
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New green synthesized reduced graphene oxide–ZrO2 composite as high performance photocatalyst under sunlight
K. Gurushanthaa,
K. S. Anantharaju*bc,
L. Renukaac,
S. C. Sharmadc,
H. P. Nagaswarupaa,
S. C. Prashantha a,
Y. S. Vidya*e and
H. Nagabhushana*f
a,
Y. S. Vidya*e and
H. Nagabhushana*f
aDepartment of Science, Research Center, East West Institute of Technology, Bengaluru – 560091, India
bDepartment of Chemistry, Dayananda Sagar College of Engineering, Shavige Malleshwara Hills, Kumaraswamy Layout, Bengaluru – 560078, India. E-mail: iamananthkurupalya@gmail.com
cDr. D. Premachandra Sagar Centre for Advanced Materials, DSCE, Bangalore – 560078, India
dDepartment of Mechanical Engineering, Jain University, Advisor, Jain Group of Institutions, Bengaluru – 560069, India
eDepartment of Physics, Lal Bahadur Shastry Government First Grade College, Bengaluru – 560032, India. E-mail: vidyays.phy@gmail.com
fProf. C.N.R. Rao Centre for Advanced Materials, Tumkur University, Tumkur – 572103, India. E-mail: bhushanvlc@gmail.com
First published on 22nd February 2017
Abstract
We report a green route method for the effective reduction of graphene oxide using Cinnamon extract. ZrO2 nanomaterial synthesized via facile green induced method and zirconia–reduced graphene oxide nanocomposite (NC) synthesized by reflux method. The structural properties of the prepared NC had been characterized using PXRD, SEM, TEM, Raman, PL and XPS analysis. Further, the excellent photocatalytic properties of NC for the decomposition of reactive blue 4 compared to commercial P25 can be attributed to its excellent electrical properties and well connected arrangement derived from the structural reward of the ZrO2 and rGO support. This results in significant electron-acceptor/transport of graphene materials that effectively decrease the photo-generated electron–hole recombination thereby enhancing the absorption of light. It opens new window to use this eco-friendly, simple and low cost method to synthesize reduced graphene-based composite materials.
1. Introduction
Recently, the main concerns for the environmentalists are water soluble and colored organic compounds such as dyes. The aquatic system present in the water are severely affected by the dye manufacturing, coloring and textile industrial residues which are highly toxic, carcinogenic to human beings.1 The major source for water pollution, eutrophication and perturbation of aquatic ecosystem is due to release of dyes into the water. In the recent years, tremendous work involved in the rectification of these environmental issues mainly highlights the photocatalytic process which is the best candidate for the efficient dye remediation under light illumination. The study of photocatalytic technology can't be separated with photocatalytic materials. Environmental friendly ZrO2 is one among the fundamental transition metal oxide with less toxicity and high chemical inertness.2 In addition, it is biocompatible, cost-effective with good thermal stability and electrochemical nature. It serves as an excellent material as an active electrode for the electro catalysis of critical analyses because of its excellent electro catalytic properties.3Currently, majority of the materials used in photocatalysis are semiconductor materials, such as TiO2, ZnO and ZrO2, etc., as the core body of the catalyst. Out of these, ZrO2 is the only metallic oxide which has both oxidizability and reducibility due to its acidic and basic nature. Since ZrO2 is a kind of p-type semiconductor,4 it easily produces oxygen holes, as the carrier of the catalyst, hence generating interaction with active components. Thus compared to other materials it has more excellent performance as a photocatalyst. Although as a single semiconductor, zirconia has high carrier recombination rate and low quantum efficiency which delimit its practical application to some extent. Due to wide band gap, zirconia can only absorb UV light which constitutes less than 4% of the solar spectrum. Thus, its large scale applications get limited. At present, most of the researches focus on the modification of ZrO2 by doping, compounding carbon nano tubes and several other techniques to enhance its photocatalytic activity.5 The last approach is the best way and these composites have been prepared by many researchers to enhance their photocatalytic activity under visible light.
