows: 500 ppm NO, 500 ppm NH3, 5 vol% O2, 5 vol% H2O (when used) and balance N2. The total flow rate was 100 mL min−1 and 100 mg sample was put to use in a fixed-bed quartz reactor tube to get a space velocity of 60000 mL g−1 h−1. Before the mixture reactant gases were fed to the system, a preprocessing for the samples at 200 °C for 0.5 h was implemented to remove existent impurities, using high-purity N2. The concentration of NO at designated temperature during the reaction was detected by an online Thermo fisher IS10 FTIR spectrometer equipped with a 2 m path-length gas cell (250 mL volume), through which NO conversion of the samples could be calculated in accordance with the following equations:

3 Results and discussion
3.1 Morphology and structure
As presented in Fig. S1 (ESI†), uniform carbon spheres (CSs) with size of about 600 nm were obtained under hydrothermal condition. During hydrothermal process, dehydration, intermolecular cross-linking and nucleus growth are supposed to occur. As a result, carbon spheres with abundant superficial active functional groups (FT-IR, Fig. S2†) and accessible pore channels were formed.29 Thus, target ions could be trapped easily on the surface of carbon spheres due to electrostatic force and diffuse inward through the channels. Sequentially, hollow structures were obtained after the following calcination process to remove the template. It was widely reported that different constituents and situations could give birth to varied hollow structures.26,30,31 In the present work, mono-component and hybrid oxide hollow structures of manganese and cerium were prepared to explore the interaction between the constituents. Furthermore, the calcination rates were investigated for the tunable inner structures of the hybrid oxide hollow spheres.The morphologies of as-synthesized samples were observed by SEM and TEM characterizations. As exhibited in Fig. 1b–d, after the calcination process under different conditions, multi-shell CeO2–MnOx hollow spheres (MSCMHSs) with sphere-in-sphere morphology were obtained. It was interesting that the inner structures of the hollow spheres could be controlled by changing the calcination rate, i.e., single-shell (2 °C min−1), double-shell (5 °C min−1) and triple-shell (10 °C min−1). The average diameter of the obtained MSCMHSs was about 400 nm, and the average shell thickness was 40 nm. Specially, the external, middle and internal shells of the triple-shell hollow spheres were approximately estimated as 400 nm, 150 nm and 50 nm, respectively. To further explore the elemental distribution of the multi-shell hollow spheres, HAADF-STEM was conducted on a single triple-shell hollow sphere as an example. Element mapping analysis in Fig. 2a showed that the triple-shell hollow structure could be discriminated clearly, in which the elements of Mn, Ce and O were distributed uniformly. It is worthy to be noted that the homogeneous elemental distribution of Mn and Ce was favorable to the synergistic effects between them. The HRTEM image and inset SAED pattern of triple-shell hollow sphere (Fig. 2b) demonstrated that the shell was polycrystalline. The lattice fringes with d spacing of 0.31 nm, 0.28 nm and 0.19 nm were ascribed to (111), (200), (220) crystal planes of fluorite-type CeO2, respectively. Moreover, the lattice fringe of 0.25 nm was indexed to (202) crystal plane of Mn3O4. The diffraction rings and the inset XRD patterns accorded well with the crystalline phase results.
 | ||
| Fig. 1 TEM images of CeO2–MnOx hollow spheres with various shell number obtained at different heating rates: (a) before calcination; (b) single-shell, 2 °C min−1; (c) double-shell, 5 °C min−1; (d) triple-shell, 10 °C min−1. Insets show the corresponding individual hollow sphere. | ||
 | ||
| Fig. 2 (a) HAADF-STEM mapping images and (b) HRTEM image of the triple-shell hollow sphere. Insets are the corresponding XRD pattern and selected area electron diffraction (SAED) image. | ||
To shed light on the possible forming process of MSCMHSs, the morphology of CSs@CeO2–MnOx precursor before calcination was investigated. For that matter, the solvent-thermal treatment was believed as the critical step for MSCMHSs formation, which could efficiently enhance the adsorption and diffusion of manganese and cerium cations into the carbon spheres through the pore channels. As shown in Fig. S3† for the SEM image of the precursor after the solvent-thermal process, the spheres were uniformly dispersed with the size of approximately 600 nm as carbon spheres. The rough surface of the spheres containing tiny particles indicated the deposition of CeO2–MnOx precursor on the carbon spheres. Corresponding TEM image (Fig. 1a) displayed the core–shell architecture with the evident interface between CSs and CeO2–MnOx precursor. Notably, the MSCMHSs (Fig. 1b–d) exhibited smaller diameters than CSs@CeO2–MnOx precursor, which revealed a shrinked process in accompany with combustion of carbon spheres during calcination. Besides, TG-DTA curves of the precursor were manifested in Fig. 3. Effects of different heating rates on the thermal process were investigated. There were two main exothermal peaks in the DTA curves for all cases. The exothermal peak around 250 °C and the sharp peak concentrated at around 320 °C derived from the combustion of carbon spheres.11 Apparently, with the increasing heating rate, the intensity of exothermal peak for carbon spheres combustion increased. In particular, when the heating rate rose to 10 °C min−1 eventually, greatly enhanced exothermal intensity for carbon spheres combustion was observed. The results indicated that higher heating rate resulted in faster combustion of carbon spheres. As was reported,32 the construction of multi-shell structure was mainly determined by the rate differences between oxide shell formation and carbon shrinkage, which might cause separation. Therefore, with the increasing heating rate, a kind of heterogeneous contraction existed and facilitated the combustion of carbon spheres as well as the diffusion, penetration and redistribution of cations inward the carbon spheres. Thus, accompanied by different degrees of crystallization and penetration for manganese and cerium cations, controllable internal structures were obtained through the non-equilibrium calcination process,21,33 with the removal of carbon spheres. The schematic diagram for synthesis of the MSCMHSs was illustrated in Scheme 1.
