
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of polystyrene–polyolefin multiblock copolymers by sequential coordination and anionic polymerization†
Dong Hyun Kim,
Seung Soo Park,
Su Hyun Park,
Jong Yeob Jeon,
Hyo Bo Kim and
Bun Yeoul Lee *
*
Department of Molecular Science and Technology, Ajou University, Suwon 443-749, South Korea. E-mail: bunyeoul@ajou.ac.kr; Tel: +82-31-219-1844
First published on 18th January 2017
Abstract
Block copolymers of polyolefins (PO) and polystyrene (PS) are attractive materials that are not synthesized directly from the olefin and styrene monomers. A strategy for construction of PS-b-PO-b-PS triblock units directly from the olefin and styrene monomers is disclosed herein. PO chains (ethylene/1-octene or ethylene/1-pentene copolymers) were grown from dialkylzinc species bearing the α-methylstyrene moiety (i.e., [4-(isopropenyl)benzyl]2Zn) by ‘coordinative chain transfer polymerization (CCTP)’ using a typical ansa-metallocene catalyst, rac-[Me2Si(2-methylindenyl)2]ZrCl2 activated with modified-methylaluminoxane (MMAO). PS chains were subsequently grown from the Zn-alkyl sites and from the α-methylstyrene moieties of the resulting PO chains by switching to anionic polymerization. When nBuLi(tmeda)2 was fed into the system as an initiator in a quantity fulfilling the criterion [Li] > [Zn] + [Al in MMAO], nBuLi(tmeda)2 successfully attacked the α-methylstyrene moieties to initiate the anionic styrene polymerization at both ends of the PO chains generated in the CCTP process. However, in model studies, the attack of nBuLi(tmeda)2 on α-methylstyrene in the presence of (hexyl)2Zn consumed two molecules of α-methylstyrene per nBuLi to afford mainly R–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li, where R is either an nbutyl or hexyl group originating from nBuLi or (hexyl)2Zn, respectively. This observation suggests that the block copolymer does not simply comprise the PS-b-PO-b-PS triblock, but instead comprises a multiblock containing PS-b-PO-b-PS units. The molecular weight of the polymer increased after performing anionic polymerization. Even though the phase separation of the PS and PO blocks observed in the TEM images is less regular in the multiblock copolymers, the elastomeric property of the multiblock copolymers observed in the hysteresis testing is better than that of the diblock analogue.
Introduction
Block copolymers comprised of polyolefins (PO) and polystyrene (PS) are attractive materials that have drawn significant attention in industry and academia.1–6 The triblock copolymer, PS-block-poly(ethylene-co-1-butene)-block-PS (SEBS) is a thermoplastic elastomer. Glassy PS blocks form spherical domains that are uniformly distributed in a rubbery poly(ethylene-co-1-butene) matrix. SEBS is commercially produced on a scale of several hundred thousand ton per year and advertised to be the material of choice for production of soft and strong compounds for handles and grips, elastic components in diapers, oil gels for telecommunications and medical applications, impact modifiers of engineering thermoplastics, and flexibilizers and tougheners for clear polypropylene. The unique and attractive properties have led to a massive increase in its demand, but its use is limited by the high manufacturing cost. In industry, SEBS is not produced directly from olefin and styrene monomers, but, instead, is generated via hydrogenation of PS-block-poly(1,4-butadiene)-block-PS (SBS), which is produced by anionic living polymerization of 1,4-butadiene and styrene (Scheme 1(a)).7–10 The hydrogenation process is not an easy task and is a significant contributor to the manufacturing cost in the production of SEBS. Hence, preparation of PO–PS block copolymers directly from olefin and styrene monomers is a worthwhile and formidable pursuit.11–14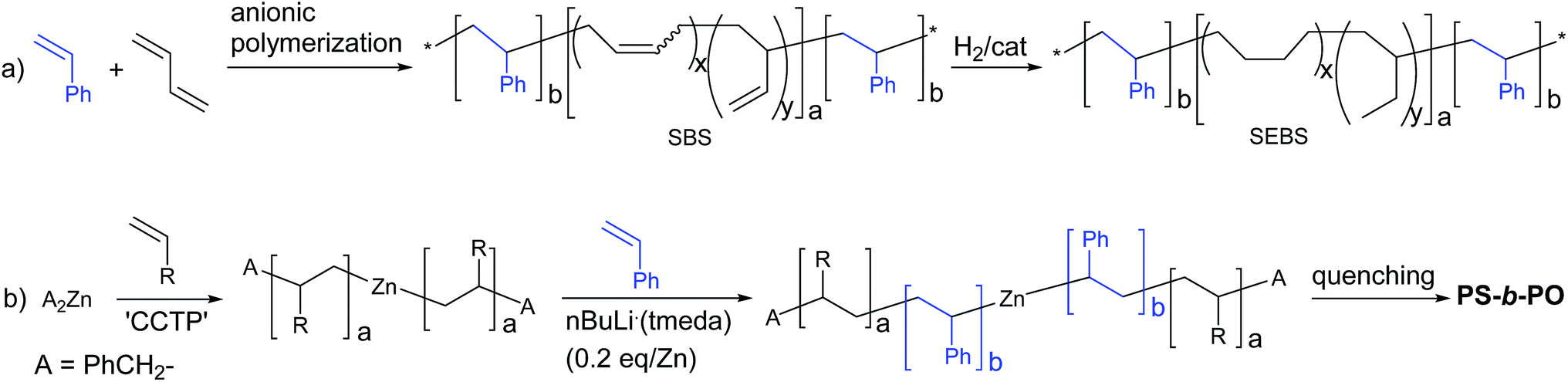 | ||
| Scheme 1 Preparation of PO–PS block copolymers. | ||