About relevant literature of ZrO2, the photodegradation of textile dyes like reactive yellow, rhodamine B, 2,2,4-diphenoxyacetic under UV light has been reported earlier.6–9 Also photocatalytic activity for the degradation of ethylene and 4-chlorophenol under UV light by TiO2–ZrO2 binary oxides has been studied.10–12 The increased activity under UV light in the above cases is ascribed to either stability of catalyst/increase in surface area or transfer of charges from ZrO2 to TiO2 hence preventing the electron–hole recombination. Recently, the photodegradation of phenol under UV light by mixed oxide TiO2–ZrO2 catalyst has been proved to be very efficient compared to P25. Here primarily fast transfer of photogenerated electrons from the conduction band of ZrO2 to TiO2 takes place effectively under UV light which is mainly due to its small particle size, high surface area and abundant surface OH groups.13
In the past few years, many researchers have been attracted by the improved photocatalytic activity of carbon-based NCs. Hence in order to prepare these composites, graphene, an extremely attractive component has been particularly chosen14,15 because of its large specific surface area and an extensive two-dimensional π–π conjugation structure that has excellent electron conductivity.16 Carriers in pristine graphene sheets have been already studied to behave as massless Dirac fermions.17 When combining TiO2 nanocrystals with graphene, excited electrons of TiO2 could transfer from the conduction band (CB) to graphene through a percolation mechanism.18 The heterojunction formed at the interface (termed Schottky barrier) separates the photoinduced electron–hole pairs, thereby inhibiting charge recombination.16 First time, Kamat and co-workers in 2010 have demonstrated the enhanced photocatalytic activity of graphene-based semiconductor–metal composites.18 After which, Zhang et al.,19 Shen et al.,20 and Zhou et al.,21 performed an one-step hydrothermal methods to prepare graphene–TiO2 hybrid materials and showed that the composites exhibited enhanced photoactivity towards organic degradation over bare TiO2. Fan et al.,22 fabricated P25–graphene composites by three different preparation methods, i.e., UV-assisted photocatalytic reduction, hydrazine reduction and hydrothermal method which had better photocatalytic performance for H2 evolution from methanol aqueous solution as compared to pure P25. Graphene (GR)–TiO2 mesocrystal composites were synthesized by a facile template-free process based on the sol–gel and solvothermal combination method for the enhancement in visible light photocatalytic performance.23 Bearing these results in mind, the study on the use of ZrO2–reduced graphene composites on the photodecomposition of Reactive Blue 4 (RB 4) is still in its infancy to the best of our knowledge. This study is intended to examine the role of reduced graphene in the composite towards the photodecomposition of RB 4 under sunlight irradiation.
Our group already demonstrated green assisted solution combustion synthesis.24–26 Since the plant based synthetic routes are environmental friendly, cost effective, energy saving and less time consumption. Cinnamon (Cinnamomum zeylanicum), a common spice used in various countries is well known for its various activities such as antiparasitic, antioxidant, antimicrobial and it was found to reduce blood glucose, blood pressure and serum cholesterol.27,28 In traditional medicine for treating various ailments in many countries, the bark of Cinnamon has been extensively used. Presence of these health benefit compounds such as cinnamaldehyde, eugenol and their derivatives29 are mainly responsible for the medicinal properties of Cinnamon and help them to act as a potential antioxidant. Reduction of GO to rGO is attained by these antioxidant which acts as a very good reducing agent. Therefore, an attempt has been made to assess the effectiveness of Cinnamon extract towards the GO reduction.
In the present investigation, GO is reduced to rGO using green induced method. Further, a simple reflux method has been adopted to prepare ZrO2–reduced graphene oxide (ZrO2/rGO) NC. The Cyclic Voltammetry (CV) and Electrochemical Impedance Studies (EIS) were carried out for ZrO2 NM and ZrO2/rGO NC to find the capacitance and charge transfer resistance. The results obtained from the electrochemical studies were correlated with photocatalytic and photoluminescence studies. The photoactivity of ZrO2/rGO NC was compared to commercial P25 and studied by evaluating its performance in the photodecomposition of RB 4 dye under sunlight illumination. The ZrO2/rGO NC was shown to exhibit excellent photocatalytic activity as compared to ZrO2 NMs and commercial P25.
2. Experimental
All the used chemicals were procured from S. D. Fine Chemicals Company and graphite flakes were received from Sigma Aldrich Company. Cinnamon was purchased from local market.2.1 Preparation of Cinnamon extract
In a round bottomed flask, powdered Cinnamon bark and double distilled water were added in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. It was refluxed for 5 hours with constant stirring at a temperature of 100 °C. The obtained extract was filtered, concentrated and dried.
:10. It was refluxed for 5 hours with constant stirring at a temperature of 100 °C. The obtained extract was filtered, concentrated and dried.