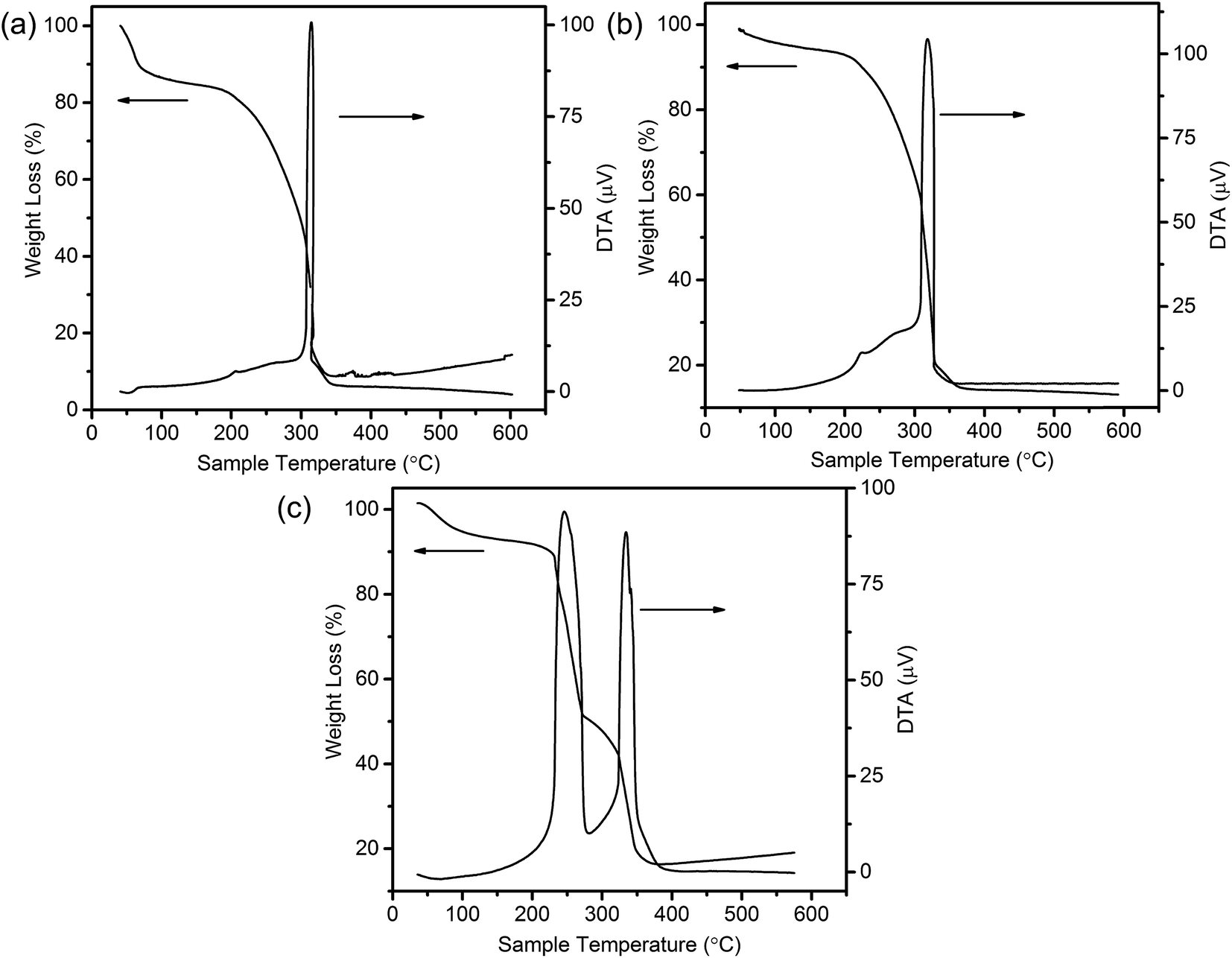 | ||
| Fig. 3 TG-DTA curves of CSs@CeO2–MnOx precursor with different heating rates of (a) 2 °C min−1, (b) 5 °C min−1 and (c) 10 °C min−1. | ||
 | ||
| Scheme 1 Schematic diagram for synthesis of the MSCMHSs. | ||
Additionally, the hollow spheres with single component (i.e., Mn2O3 and CeO2; confirmed by XRD patterns in Fig. 4) under calcination rate of 5 °C min−1 were synthesized (Fig. S4†). We could observe from the SEM image (Fig. S4a†) that Mn2O3 showed ruptured spherical structure with holes on the shell, which might be caused by airflow impact during calcination process. The ruptured structure and TEM image (Fig. S4c†) demonstrated its hollow characteristic, and it was noteworthy that double-shell hollow structures appeared. On the contrary, CeO2 sample (Fig. S4b†) exhibited smoother surface and more complete spherical in shape with respect to those of Mn2O3, indicating stronger resistant ability to airflow impact for CeO2. However, corresponding TEM image (Fig. S4d†) suggested that no double-shell structures were formed in CeO2 hollow spheres. The result showed distinct behaviors between Mn2O3 and CeO2 during the removing process of the templates. Otherwise, in the identical situations, CeO2–MnOx hybrid oxide possessed more complete spherical double-shell hollow structures than Mn2O3 and CeO2, which indicated that hybrid oxide was conducive to forming complete multi-shell hollow structures due to synergistic interaction. Besides, considering of the structural properties of Mn2O3 and CeO2 hollow spheres, it was proposed that the diffusivity between manganese and cerium elements inward carbon spheres was different. Occasionally, HAADF-STEM mapping images of one triple-shell hollow sphere with non-dispersed particles were captured. As shown in Fig. S5,† an aggregation state was detected from the non-dispersed particles outside the shells, which mainly consisted of O and Ce, with scarcely any Mn element. Therefore, combined with the widespread homogeneous dispersion of the elements in triple-shell hollow spheres, it was deduced that, under the synthesis process, the existence of Mn element was synergistically helpful for CeO2 to diffuse inward carbon spheres, and then the multi-shell hollow structure was formed.