Recently, we disclosed a novel method for direct preparation of PO–PS diblock copolymers using olefin and styrene monomers, where coordination and anionic polymerizations were sequentially performed in one-pot (Scheme 1(b)).15 The polyolefin chains were grown from dialkylzinc sites by the so-called ‘coordinative chain transfer polymerization (CCTP)’,16–20 which is a useful tool for precise architectural design of polyolefin chains.21–32 The PS chains were subsequently grown from the zinc sites attached to the PO chains by feeding styrene monomer and a sub-stoichiometric amount of nBuLi(tmeda) (tmeda: N,N,N′,N′-tetramethylethylenediamine) initiator (0.2 eq./Zn) into the system. Reversible formation of zincate species via the reaction between the dialkylzinc species and the chain-growing alkyllithium sites may be responsible for the growth of PS chain from the zinc sites. Coordination of tmeda enhances the reactivity of the alkyllithium species, playing a key role in the formation of the zincate species in the aliphatic hydrocarbon solvent in which the anionic polymerization is performed. In this work, we report the preparation of PS–PO multiblock copolymers that mimic the chain structure of SEBS (Scheme 2). Preparation of PO-based block copolymers is currently highly topical.33–43
 | ||
| Scheme 2 Strategy for direct preparation of PS-block-PO-block-PS using olefin and styrene monomers. | ||
Results and discussion
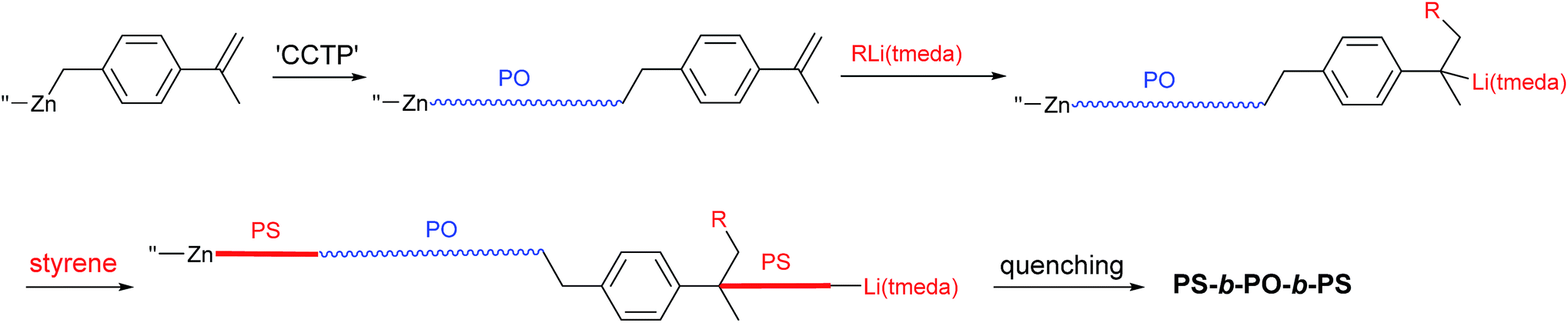
Strategy for preparation of PS-block-PO-block-PS
The present strategy for preparation of the PS-block-PO-block-PS copolymer is summarized in Scheme 2. The PO chains were grown from dialkylzinc species bearing an α-methylstyrene moiety, i.e., [4-(isopropenyl)benzyl]2Zn, by the established CCTP method, and the PS chains were subsequently grown from both the zinc sites and the α-methylstyrene moieties by switching to anionic polymerization. The prerequisite for success of the strategy is that the α-methylstyrene moiety should remain intact during the coordination polymerization process. The styrene monomer readily acts as a chain terminating agent or becomes incorporated into polymer chains during the coordination polymerization,11,12 but, α-methylstyrene does not interfere in the ethylene/1-octene copolymerization carried out using the typical ansa-metallocene catalyst rac-[Me2Si(2-methylindenyl)2]ZrCl2 activated with modified-methylaluminoxane (MMAO); the polymerization behaviour did not change in the presence or absence of α-methylstyrene.The key material in the strategy, [4-(isopropenyl)benzyl]2Zn, was prepared in a straightforward manner from 4-(chloromethyl)benzoyl chloride (Scheme 3). Thus, addition of two equivalents of MeMgBr to 4-(chloromethyl)benzoyl chloride (p-ClC(O)C6H4CH2Cl) and subsequent treatment of the resulting tert-benzylic alcohol (4-(Me2C(OH))C6H4CH2Cl) with the weak acid KHSO4 at 140 °C afforded 4-(isopropenyl)benzyl chloride in good yield (76%).44 Generation of the Grignard reagent using the prepared 4-(isopropenyl)benzyl chloride and subsequent addition of half equivalent of ZnCl2 in diethyl ether afforded the desired [4-(isopropenyl)benzyl]2Zn. Purification of dibenzylzinc derivatives is sometimes problematic, but in this case, extremely pure [4-(isopropenyl)benzyl]2Zn was obtained through recrystallization in hexane.45–47
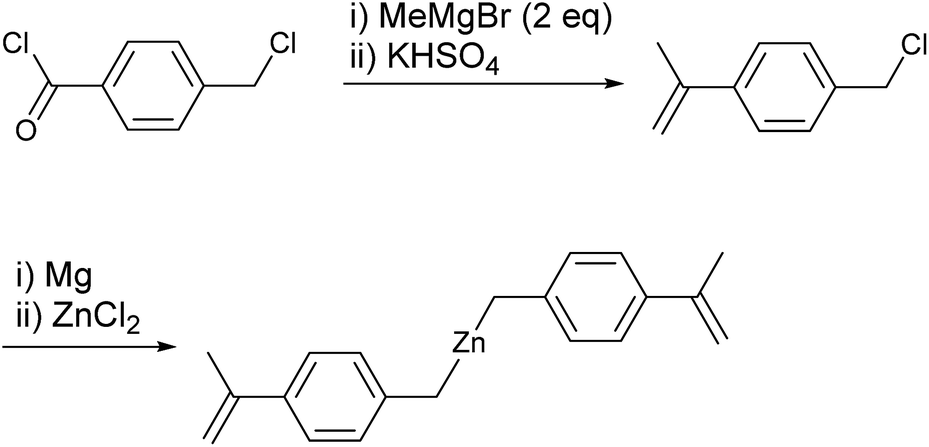 | ||
| Scheme 3 Synthesis of [4-(isopropenyl)benzyl]2Zn. | ||
In the previous studies focusing on the preparation of PO–PS diblock copolymers, a minimal amount of nBuLi(tmeda) ([Li]/[Zn] = 0.2) was used to minimize the generation of PS homopolymers (Scheme 1(b)); however, in the present strategy to prepare PS-b-PO-b-PS triblock copolymers (Scheme 2), a significant amount of RLi(tmeda) ([Li]/[Zn] = 2) should be fed into the system to attack the α-methylstyrene moieties. Gratifyingly, PS chains were effectively grown from almost all the Zn-alkyl sites, even when the [Li]/[Zn] ratio was as high as 1.0, 1.5, and 2.0. The ([PS growth sites] − [Li])/[Zn] values determined from the measured PS-Mn values (i.e., ([PS growth sites] − [Li])/[Zn] = {[styrene]/DP − [nBuLi(tmeda)]}/[Zn], where DP = PS-Mn/104) were close to the value of 2.0 that is expected when PS chain growth occurs from all the Zn-alkyl sites (1.89, 1.97, and 1.82 for [Li]/[Zn] feed ratios of 1.0, 1.5, and 2.0, respectively) (2–4 in Table 1). Additionally, the molecular weight distributions were narrower at high feed ratios of nBuLi(tmeda) (Mw/Mn, 1.26, 1.27, and 1.23 for [Li]/[Zn], 1.0, 1.5, and 2.0, respectively) compared with the Mw/Mn value (1.52) observed when the [Li]/[Zn] ratio was low at 0.20 (entry 1). Addition of two equivalents tmeda per nBuLi afforded similar results at [Li]/[Zn] ratios of 1.0 and 1.5; PS chains were effectively grown from most of the Zn-alkyl sites with narrow molecular weight distributions (([PS growth sites] − [Li])/[Zn], 2.12 and 1.73; Mw/Mn, 1.32 and 1.29; entries 5 and 6). However, growth of the PS chain from the zinc sites was not very effective when the [Li]/[Zn] ratio was high at 2.0 (([PS growth sites] − [Li])/[Zn] = 1.23) (entry 7).