2.2 Preparation of GO
GO was prepared in the laboratory by adding 5 g of graphite and 2.5 g of sodium nitrate in 120 mL of 95% sulfuric acid.30 This solution was placed in an ice bath and stirred for half an hour followed by the addition of 15 g of potassium permanganate with vigorous stirring at temperature below 20 °C. By adding 150 mL of distilled water slowly, the stirring was continued overnight. The temperature was increased rapidly from 20 to 98 °C and 50 mL of 30% H2O2 was added. The product obtained was washed with 5% HCl and then with deionized water.31 Finally, the obtained samples were collected after drying.2.3 Preparation of rGO
The above prepared GO (80 mg) was added to 50 mL of distilled water and sonicated for about 40 min. Cinnamon extract was mixed with the suspension and refluxed for 45 min, hence reducing GO to rGO.32 The final product was washed with distilled water, dried and stored.2.4 Preparation of ZrO2 nanomaterial
Solution combustion synthesis was carried out by using zirconyl nitrate (ZrO(NO3)2) as oxidizer and Cinnamon extract as fuel. Stoichiometric ratio of oxidizer and fuel were taken in a cylindrical crucible. This reaction mixture was placed in a muffle furnace preheated at 450 °C. Initially, the mixture boils and undergoes dehydration followed by liberation of large amount of gases with formation of nanomaterials. The obtained ZrO2 nanomaterial (NM) was collected.2.5 Preparation of ZrO2/rGO NC
100 mg of prepared ZrO2 was taken in 100 mL of water and added into a round bottom flask along with refluxing for about half an hour. 20 mg of prepared rGO was dispersed in 2 litre of water and stirred for half an hour. rGO solution was added dropwise to ZrO2 solution with constant stirring and mixture was stirred for about 24 hours to attain ZrO2/rGO NC. The schematic diagram for synthesis of ZrO2/rGO is shown in Fig. 1.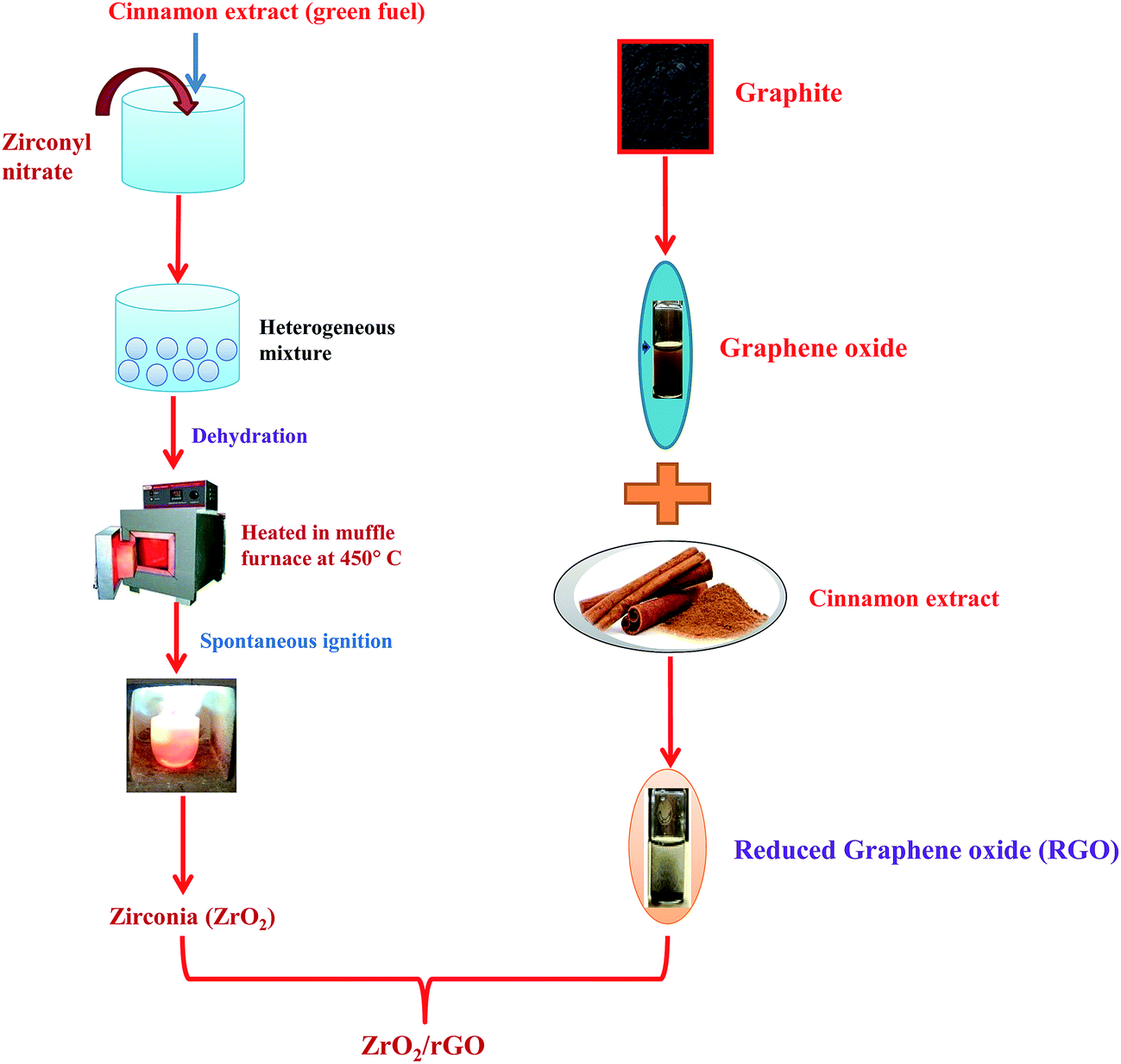 | ||
| Fig. 1 Schematic diagram for synthesis of ZrO2/rGO NC. | ||
2.6 Characterization
SEM (Scanning Electron Microscopy), TEM (Transmission Electron Microscopy) and high resolution TEM were performed to find the morphology of the synthesized samples using SEM, Hitachi-3000 and TEM, TECNAIF-30 respectively. UV-Vis absorption spectra (SL 159 ELICO UV-Visible spectrophotometer) determined the energy band gap (Eg). Using CuKα (1.541 Å) radiation at a scan rate of 2° min−1 with nickel filter in the 2θ range 20–70°, the crystalline size and phase present in the synthesized samples were analyzed by using room temperature Shimadzu Powder X-ray diffractometer (PXRD). With the help of Horiba Flurolog Spectrofluorometer, PL emission spectra of the samples were recorded at room temperature. Raman spectrophotometer (Model: Lab RAM HR) had been utilized to analyze Raman spectra with an exposure time of about 5 s with number of repetitions as three times (spot size: 100 (1l); long working distance: 50 (2l); power: 3 mW without filter). Using AXIS