 | ||
| Fig. 4 XRD patterns of the samples. | ||
The crystal structures of the samples were characterized by XRD, and the obtained patterns were displayed in Fig. 4. It was noted that the diffraction peaks of single component oxides of manganese and cerium were assigned to the characteristic reflections of Mn2O3 (bixbyite-C, JCPDS 31-0825) and CeO2 (cubic fluorite, JCPDS 04-0593), respectively. While the hybrid multi-shell CeO2–MnOx hollow spheres (single, double and triple referred to the hollow structured samples with corresponding shells, respectively) all exhibited cubic fluorite-type CeO2 phase (JCPDS 04-0593) together with a weak diffraction peak of tetragonal hausmannite Mn3O4 (JCPDS 08-0017) phase at around 36.0°. The result was in good consistency with the research finding upon CeO2–MnOx composites that when mole fraction of Mn was over than 50%, there appeared the crystalline phase of MnOx.16 Discrepancy of manganese crystalline phase for MSCMHSs (Mn3O4: Mn2+, Mn3+) and mono-component Mn2O3 hollow spheres (Mn3+) might be caused by synergistic interactions between Mn and Ce elements (Mn3+ + Ce3+ ↔ Mn2+ + Ce4+). Meanwhile, the constituents and calcination rates also had an effect on the crystallinity of the samples. For the identical calcination rates, Mn2O3 and CeO2 hollow spheres both exhibited higher crystallinity compared with double-shell CeO2–MnOx hollow spheres. While with the identical constituents for MSCMHSs, higher calcination rates resulted in higher crystallinity. As listed in Table 1, crystallite sizes of samples were calculated by Scherrer equation based on the main diffraction peaks. It could be observed that the crystallite sizes for MSCMHSs were much smaller than that of Mn2O3 and CeO2 hollow spheres due to the broader diffraction peaks. Furthermore, crystalline structure of the traditional CeO2–MnOx nanoparticles (NP) was investigated as well. As discerned in Fig. 4, except for the CeO2 and Mn3O4 phase, NP possessed extra characteristic lines ascribed to Mn2O3 phase compared to MSCMHSs, indicating that template-assisted method could promote the dispersity of manganese species. Besides, the crystallite size of NP was estimated to be slightly larger than MSCMHSs.
| Samples | BET surface area (m2 g−1) | Average pore diameter (nm) | Crystallite size (nm) |
|---|---|---|---|
| Mn2O3 | 22.6 | 3.50 | 28.8 |
| Single | 36.0 | 33.0 | 7.60 |
| Double | 30.0 | 31.5 | 6.50 |
| Triple | 35.0 | 30.8 | 5.20 |
| NP | 43.5 | 30.0 | 14.9 |
| CeO2 | 78.4 | 24.0 | 9.50 |
3.2 Catalytic performance results
Selective catalytic reduction of NO with NH3 was utilized to evaluate the catalytic performances of MSCMHSs as well as NP samples. The reaction was carried out at the space velocity of 60000 mL g−1 h−1 over the temperature range from 100 °C to 275 °C. As disclosed in Fig. 5a, the NO conversion of all samples increased as the temperature increased and then decreased with different activated temperature windows. The MSCMHSs reached more than 90% conversion at temperatures as low as 125 °C, and it was noteworthy that hollow spheres with different number of shells exhibited different catalytic abilities. Among them, the triple-shell hollow spheres with a maintained conversion at 100% from 150 °C to 250 °C had the best performance. The NO conversion of double-shell hollow spheres was similar to that of the triple-shell ones from 100 °C to 200 °C but then decreased with the temperature increasing. Moreover, the single-shell hollow spheres showed the worst activity compared with the double-shell and triple-shell hollow spheres. However, it still displayed superior performance in contrast with NP, which could just achieve 84.3% conversion as the maximum activity value at 200 °C. Thus, an apparent enhancement in NO conversion for MSCMHSs than NP was observed within the range of testing temperature, indicating the positive role of multi-shell hollow structures in the reaction of NH3-SCR, especially at low temperatures (100–150 °C). Otherwise, the N2 selectivity of the samples declined gradually along with the rising temperature (Fig. 5b), this may due to the strong redox behavior of Mn-based catalysts, which could lead to the oxidation of NH3 by O2 to the by-product, for instance, NO2 and N2O.34
 | ||
| Fig. 5 (a) NO conversion and (b) N2 selectivity of the samples as a function of temperature. | ||
As discussed above, the catalytic performances of the samples ranked as the order of triple-shell > double-shell > single-shell > NP. Interestingly, the order of performances showed an inversed correlation with the crystalline sizes of the corresponding samples, just as the reports that smaller crystalline size was good for the enhanced activities.35,36 Moreover, the superior catalytic activity of MSCMHSs might be attributed to the synergetic effect between active species of Mn and Ce elements.