| Entry | Initiator | [Li]/[Zn] | Timeb (h) | Mnc (Da) | Mw/Mn | ([PS growth sites] − [Li])/[Zn]d |
|---|---|---|---|---|---|---|
| a Polymerization conditions: styrene in methylcyclohexane (25 wt%, 2.5 g, 24 mmol), (hexyl)2Zn (11.3 mg, 0.048 mmol, [styrene]/[Zn] = 500), 90 °C.b Time not optimized for full conversion.c Measured by GPC at 40 °C using THF eluent and PS-standards.d Calculated as ([styrene]/[Zn]/DP − [nBuLi(tmeda)])/[Zn], where DP = Mn/104. | ||||||
| 1 | nBuLi(tmeda) | 0.20 | 2.0 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
1.52 | 1.87 |
| 2 | nBuLi(tmeda) | 1.0 | 2.0 | 18000 |
1.26 | 1.89 |
| 3 | nBuLi(tmeda) | 1.5 | 1.0 | 15000 |
1.27 | 1.97 |
| 4 | nBuLi(tmeda) | 2.0 | 1.0 | 13600 |
1.23 | 1.82 |
| 5 | nBuLi(tmeda)2 | 1.0 | 1.0 | 16700 |
1.32 | 2.12 |
| 6 | nBuLi(tmeda)2 | 1.5 | 1.0 | 16100 |
1.29 | 1.73 |
| 7 | nBuLi(tmeda)2 | 2.0 | 1.0 | 16100 |
1.18 | 1.23 |
Initiation of α-methylstyrene
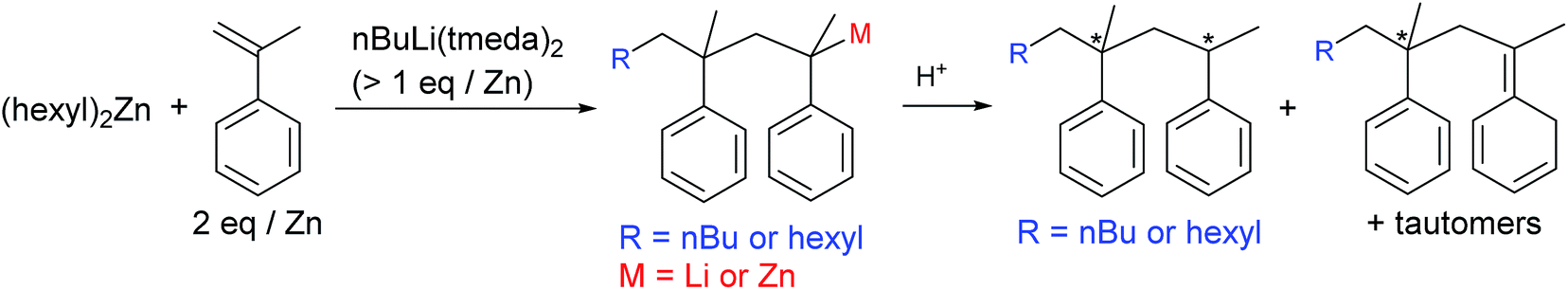
It was reported that tert-BuLi reacts with α-methylstyrene in aliphatic hydrocarbon solvent to form a 1:1 adduct (tBu–CH2C(Ph)(Me)Li).48 Less reactive nBuLi does not react with α-methylstyrene, but nBuLi(tmeda) rapidly attacks α-methylstyrene; 1H NMR studies in deuterated cyclohexane indicate that the 1:1 adduct (nBu–CH2C(Ph)(Me)Li) and the 1:2 adduct (nBu–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li) were generated (Fig. 1(a)). The xylyllithium species p-MeC6H4CH2Li(tmeda), which is generated cleanly through α-lithiation of p-xylene using nBuLi(tmeda), also attacks α-methylstyrene. Analyses of the generated organolithium species itself and the protonated organic compounds (two spots in TLC studies) by 1D and 2D NMR also indicated formation of the 1:1 and 1:2 adducts (MeC6H4CH2–CH2C(Ph)(Me)Li and MeC6H4CH2–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li) (see ESI†). Higher adducts, 1:3 adduct, etc., were not generated; that is, α-methylstyrene can only be inserted successively twice, but further insertion of three or more units is not allowed. These observations agree with the fact that homopolymerization of α-methylstyrene is not feasible, especially at a high temperature due to its low ceiling temperature (Tc, 66 °C),49–51 and that the maximum α-methylstyrene content attainable in α-methylstyrene/styrene anionic copolymerization is limited to ca. 70 wt% (i.e., 2:1 α-methylstyrene/styrene mole ratio).52
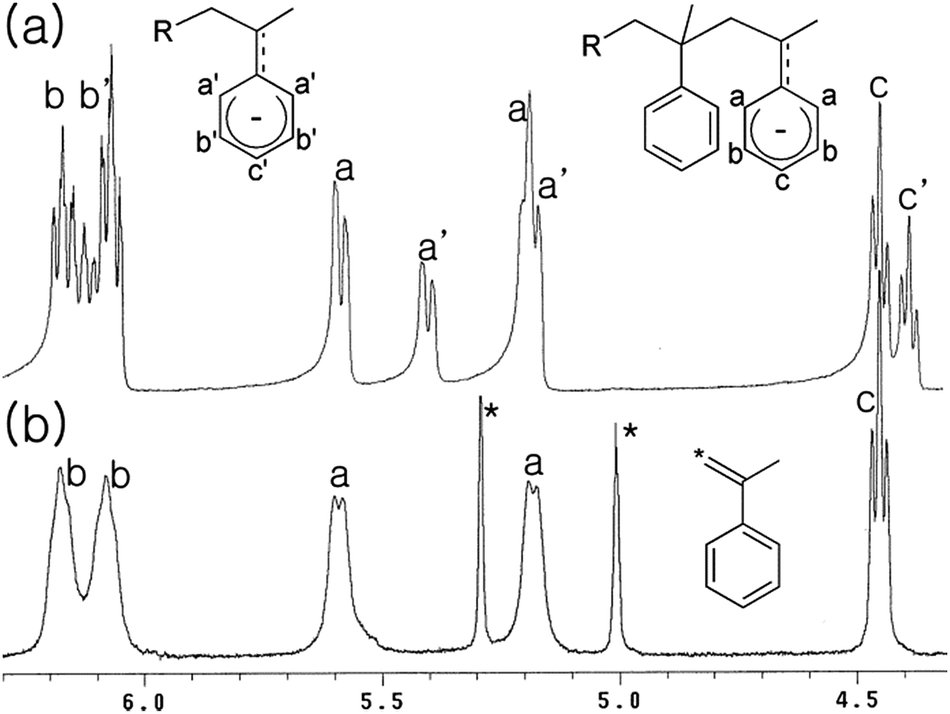 | ||
| Fig. 1 1H NMR spectra for the reactions of α-methylstyrene with nBuLi(tmeda) (a) and nBuLi(tmeda)2 in the presence of (hexyl)2Zn (b). | ||
For success of the present strategy, the alkyllithium species should be able to attack the α-methylstyrene moieties in the presence of dialkylzinc species. Disappointingly, the reactivity of tert-BuLi, nBuLi(tmeda), and p-MeC6H4CH2Li(tmeda) toward α-methylstyrene was extinguished in the presence of an equivalent amount of (hexyl)2Zn; formation of Li–Zn aggregates may reduce the reactivity. However, when [Li] > [Zn], consumption of α-methylstyrene was observed, with generation of the characteristic orange-red colour of the styryl anion. However, the reaction rate was rather slow; only ∼50% consumption of α-methylstyrene was observed at the reaction time of 30 min using the feed ratio of [nBuLi(tmeda)]:[(hexyl)2Zn]:[α-methylstyrene] = 1.5:1:2. Addition of nBuLi(tmeda)2 instead of nBuLi(tmeda) leads to acceleration of the reaction rate and almost all the α-methylstyrene was consumed within 30 min. In the 1H NMR spectrum, only signals assignable to the 1:2 adduct (R–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li) were observed, with no appearance of the signals assignable to the 1:1 adduct (R–CH2C(Ph)(Me)Li) (Fig. 1(b) and Scheme 4). After quenching with acidic water, full analyses of the protonated organic compound (one spot in TLC analysis) using NMR and mass spectral data further supported primary generation of 1:2 adduct attached via either n-butyl or hexyl group (i.e., R = nBu or hexyl in R–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li) originating from nBuLi or (hexyl)2Zn, respectively (see ESI†). When styrene monomer was added to the generated the R–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li species in the NMR cell, rapid and full consumption of styrene monomer was observed, indicating the generated lithium species acts well as an initiator for the styrene anionic polymerization.