ULTRA from AXIS 165, XPS (X-ray photoelectron spectroscopy) analysis were performed with Kratos patented magnetic immersion lens, spherical mirror and analyzer charge neutralization system. A total organic carbon (TOC) analyzer (Shimadzu) was used to determine the TOC. Fourier transform infrared spectroscopy (FTIR) studies for the synthesized samples were performed with a Perkin Elmer FTIR spectrometer (Spectrum-1000).2.7 Photocatalytic procedure
Photocatalytic experiments were carried out under UV light irradiation in the laboratory and also under direct sunlight. The experiment was carried out in a photocatalytic reactor with surface area 176.6 cm2. In the photocatalytic reactor, dye solution of known ppm was mixed with required amount of synthesized catalyst (ZrO2/rGO). Before starting the experiment, the mixture was stirred for half an hour continuously in order to attain adsorption/desorption equilibrium. Once experiment starts, 5 mL of suspension was withdrawn from photocatalytic reactor at regular intervals. All the experiments were conducted between 11 a.m. to 2 p.m. in the month of May when the solar intensity fluctuations were minimal. The average solar intensity in a day was found to be 0.753 kW m−2. In order to avoid the error arising due to changes in the solar intensities, the experiments were carried out simultaneously to ensure similar exposure to sunlight irradiation and their activities were compared. Finally the suspension was centrifuged and analyzed in UV-Visible spectrophotometer. The amount of dye adsorbed was calculated from the difference in the absorbance before and after the decomposition and tabulated.3. Results and discussions
3.1 Morphological studies
Fig. 2a–c shows the SEM images of ZrO2 NM and ZrO2/rGO NC. It could be observed that the crystals are irregular in shape/size and slight agglomeration is observed (Fig. 2a). Here, rGO helps ZrO2 NM to disperse more smoothly in ZrO2/rGO NC. Thus, the separation between the agglomerated NPs slightly increases (Fig. 2b and c at different magnification). In these spaces, the rGO sheets exist in the form of flakes. The material was found to be porous with black appearance, which helps in the increased photocatalytic performance. This is analogous with the study carried out by Mao et al., demonstrated black TiO2 can more efficiently harvest solar light for photocatalysis.33 | ||
| Fig. 2 Scanning electron microscopy of (a) ZrO2 NM (b & c) ZrO2/rGO NC. | ||
The microstructure of ZrO2/rGO NC is studied by TEM (Fig. 3a and b). Abundant ZrO2 nanocrystals are densely deposited on the wrinkled graphene sheets due to preferential heterogeneous nucleation and interfacial interactions. As shown in Fig. 3a and b, the wrinkled paper like structure of the 2D graphene sheet can be clearly observed. The HRTEM image (Fig. 3d) of the wrinkled part obtained from the area marked by the red rectangle in Fig. 3a depicts that there were approximately 18 graphene layers.