To understand the H2O resistance performance of the samples, the NH3-SCR reaction under the condition with water at 150 °C was performed. As displayed in Fig. 6a, after the H2O was added into the mixture reactant gases, a decrease from 80% to less than 40% of NO conversation for NP samples was detected. At the same time, the NO conversation of the MCMSHSs declined as well but maintained at around 70% for more than 14 h, exhibiting better H2O resistance performance than NP samples. It was obvious that the addition of H2O caused deactivation of the samples in some degree, which could be explained that the adsorption of H2O on the surface of the catalyst led to the formation of hydroxyl, resulting in a decline in activity.37–39 Besides, an enhancement in N2 selectivity (Fig. 6b) was also observed among the samples after the addition of H2O, and this could be ascribed to the inhibition of H2O to the oxidation of NH3 by O2.38 To further investigate the key factor for the improved NH3-SCR activity and the effects of special multi-shell hollow structures on the properties of the catalysts, characterizations of different samples were deeply examined.
 | ||
| Fig. 6 (a) NO conversion and (b) N2 selectivity of the samples as a function of temperature with the presence of water at 150 °C. | ||
3.3 N2-Sorption characterization (BET)
N2-Sorption isotherms were measured to get insights into the textural properties of the samples. It could be seen from Fig. 7a that all the isotherms of hollow spheres belonged to typical type-IV in the light of IUPAC classification, indicating the existence of mesoporous structure.40 The observed H2-type hysteresis loops of CeO2 hollow spheres manifested uniform pore structures. However, Mn2O3 and MCMSHSs possessed H3-type hysteresis loops, which might symbolize an irregular pore size distribution, just in keeping with pore distribution curves in Fig. 7b. It was noted that MSCMHSs samples showed similar broad pore size distribution with the average pore diameters being at around 30.0 nm. While Mn2O3 and CeO2 hollow spheres exhibited narrower pore size distributions and smaller average pore diameters centered at 3.50 nm and 24.0 nm, respectively. As summarized in Table 1, there were no significantly differences among single-shell (36.0 m2 g−1), double-shell (30.0 m2 g−1) and triple-shell (35.0 m2 g−1) hollow spheres, which were smaller than 78.4 m2 g−1 of CeO2 but larger than 22.6 m2 g−1 of Mn2O3. Thus, compared to mono-component oxides, the composite of manganese and cerium resulted in modifications on not only the inner structures but also the textural properties. | ||
| Fig. 7 (a) The N2 adsorption/desorption isotherms and (b) BJH pore distribution curves of the hollow spheres (inset denotes CeO2 hollow spheres). | ||
The adsorption–desorption isotherms and pore distribution curves of CeO2–MnOx nanoparticles (NP) was shown in Fig. 8. It was observed that, compared with MSCMHSs, NP showed a narrower pore size distribution while the same main distribution location. It could be explained by the removal of carbon spheres for the precursor during calcination, which might cause non-uniform pore size distribution of the obtained MCMSHSs, containing interstitial pores between the particles. Generally, the surface area of catalysts acted an important role in catalytic performance. However, despite the larger surface area (43.5 m2 g−1), NP exhibited poorer activity compared to MSCMHSs, which suggested that multi-shell hollow structures played more crucial roles in catalytic performances.
 | ||
| Fig. 8 (a) The N2 adsorption/desorption isotherms and (b) BJH pore distribution curves of the composite CeO2–MnOx oxides. | ||
3.4 Bulk and surface analyses of the catalysts (XRF and XPS)
XRF characterization was performed to discover the bulk composition of the samples, and the result was summarized in Table 2. It could be found that the actual ratios of the two metal elements were approximately the same as the theoretical value for each sample, indicating the feasibility of the preparation process with negligible mass losses.XPS was conducted to investigate the surface composition and existential state of active species on the samples. Herein, XPS spectra of the MSCMHSs and NP in Ce 3d, Mn 2p and O 1s regions were displayed in Fig. 9, with the peaks being fitted to several designated peaks based on the Gauss–Lorentz curves. The complicated Ce 3d spectrum of all the samples was separated into eight components with the allocation defined in Fig. 9a. The series of peaks labeled u were attributed to Ce 3d3/2 spin–orbit states, whereas those labeled v belonged to Ce 3d5/2 spin–orbit states. Specially, the two peaks signed as u′ and v′ represented Ce3+ species, and the other six peaks (u, v, u′′, v′′, u′′′ and v′′′) were ascribed to Ce4+ species.41–43 It was obvious that both Ce3+ and Ce4+ species were co-existed in all the samples and the Ce4+ was the predominant chemical valence. In general, the existence of Ce3+ often led to the increase of active oxygen species on the surface of catalysts, which were in favor of the redox reaction.44 Based on the fitting areas of the peaks, the ratios of Ce3+/(Ce4+ + Ce3+) of the samples were calculated and listed in Table 2, which ranked in the order of triple-shell > double-shell > single-shell > NP. The results suggested that, with the multi-shell hollow structures, MSCMHSs possessed more active Ce3+ species than NP, thus leading to the enhanced catalytic performance.