 | ||
| Scheme 4 Reaction of nBuLi(tmeda)2 with α-methylstyrene. | ||
Preparation of multiblock copolymers
Based on the aforementioned background studies, preparation of multiblock copolymers containing PS-b-PO-b-PS units was attempted (Table 2). The olefin polymerization was performed using a typical ansa-metallocene catalyst (rac-[Me2Si(2-methylindenyl)2]ZrCl2) activated with MMAO, which was reported to be effective in the CCTP with suppression of the undesirable β-elimination process.15,53,54 Olefin polymerization was performed by feeding ethylene gas into the system containing 1-octene or 1-pentene monomer. The catalyst worked efficiently, leading to consumption of almost all of the α-olefin monomers, once the zinc species [4-(isopropenyl)benzyl]2Zn is sufficiently pure. The molecular weights of the generated POs (Mw, 115–147 kDa) were similar to the Mw (140 kDa) observed for PO generated in the presence of (benzyl)2Zn, indicating that [4-(isopropenyl)benzyl]2Zn works as effectively as (benzyl)2Zn in the CCTP while the α-methylstyrene moieties do not interfere in the olefin polymerization (entry 4 versus others).| Entry | RLi(tmeda)2 (μmol) | Consumed monomers | Homo-PSc (g; %) | PS-Mn (PDI)d (kDa) | Homo-PS growth sitese (μmol) | PO-Mw (PDI)f (kDa) | Block copolymer-Mw (PDI)f (kDa) | ||
|---|---|---|---|---|---|---|---|---|---|
| 1-Alkene (g) | C2H4 (g) | Styreneb (g) | |||||||
| a Polymerization conditions: rac-[Me2Si(2-methylindenyl)2]ZrCl2 (1.0 μmol), MMAO (200 μmol-Al), initial ethylene charge at 30 bar and then regulation at 10–20 bar, 120 °C, 20 min for the first step CCTP; [styrene]/[Zn] = 500, 120–130 °C, 1.5 h (complete conversion of styrene) in the second step anionic polymerization.b Values in parentheses indicate mol% of additional divinylbenzene per styrene.c Filtrate portion extracted with acetone and chloroform (2:1 weight ratio); percentage value = (extracted PS weight)/(consumed styrene weight).d Measured with GPC at 40 °C eluting with THF using PS-standards.e (Homo-PS weight)/PS-Mn.f Measured with GPC at 160 °C eluting with 1,2,4-trichlorobenzene using PS-standards.g (Benzyl)2Zn was used instead of [4-(isopropenyl)benzyl]2Zn. |
|||||||||
| 1 | nBu; 300 | C8; 10.0 | 15.4 | 10.4 | 4.4 (42) | 14.3 (1.82) | 310 | 129 (4.06) | 143 (2.60) |
| 2 | nBu; 400 | C8; 10.0 | 19.0 | 10.4 | 4.6 (44) | 11.4 (1.66) | 400 | 135 (4.57) | 150 (2.92) |
| 3 | nBu; 500 | C8; 10.0 | 18.4 | 10.4 | 4.6 (45) | 14.5 (1.46) | 320 | 115 (3.02) | 145 (2.51) |
| 4g | nBu; 500 | C8; 10.0 | 17.4 | 10.4 | 5.5 (53) | 10.7 (1.65) | 510 | 140 (3.55) | 148 (3.37) |
| 5 | nBu; 600 | C8; 10.0 | 18.0 | 10.4 | 5.7 (55) | 11.2 (1.66) | 510 | 134 (3.76) | 155 (2.68) |
| 6 | Benzyl; 400 | C8; 10.0 | 16.7 | 10.4 | 5.4 (52) | 12.0 (1.56) | 450 | 133 (3.33) | 138 (2.86) |
| 7 | Benzyl; 500 | C8; 10.0 | 20.1 | 10.4 | 5.0 (48) | 14.3 (1.65) | 350 | 142 (3.61) | 154 (3.19) |
| 8 | nBu; 500 | C8; 10.0 | 19.3 | 10.4 (0.25) | 3.1 (30) | 16.1 (1.54) | 147 (2.96) | 162 (3.68) | |
| 9 | nBu; 500 | C8; 10.0 | 18.3 | 10.4 (0.50) | 3.1 (29) | 11.3 (1.79) | 132 (3.15) | 157 (2.54) | |
| 10 | nBu; 500 | C8; 10.0 | 19.1 | 10.4 (0.75) | 2.0 (19) | 11.1 (1.56) | 136 (3.09) | 146 (2.42) | |
| 11 | nBu; 500 | C8; 10.0 | 19.8 | 10.4 (1.0) | 1.5 (14) | 10.1 (1.66) | 130 (3.05) | 155 (3.28) | |
| 12 | nBu; 500 | C5; 10.0 | 15.8 | 10.4 | 5.0 (48) | 14.5 (1.51) | 340 | 119 (3.65) | 134 (2.98) |
| 13 | nBu; 500 | C5; 10.0 | 16.2 | 10.4 (0.25) | 3.7 (36) | 14.8 (1.74) | 126 (3.22) | 140 (3.20) | |
| 14 | nBu; 500 | C5; 10.0 | 19.0 | 10.4 (0.50) | 2.8 (27) | 11.5 (1.78) | 129 (2.92) | 153 (3.19) | |
Anionic polymerization of styrene was subsequently performed in one-pot by successive feeding of nBuLi(tmeda)2 and styrene monomer into the system with a lag time of 1 h. The time lag was introduced to allow nBuLi(tmeda)2 to attack the α-methylstyrene moieties fully. nBuLi forms 1:1 aggregates with alkylaluminum species (MMAO), preventing initiation of the anionic polymerization when [Li] < [Al]. However, when the [Li]/[Al] ratio exceeds 1.0, styrene polymerization is initiated, but the PS-chains grow from all of nBuLi fed into the system, including nBuLi units trapped by alkylaluminum species and dialkylzinc sites.55 PS chains grown from nBuLi form the undesired PS homopolymer (homo-PS), while those grown from Zn sites form the desired block copolymers. Attack of nBuLi(tmeda)2 on the α-methylstyrene moieties that are attached to the end of PO chains results in formation of PS-b-PO-b-PS triblock units. In this one-pot process, a substantial amount of nBuLi(tmeda)2 is needed for the reaction between nBuLi(tmeda)2 and α-methylstyrene (i.e., [Li] > [Al] + [Zn]) since some portion of the nBuLi(tmeda)2 fed into the system remains unreacted with α-methylstyrene moieties by becoming trapped by aggregation with the Al and Zn sites; consequently, formation of a substantial amount of PS-homopolymers is inevitable.
The PS homopolymer can be selectively extracted from the generated block copolymers using chloroform and acetone (1:2 w/w). When the amount of nBuLi(tmeda)2 (300 μmol) was less than ‘[Zn] + [Al]’ (i.e., 200 + 200 = 400 μmol), nBuLi(tmeda)2 did not attack the α-methylstyrene moieties, as indicated by the lack of appearance of the characteristic orange colour of the styryl anion. The number of homo-PS growth sites calculated as ‘(homo-PS weight)/PS-Mn’ was 310 μmol, which is consistent with the amount of nBuLi(tmeda)2 fed into the system. This agreement indicates that PO-b-PS diblock copolymers were generated and the α-methylstyrene moieties did not participate in the anionic styrene polymerization (entry 1). However, when the amount of nBuLi(tmeda)2 fed into the system was 500 μmol, which exceeds the sum of [Zn] and [Al] (i.e., [Li] > [Zn] + [Al]), the colorless solution became red-orange, indicating generation of the styryl anion via attack of nBuLi(tmeda)2 on the α-methylstyrene moieties; the homo-PS growth sites (320 μmol) were less than the amount of nBuLi(tmeda)2 used as feed (500 μmol) (entry 3). When the zinc species do not bear α-methylstyrene moieties, i.e., when (benzyl)2Zn was used instead of [4-(isopropenyl)benzyl]2Zn, the quantity of homo-PS growth sites (510 μmol) again agreed well with the amount of nBuLi(tmeda)2 used as feed (500 μmol) (entry 4). In all other cases where [Li] > [Zn] + [Al], some portion of the fed lithium species reacted with the α-methylstyrene moieties, and the quantity of homo-PS growth sites was less than the amount of the lithium species used as feed (entries 5, 7, and 12). As the amount of nBuLi(tmeda)2 fed into the system increased from 300 to 400, and 500 μmol, the polydispersity (PDI, Mw/Mn) of the extracted homo-PS gradually decreased from 1.82 to 1.66 and 1.46; however, the PDI increased (1.66) with a further increase of the nBuLi(tmeda)2 feed to 600 μmol (entries 1–3 and 5). In all cases, the polymerization rate was fairly high and the styrene monomers were completely consumed within 1.5 h.