 | ||
| Fig. 3 TEM (a & b), HRTEM (c & d), and SAED pattern (e) of ZrO2/rGO NC. | ||
The graphene plays a vital role in supporting ZrO2 nanoparticles dispersion on its surface while ZrO2 facilitates to avoid the agglomeration of rGO. The crystallite sizes of the deposited ZrO2 nanocrystals are normally 14–18 nm, which is in good agreement with the PXRD results. The intimate interfacial contact between rGO and ZrO2 NC can be observed in HRTEM image (Fig. 3c and e) which shows the lattice of both graphene and ZrO2. Since that the transfer process of charge carriers in rGO–ZrO2 NC is related to the interfacial interaction between graphene and ZrO2 NC. This leads to the expectation for improved photocatalysis process due to good charge transfer property. The growth direction of the ZrO2 NC is parallel to the lattice fringes and the d spacing of the (−111) plane is about 0.3368 nm which is close to the lattice spacing of the monoclinic ZrO2. Considering our PXRD study and close observation of the SAED images (Fig. 3f), it can be concluded that the diffraction rings signify the monoclinic ZrO2 structure. The SAED pattern of the NC demonstrates the crystalline nature.
3.2 X-ray photoelectron spectroscopy
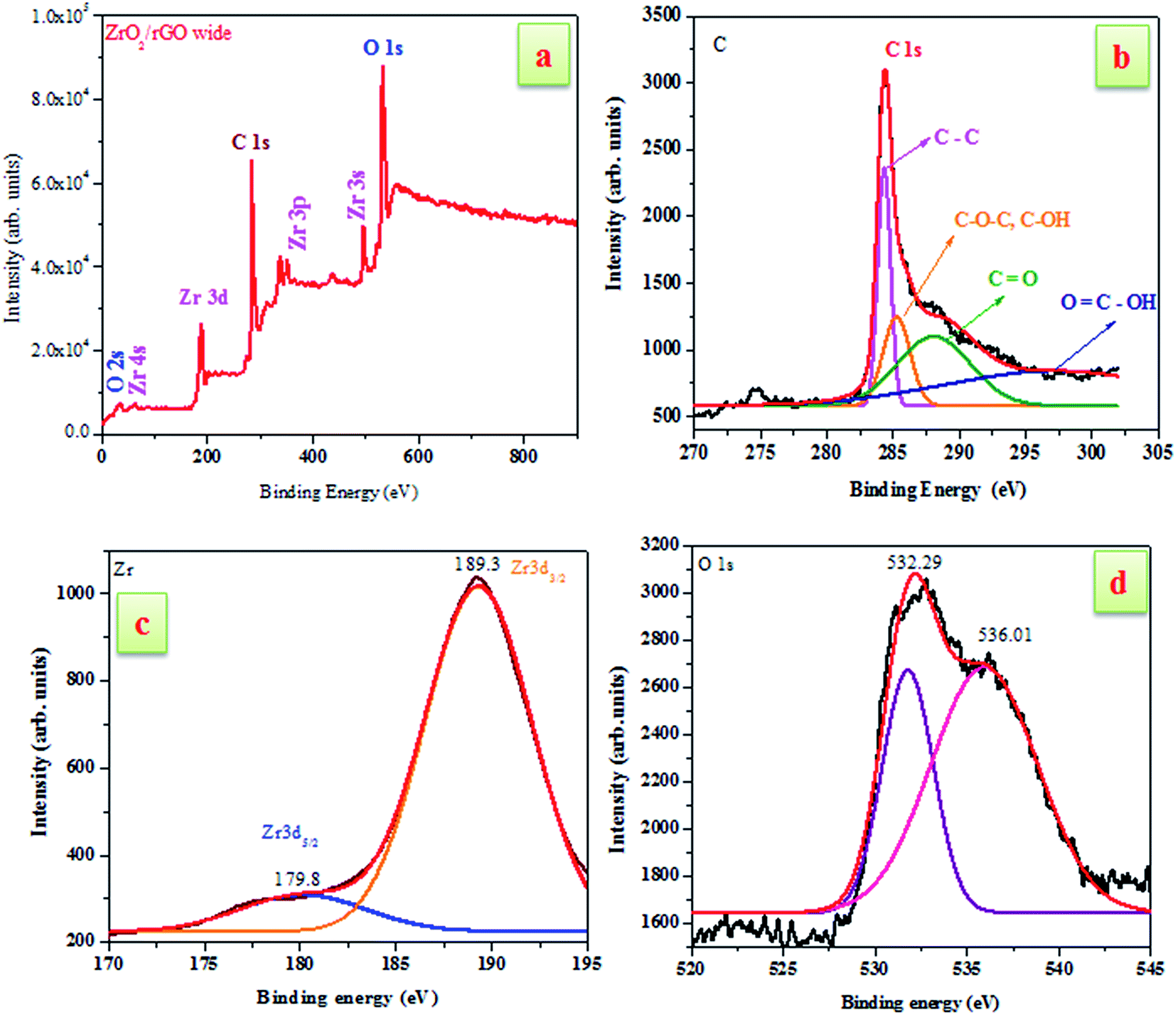
To investigate the elemental composition of ZrO2/rGO NC, XPS measurements have been carried out (Fig. 4). The existence of carbon and ZrO2 was confirmed by XPS analysis of the ZrO2/rGO composite as shown in Fig. 4a. The presence of Zr, oxygen and carbon elements in the sample were indicated by Zr 3d, O 1s and C 1s characteristic peaks in the wide scan XPS spectrum of ZrO2/rGO NC. The existence of graphene sheets were confirmed by the signal for C 1s obtained at the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carboxyl groups, 296.4 eV), CO (carbonyl groups, 288.1 eV), C–OH/C–O–C (hydroxyl and epoxy groups, 285.3 eV) and CC/C–C (284.4 eV) (see Fig. 4b).