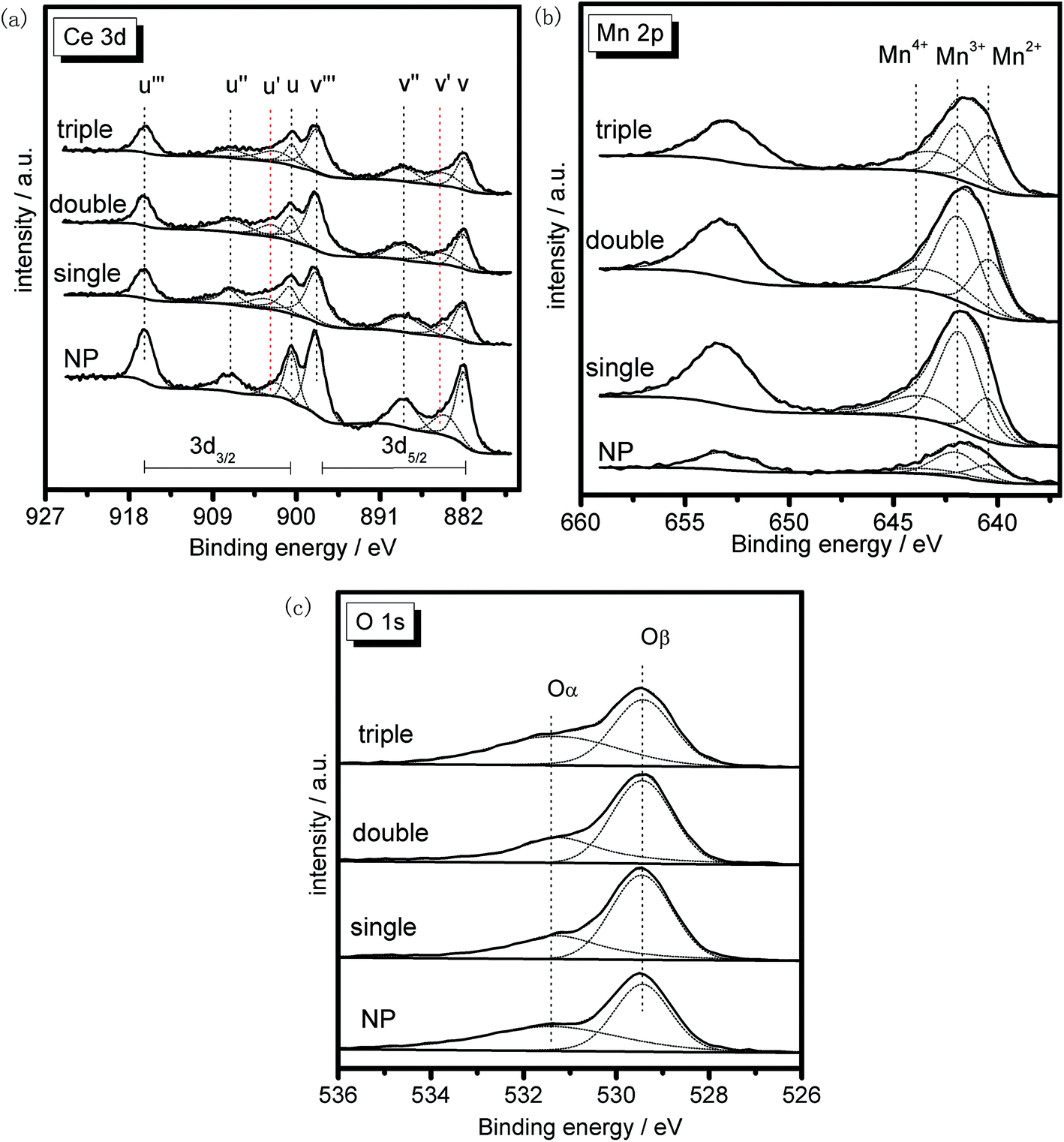 | ||
| Fig. 9 XPS spectra of the samples in (a) Ce 3d, (b) Mn 2p and (c) O 1s regions. | ||
Fig. 9b exhibited two main peaks in Mn 2p spectrum of the samples at about 653.2 eV and 641.5 eV. There were mixed valences in manganese species on the surface of the samples, and the binding energies were overlapped. The peak at 653.2 eV was attributed to Mn 2p1/2 spin–orbit states, while the peak at 641.5 eV was due to Mn 2p3/2 spin–orbit states. As for Mn 2p3/2 spectrum, the peak was fitted to three characteristic peaks representing Mn2+ (640.7 eV), Mn3+ (642.1 eV) and Mn4+ (643.6 eV) species, respectively.45,46 The existence of Mn2+ species for MSCMHSs proved the synergistic interaction between manganese and cerium oxide species, which was in good conformity with the results of XRD. Previous studies revealed that manganese species of higher oxidation state usually acted more important roles in redox reaction.47 From Table 2, it was observed that the surface Mn4+ concentration of MSCMHSs were higher than NP. Especially, the triple-shell hollow spheres showed higher Mn4+ concentration than double-shell and single-shell hollow spheres. The result indicated the superiority of multi-shell hollow structures on surface active species, which was evidenced by the catalytic performances.