The fraction of extracted homo-PS declined when a small amount of divinylbenzene (0.25–1.0 mol% per styrene), which may connect the homo-PS chains with the block copolymer chains during the course of anionic polymerization, was used as the feed. In fact, by feeding divinylbenzene in quantities of 0.25, 0.50, 0.75, and 1.0 mol%, the fraction of homo-PS fraction gradually declined from 45% to 30%, 29%, 19%, and 14%, respectively (entries 8–11). The extracted homo-PS Mn values gradually decreased from 16.1 to 11.3, 11.1, and 10.1 kDa with an increase of the amount of divinylbenzene from 0.25 mol% to 0.50, 0.75, and 1.0 mol%.
Formation of the block copolymers was evident from comparison of the molecular weights of the samples before and after anionic polymerization (i.e., PO-Mw and block copolymer-Mw). The refractive index (RI) detector response of the GPC instrument is opposite for the PO and PS samples (see ESI†), which complicates the analyses, but the retention time should be shortened by attaching the PS-blocks, which leads to increase of the molecular weight, even though the actual value of the increment may not be fully reliable. When [Li] was less than ‘[Al] + [Zn]’, where PO-b-PS diblock copolymers are the main species generated, the Mw increased from 129 to 143 kDa after anionic polymerization (entry 1); the increment of the Mw values (ΔMw) was 14 kDa, which is almost similar to the measured molecular weight of homo-PS (Mn, 14.3 kDa). In the GPC curves plotted in the log(molecular weight) scale (Fig. 2(a)), the shift is substantial at the low molecular end while it is insubstantial at the high molecular weight end, and the molecular weight distribution is narrowed after the anion polymerization (Mw/Mn, 2.60 versus 4.06). In the step of anion polymerization, the molecular weight distribution of PS fraction is not as narrow as that in the living polymerization (Mw/Mn 1.82) and some low molecular weight portion below 10 kDa is observed in the GPC curve of the generated block copolymer. When the amount of nBuLi was 500 μmol to meet the criterion ‘[Li] > [Al] + [Zn]’, where PO–PS multiblock copolymers are generated by the attack of nBuLi on the α-methylstyrene moieties (entry 3), ΔMw was significant, i.e., 30 kDa, which is almost double of the Mn value obtained for homo-PS (14.5 kDa). In the GPC curves plotted in the log(molecular weight) scale (Fig. 2(b)), the shift is substantial at both the low molecular and the high molecular weight ends, which suggests that the generated copolymer is not a simple triblock but, instead, a multiblock copolymer. In contrast, ΔMw was marginal (8 kDa) when (benzyl)2Zn was used instead of [4-(isopropenyl)benzyl]2Zn under the identical conditions; the α-methylstyrene moieties must play a role in obtaining the high ΔMw value. The ΔMw values were relatively small (5 or 12 kDa) when benzyl-Li(tmeda)2 was fed as an initiator in amounts of 400 and 500 μmol; benzyl-Li(tmeda)2 is not as good an initiator as nBuLi(tmeda)2 for successful execution of the strategy. When divinylbenzene is used, much higher ΔMw values are expected due to connection of the growing block copolymer chains; however, the observed ΔMw values were not so very high (10–25 kDa). The polymerization solution turned turbid at an early stage of the anionic polymerization, indicating that the generated block copolymers are insoluble and form micelles. In this situation, connection between the growing block copolymer chains may not be feasible. In the case of ethylene/1-pentene copolymerization, the molecular weight also increased after anionic polymerization (ΔMw, 14–24 entries 12–14). In most cases, the molecular weight distributions became narrow after anionic polymerization.
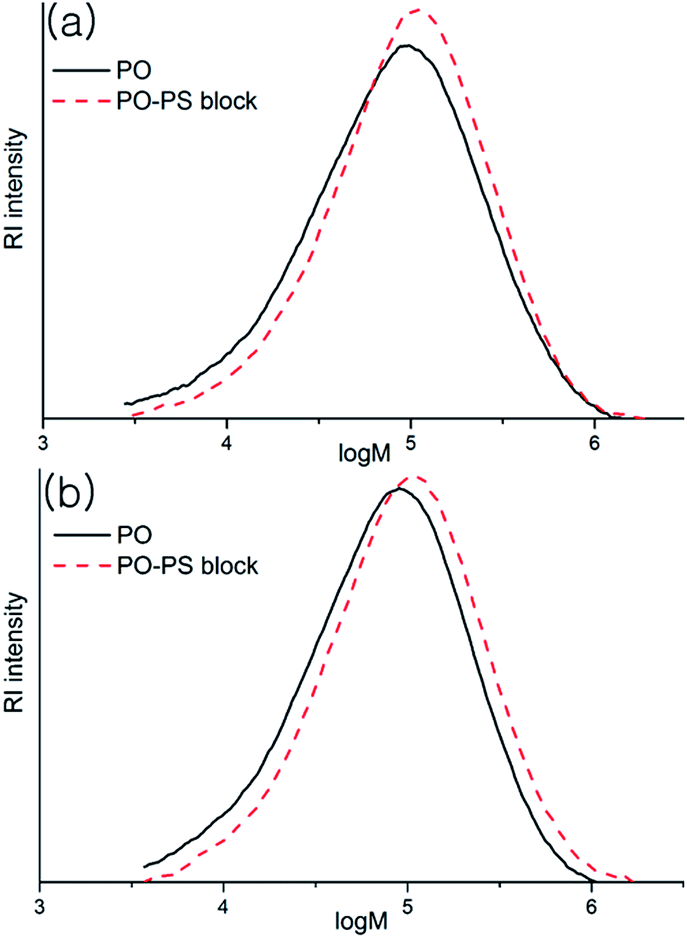 | ||
| Fig. 2 GPC curves before and after the anionic polymerization for the generation of PO–PS diblock copolymer (a, entry 1 in Table 2) and multiblock copolymer (b, entry 3 in Table 2). | ||
Formation of the block copolymers was evident from investigation of the TEM images of the thin films after staining with RuO4. Because PS is immiscible with PO, phase separation occurs and the PS domains selectively stained by RuO4 are clearly seen as a dark image. In contrast with the image of the PO–PS blend (Fig. 3(a)), the PO-b-PS diblock copolymers generated under the condition [Li] < [Al] + [Zn] (entry 1) formed spherical PS domains with an average 40 nm; these domains were regularly distributed in the PO matrix (Fig. 3(b)). However, the shape of the PS domains was rather blurred and irregular for the multiblock copolymers generated by the attack of nBuLi(tmeda)2 on the α-methylstyrene moieties under the condition [Li] > [Al] + [Zn] (Fig. 3(c)). When divinylbenzene was fed in the system, the shape of the PS domains became much more blurred and irregular. Specifically, large domains were observed when the amount of divinylbenzene feed was high (0.50 and 0.75 mol% per styrene) (Fig. 3(e) and (f)). The PS-domains are very regularly distributed with uniform sizes in the case of the related triblock copolymer SBS.56 Generation of multiblock instead of triblock copolymers may deteriorate the regularity of the phase separation of PS and PO blocks.