O (carboxyl groups, 296.4 eV), CO (carbonyl groups, 288.1 eV), C–OH/C–O–C (hydroxyl and epoxy groups, 285.3 eV) and CC/C–C (284.4 eV) (see Fig. 4b).
 | ||
| Fig. 4 XPS spectra of ZrO2/rGO NC (a) survey spectrum (b) C 1s (c) Zr 3d (d) O 1s peaks. | ||
Furthermore, the spin–orbit splitting of the Zr 3d components, Zr 3d3/2, Zr 3d5/2 with a peak separation of 2.4 eV results in the peaks located at 179.8 and 189.3 eV (see Fig. 4c). The peak of the Zr 3d of the nanocomposite showed peaks centered at 179.8 eV is attributed to the formation of a Zr–C bond in the composite.34 Additionally, the presence of multi-component oxygen species in the surface is understood by the presence of oxygen vacancies of O1s which are asymmetric (Fig. 4d). The curve was deconvoluted into two separate peaks located at 532.29 and 536.01 eV. In the spectrum, there arises a high binding energy component (HBEC) peak located at 536.01 eV and low binding energy component (LBEC) peak located at 532.29 eV due to two different types of oxygen species. The chemisorbed oxygen and lattice oxygen were two different types of oxygen species present in HBEC and LBEC that was originated by surface chemisorbed species such as OH− and H2O. Thus, from the above explanations it was clear that ZrO2 had been incorporated into the rGO layers during synthesis.35,36
3.3 Raman studies
Raman spectroscopy is a widely used tool for the characterization of sp2 and sp3 hybridized carbon atoms in graphene based materials to differentiate the order and disorder/defects.37,38 The Raman signature of GO, rGO and ZrO2/rGO are depicted in the Fig. 5. The Raman spectrum of GO can be characterized by two main features, D band (the symmetric A1g mode) at 1353 cm−1 and G band (the E2g mode of the sp2 carbon atoms) at 1581 cm−1.39 The G band related to the