O 1s spectra of the samples (Fig. 9c) were ascribed to two overlapped peaks denoted as Oα (531.2–531.4 eV) and Oβ (529.2–529.4 eV), respectively. Peak Oα with the higher binding energy was referred to the surface chemisorbed oxygen, correlated to the surface defects of samples.48,49 While peak Oβ was associated with lattice oxygen, bonding to the manganese or cerium cations.50 Due to its remarkable mobility, Oα was regarded to be the most active oxygen species on the surface of catalysts.51 Besides, the higher content of Oα relative to total oxygen concentration was able to bring better activity in oxidation reaction of NO to NO2, which could accelerate the NH3-SCR reaction through a “fast SCR” process.52 The listed values of Oα/(Oα + Oβ) in Table 2 stated that the chemisorbed oxygen species of MSCMHSs were higher than NP just as that order of surface Ce3+ contents as well, accordant with the report that the existence of Ce3+ could trigger the formation of active oxygen species.53
As manifested in above results of surface analysis, it was worth noting that the surface active species contents of the samples, which were beneficial to redox process, possessed the identical order with their catalytic performances. Hence, it seemed that the enriched surface active species of MSCMHSs could be essential for their better activities in SCR reaction compared with NP.
3.5 Redox performance
Redox performance of the catalysts that plays an important role in NH3-SCR activity was measured by H2-temperature programmed reduction (H2-TPR) characterization. As demonstrated in Fig. 10, two series of broad reduction peaks could be obtained. In the case of lower temperature peak from 150 °C to 600 °C, two kinds of reduction were obtained, with one attributed to the step-by-step reduction of MnOx species, and the other reduction of surface CeOx species.34 Besides, the peak at higher temperature from 700 °C to 800 °C corresponded to the reduction of bulk CeOx species.54 By means of the Gauss–Lorentz fitting, the lower-temperature broad reduction peak of all the samples was fitted to three reduction peaks characterized by apparent maxima at around 249, 330 and 410 °C, respectively. As reported,16,34,55 the low-, middle-, and high-temperature reduction peak could be respectively attributed to the reduction of MnO2 into Mn2O3, Mn2O3 into Mn3O4 and Mn3O4 into MnO, overlapped by reduction peak of surface Ce4+. The discrepancy of redox performance showed that different preparation procedures brought about different interaction between manganese oxide and cerium oxide in the samples.56 Compared with NP samples, the MCMSHSs, especially triple-shell ones, exhibited more superficial manganese species in a higher oxidation state (Mn4+) and much broader reduction temperature range, which was consistent with the XPS analyses. Moreover, NP samples exhibited much higher Mn2O3 manganese species, just as shown in XRD characterization.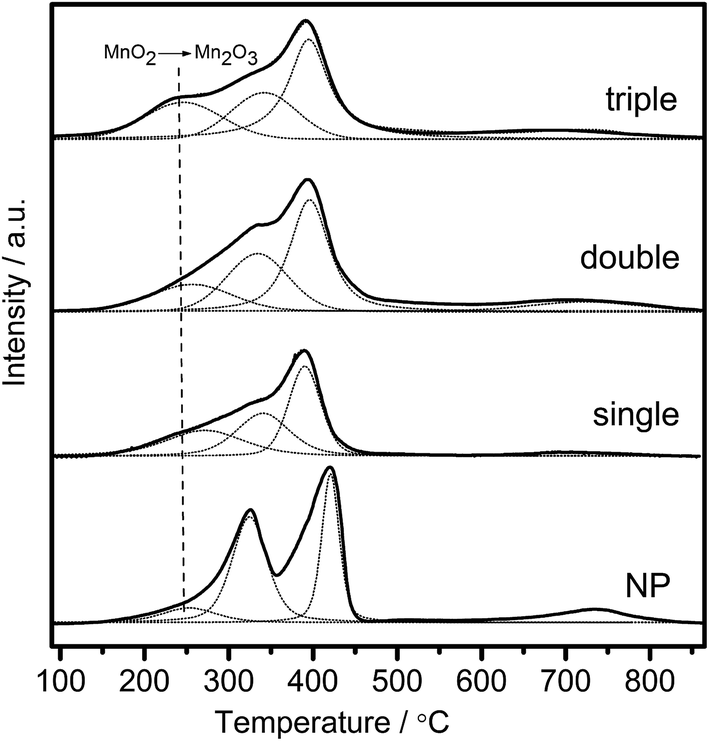 | ||
| Fig. 10 H2-TPR profiles of the samples as a function of temperature. | ||
Moreover, the effect of unique hollow spheres on gases diffusion also acted an inspiring role in catalytic performances.57 In contrast with NP, despite the smaller surface areas, it was considered that multi-shell hollow spheres with porous structures could promote gases diffusion into internal spaces. Thus, thorough contact of reactive gases with both internal and external of spheres was obtained, and then enhanced reactive surfaces were achieved. In addition, hollow spheres with more shells were demonstrated to be more effective to enhance the catalytic performances than those with fewer shells, which should be attributed to multiple collisions of reactive gases among different shells. As illustrated in Fig. 11, more effective collisions tended to appear in hollow structures with more shells, which exhibited richer surface active species as well. As a result, CeO2–MnOx catalysts with multi-shell hollow spherical morphology showed admired performances in NH3-SCR reaction. The present study demonstrated that the well-designed morphology and structure offered an excellent strategy to the creation of highly efficient catalysts.