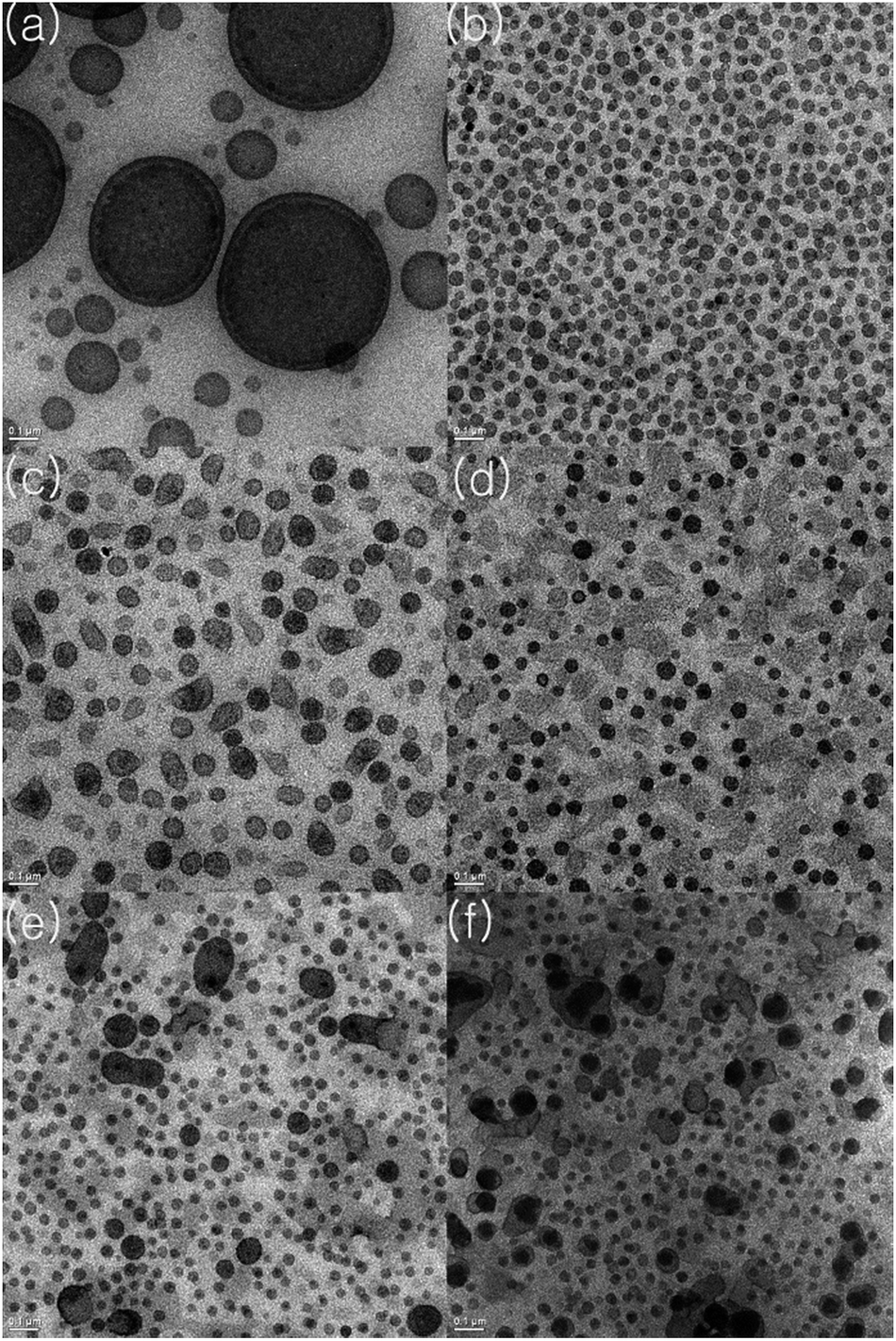 | ||
| Fig. 3 TEM images (1500 nm × 1500 nm) of thin films prepared using a blend of poly(ethylene/1-octene) and PS (a) and PO–PS block copolymers (entry 1 (b), entry 3 (c), entry 8 (d), entry 9 (e), entry 10 (f) in Table 2). | ||
The polymer samples were compressed between hot plates at 110 °C to make films with 400–500 μm thickness and the films, after cutting with 100 × 10 mm2 size, were subjected to hysteresis testing where each sample was extended to 200% strain over 10 cycles to determine elastic recovery. Both the PO–PS diblock (entry 1 in Table 2) and multiblock copolymers (entry 3 in Table 2) exhibit the elastomeric property; the first cycle results in the most significant amount of permanent deformation, followed by minimal deformation on subsequent cycles (Fig. 4). However, the multiblock copolymer shows better elastomeric property than the diblock analogue; the permanent deformation of the multiblock copolymer is significantly less than that of the diblock analogue.
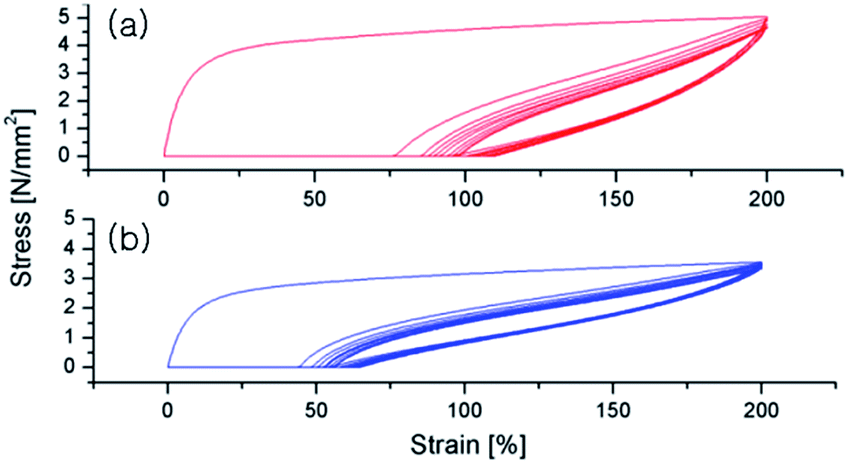 | ||
| Fig. 4 Plots of hysteresis experiment for the PO–PS diblock copolymer (a, entry 1 in Table 2) and PO–PS multiblock copolymer (b, entry 3 in Table 2). Ten cycles at 200% strain were performed. | ||
Conclusion
Dialkylzinc bearing an α-methylstyrene moiety (i.e., [4-(isopropenyl)benzyl]2Zn) was prepared for the use in so-called ‘coordinative chain transfer polymerization (CCTP)’ using a typical ansa-metallocene catalyst, rac-[Me2Si(2-methylindenyl)2]ZrCl2 activated with modified-methylaluminoxane (MMAO) to grow PO chains attached to zinc at one end and bearing the α-methylstyrene moiety at the other end. Switching to anionic polymerization by adding nBuLi(tmeda)2 in an amount to meet the criterion [Li] > [Zn] + [Al in MMAO] with subsequent addition of the styrene monomer enabled growth of PS chains from the Zn-alkyl sites and α-methylstyrene moieties, thereby generating PS-b-PO-b-PS triblock units. A PS growth site was generated from the α-methylstyrene moiety by the attack of nBuLi(tmeda)2 and the PS growth sites on the other side were generated from the Zn-alkyl sites via alkyl exchange between the zinc and lithium species. Formation of a substantial amount of PS homopolymers grown from nBuLi(tmeda)2 was inevitable (40–50%). The amount of homo-PS was reduced due to the consumption of some nBuLi(tmeda)2 by the reaction with α-methylstyrene moieties and was further significantly reduced by up to 14% upon addition of a small amount of divinylbenzene (0.25–1.0 mol% per styrene). The molecular weights increased after anionic polymerization, supporting formation of the block copolymers. However, in the model reaction of nBuLi(tmeda)2 with α-methylstyrene in the presence of (hexyl)2Zn, the 1:2 adduct, R–CH2C(Ph)(Me)–CH2C(Ph)(Me)Li, where R is either n-butyl originating from nBuLi or hexyl from (hexyl)2Zn, was generated. This observation suggests that simple PS-b-PO-b-PS triblock copolymers were not generated, but, instead, multiblock copolymers containing PS-b-PO-b-PS units might be formed. Generation of multiblock beyond triblock deteriorates the regularity of the phase separation of PS and PO blocks.