first-order scattering of the E2g mode noticed for sp2 carbon domains, while the D band is ascribed to a breathing mode of point phonons of A1g symmetry, which is a familiar feature of sp3 defects in carbon and usually can be allied with the structural defects, amorphous carbon, or edges that split the symmetry and selection rule.38 As a result, the intensity ratio of the D to the G band is usually a gauge of the disorder/defects in graphene, and a smaller intensity of ID/IG ratio can be consigned to fewer sp3 defects/disorders and better average size (or less amount) of the in-plane graphitic crystallite sp2 domains.38,40 The GO (0.442) shows lesser ID/IG ratio compared to rGO (0.452), signifying that the rGO include more defects.41 For the ZrO2–rGO composite, the ID/IG ratio is further enhanced to 0.657, signifying the formation of more sp3 defects in carbon. The probable reason for the increased sp3 defects can be credited to the strong interaction like Zr–O–C bond between the interface of ZrO2 and rGO.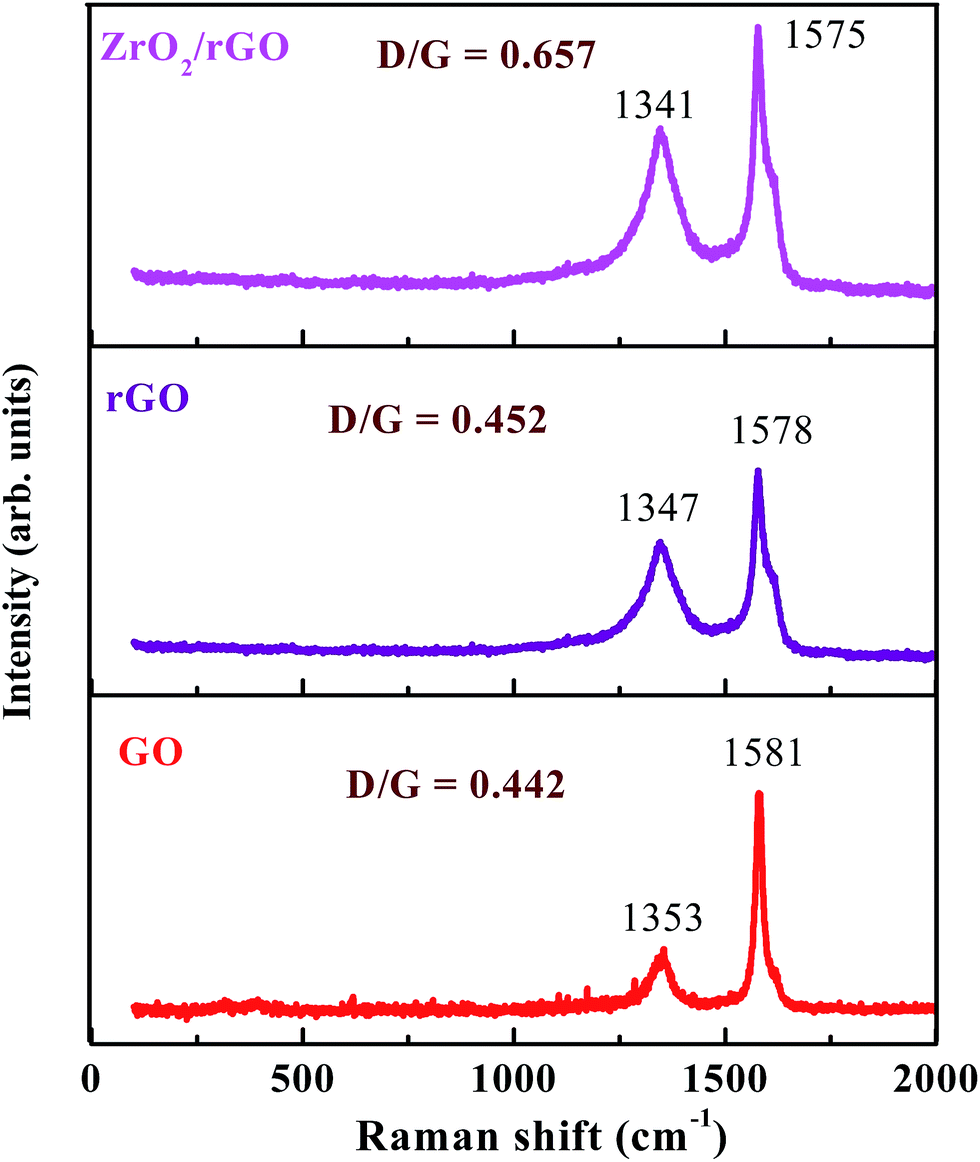 | ||
| Fig. 5 Raman spectra of GO, rGO and ZrO2/rGO NC. | ||
3.4 PXRD analysis
Fig. 6 shows the PXRD patterns of ZrO2, GO, rGO and ZrO2/rGO NC. The diffraction peaks of synthesized ZrO2 NM show the existence of both monoclinic and tetragonal mixed phase, in good agreement with the standard cards (JCPDS PDF no. 37-1484 and JCPDS PDF no. 49-1642 respectively). But the maximum crystallization takes place along the (−111) plane of monoclinic phase. The peak at ∼10° due to (001) plane for GO corresponds to the interlayer distance of 0.76 nm (Fig. 6). The fusing of many oxygenated functional groups between the carbon layers increases the interlayer distance. The synthesized GO was subjected to reduction using Cinnamon extract resulting in the shift of the diffraction from ∼10 to ∼25.6°. Thus rGO shows a major (002) peak at about 25.6°, which is ascribed to the reduction of GO sheets and restacking into an ordered crystalline structure.42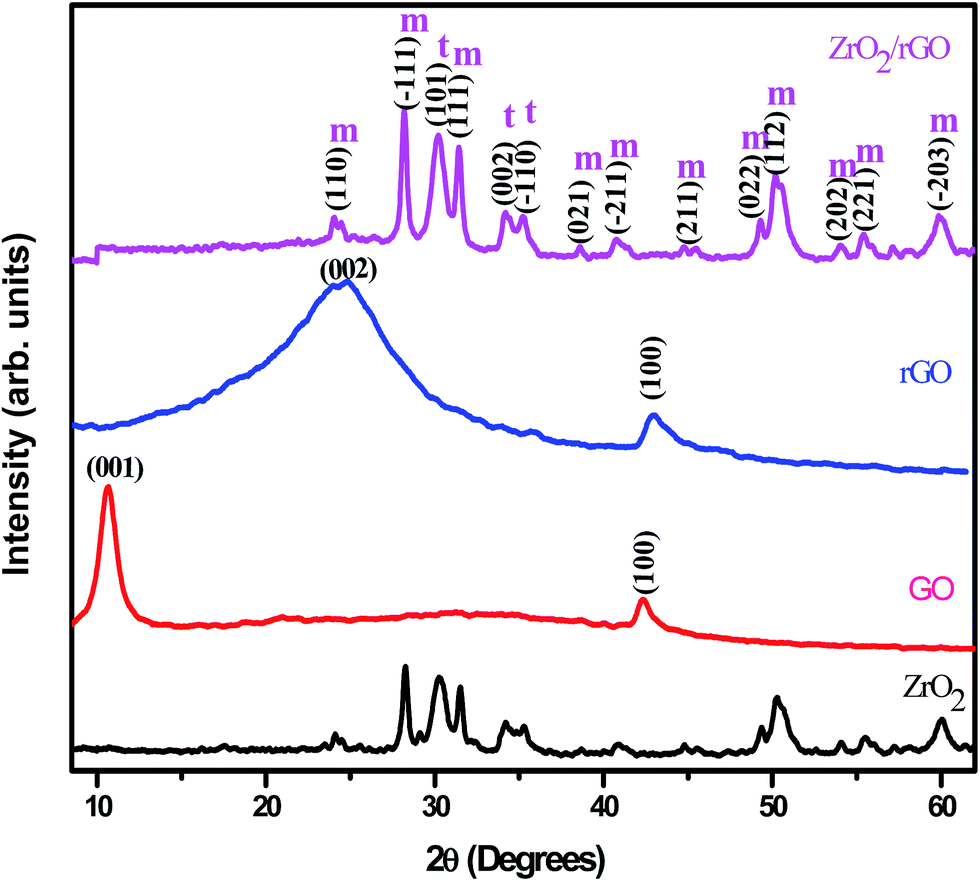 | ||