 | ||
| Fig. 11 Proposed collision processes of reactive gases against hollow spheres with different shells. | ||
4 Conclusions
MSCMHSs with the average size of 400 nm were prepared successfully by the hard template method of carbon spheres, during which it was demonstrated that different calcination rate brought hollow spheres with different inner shell structures. The composite of manganese and cerium resulted in more complete hollow structures compared to mono-component oxides, accompanied by modifications of crystalline phases, valence states and textural properties. Especially, the existence of Mn element could be helpful for CeO2 to diffuse inward carbon spheres to form multi-shell hollow structures under experimental conditions. Furthermore, superior activities of MSCMHSs for NH3-SCR reaction than traditional nanoparticles were displayed, and especially, triple-shell hollow spheres exhibited better performance than double-shell and single-shell ones, which could be attributed to the special structure and abundant surface active species, providing a new kind of material for low-temperature NH3-SCR reaction.Acknowledgements
The financial supports of the National Natural Science Foundation of China (No. 21273110, 21303082), the National High-tech R&D Program of China (863 program, No. 2015AA03A401) and Scientific Research Project of Environmental Protection Department of Jiangsu Province (2016048) are gratefully acknowledged.References
- L. Wang, F. Liu, W. Yang, H. Zhao, Y. Zheng, X. Chen and W. Dong, Mater. Lett., 2013, 107, 42–45 CrossRef CAS.
- S. Putla, M. H. Amin, B. M. Reddy, A. Nafady, K. A. Al Farhan and S. K. Bhargava, ACS Appl. Mater. Interfaces, 2015, 7, 16525–16535 CAS.
- A. Xie, J. Guo, W. Liu and Y. Yang, RSC Adv., 2014, 4, 11357 RSC.
- J. Qi, K. Zhao, G. Li, Y. Gao, H. Zhao, R. Yu and Z. Tang, Nanoscale, 2014, 6, 4072–4077 RSC.
- S. Fang, Y. Xin, L. Ge, C. Han, P. Qiu and L. Wu, Appl. Catal., B, 2015, 179, 458–467 CrossRef CAS.
- S. Andreoli, F. A. Deorsola and R. Pirone, Catal. Today, 2015, 253, 199–206 CrossRef CAS.
- C. Tang, H. Zhang and L. Dong, Catal. Sci. Technol., 2016, 6, 1248–1264 CAS.
- X. Yao, F. Gao, Q. Yu, L. Qi, C. Tang, L. Dong and Y. Chen, Catal. Sci. Technol., 2013, 3, 1355 CAS.
- R. Qu, Y. Peng, X. Sun, J. Li, X. Gao and K. Cen, Catal. Sci. Technol., 2016, 6, 2136–2142 CAS.
- P. Trogadas, J. Parrondo and V. Ramani, Chem. Commun., 2011, 47, 11549–11551 RSC.
- P. Xu, R. Yu, H. Ren, L. Zong, J. Chen and X. Xing, Chem. Sci., 2014, 5, 4221–4226 RSC.
- Z. Liu, Y. Liu, Y. Li, H. Su and L. Ma, Chem. Eng. J., 2016, 283, 1044–1050 CrossRef CAS.
- H. Hu, S. Cai, H. Li, L. Huang, L. Shi and D. Zhang, ACS Catal., 2015, 5, 6069–6077 CrossRef CAS.
- S. Cai, H. Hu, H. Li, L. Shi and D. Zhang, Nanoscale, 2016, 8, 3588–3598 RSC.
- W. Zhan, X. Zhang, Y. Guo, L. Wang, Y. Guo and G. Lu, J. Rare Earths, 2014, 32, 146–152 CrossRef CAS.
- J. Quiroz, J.-M. Giraudon, A. Gervasini, C. Dujardin, C. Lancelot, M. Trentesaux and J.-F. Lamonier, ACS Catal., 2015, 5, 2260–2269 CrossRef CAS.
- F. Arena, Catal. Sci. Technol., 2014, 4, 1890 CAS.
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS.
- G. Chen, F. Rosei and D. Ma, Adv. Funct. Mater., 2012, 22, 3914–3920 CrossRef CAS.
- Y. Wei, Y. Sun, W. Su and J. Liu, RSC Adv., 2015, 5, 26231–26235 RSC.
- L. Zhou, H. Xu, H. Zhang, J. Yang, S. B. Hartono, K. Qian, J. Zou and C. Yu, Chem. Commun., 2013, 49, 8695–8697 RSC.
- B. Liu, Q. Wang, S. Yu, T. Zhao, J. Han, P. Jing, W. Hu, L. Liu, J. Zhang, L. D. Sun and C. H. Yan, Nanoscale, 2013, 5, 9747–9757 RSC.
- C. Sun, L. Liu, L. Qi, H. Li, H. Zhang, C. Li, F. Gao and L. Dong, J. Colloid Interface Sci., 2011, 364, 288–297 CrossRef CAS PubMed.
- W. Tian, C. Zhang, T. Zhai, S. L. Li, X. Wang, M. Liao, K. Tsukagoshi, D. Golberg and Y. Bando, Chem. Commun., 2013, 49, 3739–3741 RSC.