Experimental section
General remarks
All manipulations were performed under an inert atmosphere using a standard glove box and Schlenk techniques. Methylcyclohexane and 1-octene were purchased from Sigma-Aldrich and purified over a Na/K alloy. Ethylene was purified by contact with molecular sieves and copper at 40 bar pressure. Styrene was purchased from Sigma-Aldrich. N,N,N′,N′-Tetramethylethylenediamine (tmeda) was purchased from Sigma-Aldrich and purified over CaH2. The 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded using a Varian Mercury Plus 400 instrument. The gel permeation chromatograms (GPC) were obtained in THF at 40 °C using a Waters Millennium apparatus or in 1,2,4-trichlorobenzene at 160 °C using a PL-GPC 220 system at Korea Polymer Testing & Research Institute. Transmission electron microscopes (TEM) images were recorded on an FEI Tecnai G2 F30 S-Twin instrument. Differential scanning calorimetry (DSC) data were collected from the second heating, performed at a heating rate of 10 °C min−1 using a Thermal Analysis Q10 instrument. 1-Chloromethyl-4-isopropenylbenzene was prepared by the method reported in the literature.44Synthesis of [4-(isopropenyl)benzyl]2Zn
1-Chloromethyl-4-isopropenylbenzene (6.00 g, 36.0 mmol) in diethyl ether (20 mL) was added dropwise to a flask containing magnesium (1.31 g, 54.0 mmol) in diethyl ether (40 mL). After stirring for 4 h, the remaining Mg was removed by filtration over Celite. ZnCl2 (2.70 g, 18.0 mmol) was subsequently added to the filtrate. The white solids that precipitated after stirring for three days were removed by filtration over Celite. The solvent was removed under vacuum to yield a yellowish residue, which was dissolved in methylcyclohexane (200 g). After removal of the residual solid by filtration, the white solid was isolated by evaporation of the solvent under vacuum (5.31 g, 90%); the solid was pure based on 1H and 13C NMR spectral analyses. For use in polymerization as a chain transfer agent, the isolated product was recrystallized from hexane at −30 °C two additional times. 1H NMR (400 MHz, C6D6) : δ 7.36 (d, J = 8.0 Hz, 4H), 6.86 (d, J = 8.0 Hz, 4H), 5.44 (s, 2H), 5.00 (s, 2H), 2.04 (s, 6H), 1.55 (s, 4H) ppm. 13C{1H} NMR (C6D6): δ 144.21, 143.42, 127.24, 126.19, 110.63, 23.91, 22.28 ppm.Typical procedure for anionic polymerization of styrene in the presence of (hexyl)2Zn (entry 5 in Table 1)
nBuLi (3.07 mg, 48 μmol) and tmeda (11.2 mg, 96 μmol) were successfully added to a flask containing (hexyl)2Zn (11.3 mg, 48 μmol) and methylcyclohexane (1.0 g) inside a glove box. Styrene (2.5 g, 24 mmol) dissolved in methylcyclohexane (6.5 g) was added and the anionic polymerization was performed at 90 °C for 2 h. Complete conversion of the styrene monomer was evident from the analysis of the 1H NMR spectrum of the sample. Aqueous HCl (2 N, 0.3 mL) was added and the resulting solution was stirred for 30 min at 90 °C to destroy the zinc species. The solution was filtered through a short pad of silica gel that was thoroughly washed with toluene. Toluene was removed by using a rotary evaporator to isolate PS; the isolated sample was dried in a vacuum oven at 130 °C for 1 h. The weight of isolated PS was identical to that of the styrene monomer fed into the system.Typical procedure for synthesis of PO–PS block copolymers (entry 3 in Table 2)
A bomb reactor (125 mL) was charged with a solution of MMAO (AkzoNobel, 7.0 wt%-Al in heptane, 77 mg, 200 μmol-Al) in methylcyclohexane (17.0 g). After stirring for 1 h at 100 °C using a mantle, the solution was removed using a cannula. This washing procedure was performed to clean up any catalyst poisons with the MMAO solution. The reactor was again charged with a solution of 1-octene (10.0 g) and [(4-isopropenyl)benzyl]2Zn (65.6 mg, 0.200 mmol) in methylcyclohexane (17.0 g) under an inert atmosphere. The catalyst solution was prepared by reacting rac-[Me2Si(2-methylindenyl)2]ZrCl2 (0.48 mg, 1.0 μmol) and MMAO (77 mg, 200 μmol-Al) in methylcyclohexane (2.0 g) for 40 min. After injection of the catalyst solution with a syringe at 60 °C, ethylene gas was immediately charged into the system at 30 bar. Even though the reactor was cooled in an ice bath, the temperature increased to ∼130 °C within 5 min due to the exothermic nature and the pressure gradually decreased due to the consumption of ethylene. Whenever the pressure reached 10 bar, ethylene gas was fed to a pressure of 15 bar and the temperature was self-controlled within the range of 120–130 °C. Twenty minutes after injection of ethylene gas, the stirring rate was decreased from 300 to 15 rpm by formation of a thick viscose solution. The olefin polymerization was terminated by venting the remaining ethylene gas and an aliquot of the sample was taken to measure the molecular weight of the generated PO. When the ethylene feed was disconnected, the temperature decreased. When the temperature reached 90 °C, a solution of nBuLi(tmeda)2 prepared by mixing nBuLi (32.0 mg, 0.500 mmol) and tmeda (116.2 mg, 1.00 mmol) in methylcyclohexane (2.0 g) was added to the reactor. The temperature was increased to 120 °C by heating with a mantle for 1 h under stirring, after which styrene (10.4 g, 100 mmol) was injected. The temperature was controlled in the range of 100–130 °C using a mantle. The viscosity gradually increased to reach an almost unstirrable state after 1.5 h. Full conversion of styrene and 1-octene was evident from 1H NMR analysis of an aliquot of the reaction mixture. After complete conversion of styrene, the reactor was cooled and the generated polymers were broken into pieces with scissors. The polymer pieces were dispersed in chloroform (290 g) containing HCl (2 N, 5 mL), and the resulting mixture was refluxed overnight. When acetone (580 g) was added, the block copolymers precipitated, whereas the PS-homopolymer is soluble in the resulting co-solvent of chloroform and acetone (1:2 w/w). The block copolymers were isolated by filtration, and were thoroughly dried in a vacuum oven at 80 °C for several hours. The PS-homopolymers were isolated by evaporation of the solvent in the filtrate using a rotary evaporator. The residual PS-homopolymers was redissolved in chloroform and precipitated by adding methanol. The PS-homopolymers isolated by filtration were thoroughly dried in a vacuum oven at 80 °C for several hours. The weight of consumed ethylene (18.4 g) was calculated by subtracting the weights of 1-octene (10.0 g) and styrene (10.4 g) fed into the system (both of which were completely converted to polymers) from the weight of total isolated polymers [multiblock-copolymer (34.2 g) + PS-homopolymer (4.6 g)]. The GPC and TEM studies were carried out using the isolated multiblock-copolymers after separation of the PS-homopolymer.
Sample preparation for transmission electron microscopy (TEM)
The multiblock copolymer (10 mg) was completely dissolved in toluene (5 mL) by stirring at 100 °C for 20 min. A drop of the hot solution was loaded on a carbon-coated copper TEM grid (400 mesh). After evaporation of toluene at room temperature, the sample was completely dried in a vacuum oven at 130 °C for 1 h. The sample was annealed in an oven at 150 °C for 12 h. The sample was stained with RuO4 by suspending the TEM grid coated with the film for 1 h in a closed chamber containing an aqueous solution of RuO4, which was prepared by reacting RuO2 (30 mg) with NaIO4 (0.20 g) in water (5 mL) at 0 °C for 3 h.57Acknowledgements
This work was supported by the Korea CCS R&D Center (KCRC) grant (No. 2012-0008935) and the Priority Research Centers Program (No. 2012-0006687) funded by the Korean Government (Ministry of Science, ICT and future planning).Notes and references
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS.
- W. S. Chi, S. Hwang, S. J. Lee, S. Park, Y. S. Bae, D. Y. Ryu, J. H. Kim and J. Kim, J. Membr. Sci., 2015, 495, 479–488 CrossRef CAS.
- H. Ohnogi, S. Sasaki and S. Sakurai, Macromol. Symp., 2016, 366, 35–41 CrossRef CAS.
- X. Zhao, Y. Huang, M. Kong, Q. Yang and G. Li, RSC Adv., 2014, 4, 59302–59309 RSC.
- M. Uenishi, N. Fukushima, M. Teramachi, M. Mizuta, J. Kamo and T. Tsuru, J. Appl. Polym. Sci., 2014, 131 DOI:10.1002/APP.38386.
- B. Liu, Y. Shangguan, Y. Song and Q. Zheng, J. Appl. Polym. Sci., 2013, 129, 973–982 CrossRef CAS.
- D. T. Wong, C. Wang, J. A. Pople and N. P. Balsara, Macromolecules, 2013, 46, 4411–4417 CrossRef CAS.
- A. J. Müller, A. T. Lorenzo, M. L. Arnal, A. B. de Fierro and V. Abetz, Macromol. Symp., 2006, 240, 114–122 CrossRef.
- R. C.-c. Tsiang, W.-s. Yang and M.-d. Tsai, Polymer, 1999, 40, 6351–6360 CrossRef CAS.
- R. Matmour, A. S. More, P. P. Wadgaonkar and Y. Gnanou, J. Am. Chem. Soc., 2006, 128, 8158–8159 CrossRef CAS PubMed.
- T. C. Chung and J. Y. Dong, J. Am. Chem. Soc., 2001, 123, 4871–4876 CrossRef CAS PubMed.
- J. Y. Dong and T. C. Chung, Macromolecules, 2002, 35, 1622–1631 CrossRef CAS.
- K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2008, 41, 640–649 CrossRef CAS.
- M. S. Weiser and R. Mülhaupt, Macromol. Rapid Commun., 2006, 27, 1009–1014 CrossRef CAS.
- J. Y. Jeon, S. H. Park, D. H. Kim, S. S. Park, G. H. Park and B. Y. Lee, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3110–3118 CrossRef CAS.
- A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836–3857 CrossRef CAS PubMed.
- R. Kempe, Chem.–Eur. J., 2007, 13, 2764–2773 CrossRef CAS PubMed.
- M. van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, J. Am. Chem. Soc., 2005, 127, 9913–9923 CrossRef CAS PubMed.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef CAS PubMed.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson, P. J. Maddox and M. van Meurs, Angew. Chem., Int. Ed., 2002, 41, 489–491 CrossRef CAS PubMed.
- R. Ribeiro, R. Ruivo, H. Nsiri, S. Norsic, F. D'Agosto, L. Perrin and C. Boisson, ACS Catal., 2016, 6, 851–860 CrossRef CAS.
- T. S. Thomas, W. Hwang and L. R. Sita, Angew. Chem., Int. Ed., 2016, 55, 4683–4687 CrossRef CAS PubMed.
- F. Bonnet, H. E. Dyer, Y. El Kinani, C. Dietz, P. Roussel, M. Bria, M. Visseaux, P. Zinck and P. Mountford, Dalton Trans., 2015, 44, 12312–12325 RSC.
- F. Wang, B. Dong, H. Liu, J. Guo, W. Zheng, C. Zhang, L. Zhao, C. Bai, Y. Hu and X. Zhang, Macromol. Chem. Phys., 2015, 216, 321–328 CrossRef CAS.
- P. Zinck, Polym. Int., 2016, 65, 11–15 CrossRef CAS.
- Y. Ota, T. Murayama and K. Nozaki, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2857–2861 CrossRef CAS PubMed.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS PubMed.
- J. Wei, W. Hwang, W. Zhang and L. R. Sita, J. Am. Chem. Soc., 2013, 135, 2132–2135 CrossRef CAS PubMed.
- L. Pan, K. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 12012–12015 CrossRef CAS PubMed.
- A. Valente, G. Stoclet, F. Bonnet, A. Mortreux, M. Visseaux and P. Zinck, Angew. Chem., Int. Ed., 2014, 53, 4638–4641 CrossRef CAS PubMed.
- Y. Mohammadi, M. Ahmadi, M. R. Saeb, M. M. Khorasani, P. Yang and F. J. Stadler, Macromolecules, 2014, 47, 4778–4789 CrossRef CAS.
- M. Zhang, T. W. Karjala, P. Jain and C. Villa, Macromolecules, 2013, 46, 4847–4853 CrossRef CAS.
- K. E. Crawford and L. R. Sita, ACS Macro Lett., 2015, 4, 921–925 CrossRef CAS.
- N. Hu, C. K. Mai, G. H. Fredrickson and G. C. Bazan, Chem. Commun., 2016, 52, 2237–2240 RSC.
- X. Song, Q. Ma, Z. Cai, R. Tanaka, T. Shiono and R. B. Grubbs, Macromol. Rapid Commun., 2016, 37, 227–231 CrossRef CAS PubMed.
- H. Ohtaki, F. Deplace, G. D. Vo, A. M. Lapointe, F. Shimizu, T. Sugano, E. J. Kramer, G. H. Fredrickson and G. W. Coates, Macromolecules, 2015, 48, 7489–7494 CrossRef CAS.
- Z. Zhou, M. D. Miller, D. Lee, R. Cong, C. Klinker, T. Huang, C. Li Pi Shan, B. Winniford, A. W. Degroot, L. Fan, T. Karjala and K. Beshah, Macromolecules, 2015, 48, 7727–7732 CrossRef CAS.
- S. Guo, H. Fan, Z. Bu, B. G. Li and S. Zhu, Polymer, 2015, 80, 109–114 CrossRef CAS.
- M. R. Saeb, M. M. Khorasani, M. Ahmadi, Y. Mohammadi and F. J. Stadler, Polymer, 2015, 76, 245–253 CrossRef CAS.
- B. Jiang, H. Shao, H. Nie and A. He, Polym. Chem., 2015, 6, 3315–3323 RSC.
- A. Kermagoret, A. Debuigne, C. Jérôme and C. Detrembleur, Nat. Chem., 2014, 6, 179–187 CrossRef CAS PubMed.
- C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2014, 53, 6683–6686 CrossRef CAS PubMed.
- R. Sugimoto, H. Kaneko, J. Saito, N. Kawahara, S. Matsuo and T. Matsugi, Polym. Bull., 2014, 71, 1421–1431 CrossRef CAS.
- X. Creary, M. E. Mehrsheikh-Mohammadi and S. McDonald, J. Org. Chem., 1987, 52, 3254–3263 CrossRef CAS.
- R. R. Schrock, J. Organomet. Chem., 1976, 122, 209–225 CrossRef CAS.
- R. Ramírez-Contreras, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2015, 34, 1143–1146 CrossRef.
- V. Weissig, R. Beckhaus, U. Banasiak and K. H. Thiele, Z. Anorg. Allg. Chem., 1980, 467, 61–67 CrossRef CAS.
- G. Fraenkel, M. J. Geckle, A. Kaylo and D. W. Estes, J. Organomet. Chem., 1980, 197, 249–259 CrossRef CAS.
- D. J. Worsfold and S. Bywater, J. Polym. Sci., 1957, 26, 299–304 CrossRef CAS.
- T. Fujimoto, N. Ozaki and M. Nagasawa, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2259–2274 CrossRef CAS.
- D. P. Wyman and I. H. Song, Makromol. Chem., 1968, 115, 64–72 CrossRef CAS.
- V. I. W. Stuart and D. B. Priddy, US Pat. 4704431, 1987.
- W. Spaleck, M. Antberg, J. Rohrmann, A. Winter, B. Bachmann, P. Kiprof, J. Behm and W. A. Herrmann, Angew. Chem., Int. Ed., 1992, 31, 1347–1350 CrossRef.
- C. Zhang, H. Niu and J.-Y. Dong, Polym. Bull., 2010, 65, 779–786 CrossRef CAS.
- S. Carlotti, P. Desbois, V. Warzelhan and A. Deffieux, Polymer, 2009, 50, 3057–3067 CrossRef CAS.
- P. R. Lewis and C. Price, Polymer, 1972, 13, 20–26 CrossRef CAS.
- V. Rek, N. Vranješ, M. Šlouf, I. Fortelný and Ž. Jelčić, J. Elastomers Plast., 2008, 40, 237–251 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, mass data, GPC curves, and DSC thermogram. See DOI: 10.1039/c6ra25848d |
| This journal is © The Royal Society of Chemistry 2017 |