- H. Zhao, J.-F. Chen, Y. Zhao, L. Jiang, J.-W. Sun and J. Yun, Adv. Mater., 2008, 20, 3682–3686 CrossRef CAS.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef CAS PubMed.
- S. Liu, M. Xie, X. Guo and W. Ji, Mater. Lett., 2013, 105, 192–195 CrossRef CAS.
- L. C. Liu, Q. Fan, C. Z. Sun, X. R. Gu, H. Li, F. Gao, Y. F. Chen and L. Dong, J. Power Sources, 2013, 221, 141–148 CrossRef CAS.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- M. H. Oh, T. Yu, S.-H. Yu, B. Lim, K.-T. Ko, M.-G. Willinger, D.-H. Seo, B. H. Kim, M. G. Cho, J.-H. Park, K. Kang, Y.-E. Sung, N. Pinna and T. Hyeon, Science, 2013, 340, 964–968 CrossRef CAS PubMed.
- J. Wang, H. Tang, H. Ren, R. Yu, J. Qi, D. Mao, H. Zhao and D. Wang, Adv. Sci., 2014, 1, 1400011 CrossRef PubMed.
- X. Lai, J. Li, B. A. Korgel, Z. Dong, Z. Li, F. Su, J. Du and D. Wang, Angew. Chem., 2011, 50, 2738–2741 CrossRef CAS PubMed.
- J. Guan, F. Mou, Z. Sun and W. Shi, Chem. Commun., 2010, 46, 6605–6607 RSC.
- Y. Xiong, C. Tang, X. Yao, L. Zhang, L. Li, X. Wang, Y. Deng, F. Gao and L. Dong, Appl. Catal., A, 2015, 495, 206–216 CrossRef CAS.
- P. R. Ettireddy, N. Ettireddy, T. Boningari, R. Pardemann and P. G. Smirniotis, J. Catal., 2012, 292, 53–63 CrossRef CAS.
- F. Liu, H. He, C. Zhang, Z. Feng, L. Zheng, Y. Xie and T. Hu, Appl. Catal., B, 2010, 96, 408–420 CrossRef CAS.
- S. Pan, H. Luo, L. Li, Z. Wei and B. Huang, J. Mol. Catal. A: Chem., 2013, 377, 154–161 CrossRef CAS.
- S. Xiong, Y. Liao, X. Xiao, H. Dang and S. Yang, Catal. Sci. Technol., 2015, 5, 2132–2140 CAS.
- Z. Lei, B. Han, K. Yang and B. Chen, Chem. Eng. J., 2013, 215–216, 651–657 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Watanabe, X. Ma and C. Song, J. Phys. Chem. C, 2009, 113, 14249–14257 CAS.
- P. Hartmann, T. Brezesinski, J. Sann, A. Lotnyk, J.-P. Eufinger, L. Kienle and J. Janek, ACS Nano, 2013, 7, 2999–3013 CrossRef CAS PubMed.
- S. M. Lee and S. C. Hong, Appl. Catal., B, 2015, 163, 30–39 CrossRef CAS.
- S. Yang, W. Zhu, Z. Jiang, Z. Chen and J. Wang, Appl. Surf. Sci., 2006, 252, 8499–8505 CrossRef CAS.
- X. Tang, Y. Li, X. Huang, Y. Xu, H. Zhu, J. Wang and W. Shen, Appl. Catal., B, 2006, 62, 265–273 CrossRef CAS.
- G. C. Allen, S. J. Harris, J. A. Jutson and J. M. Dyke, Appl. Surf. Sci., 1989, 37, 111–134 CrossRef CAS.
- J. S. Park, D. S. Doh and K.-Y. Lee, Top. Catal., 2000, 10, 127–131 CrossRef CAS.
- G. Carja, Y. Kameshima, K. Okada and C. D. Madhusoodana, Appl. Catal., B, 2007, 73, 60–64 CrossRef CAS.
- S. Wu, X. Yao, L. Zhang, Y. Cao, W. Zou, L. Li, K. Ma, C. Tang, F. Gao and L. Dong, Chem. Commun., 2015, 51, 3470–3473 RSC.
- B. M. Reddy, P. Bharali, P. Saikia, S.-E. Park, M. W. E. van den Berg, M. Muhler and W. Grunert, J. Phys. Chem. C, 2008, 112, 11729–11737 CAS.
- L. Jing, Z. Xu, X. Sun, J. Shang and W. Cai, Appl. Surf. Sci., 2001, 180, 308–314 CrossRef CAS.
- W. Shan, F. Liu, H. He, X. Shi and C. Zhang, Chem. Commun., 2011, 47, 8046–8048 RSC.
- Z. Zou, M. Meng, L. Guo and Y. Zha, J. Hazard. Mater., 2009, 163, 835–842 CrossRef CAS PubMed.
- H. C. Yao and Y. F. Y. Yao, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- J. Q. Torres, J.-M. Giraudon and J.-F. Lamonier, Catal. Today, 2011, 176, 277–280 CrossRef CAS.
- C. Fang, D. Zhang, L. Shi, R. Gao, H. Li, L. Ye and J. Zhang, Catal. Sci. Technol., 2013, 3, 803–811 CAS.
- C. Song, C. Wang, H. Zhu, X. Wu, L. Dong and Y. Chen, Catal. Lett., 2008, 120, 215–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25863h |
| This journal is © The Royal Society of Chemistry 2017 |