| Fig. 6 PXRD patterns of (a) ZrO2 NM (b) GO (c) rGO (d) ZrO2/rGO NC. | ||
While, the PXRD pattern of ZrO2/rGO NC is similar to diffraction peaks of host matrix except the variation in intensity. The (002) diffraction peak of graphene was not properly resolved, as the same peak at ∼25° overlapped with the (110) diffraction peak of the monoclinic ZrO2. The broadening of the diffraction peaks observed in ZrO2/rGO NC indicated the formation of ZrO2–rGO nanocrystallites. The primary size of the ZrO2 NM and ZrO2/rGO NC was estimated to be ∼14 and ∼15 nm from the Debye Scherrer's equation.
The other structural parameters such as dislocation density (δ), stacking fault were calculated using the following equation and mentioned in Table 1.
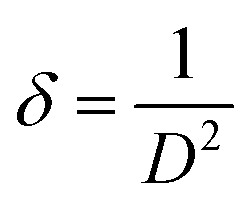 | (1) |
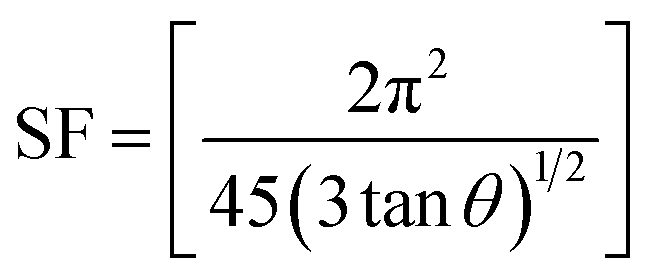 | (2) |
| Samples | FWHM (radians) | Crystallite size (nm) by Scherrer's method | Micro strain (ε) × 10−3 | SF | δ (1015 lines per m2) |
|---|---|---|---|---|---|
| ZrO2 | 0.653 | 14.7 | 2.68 | 0.438 | 4.5 |
| ZrO2/rGO | 0.627 | 15.2 | 2.56 | 0.460 | 4.3 |
3.5 FTIR studies
The presence of functional groups in the synthesized samples was analyzed by FTIR. The analysis was carried out by exposing the samples to infrared radiations in the range of 400 to 4000 cm−1. The FTIR spectra of ZrO2 NM and ZrO2/rGO NC are represented in Fig. 7. The peaks at 3443 cm−1 correspond to hydroxyl stretching and 1635 cm−1 was attributed to vibration modes from the moisture absorbed on the samples. As reported by several researchers, these two peaks are almost present in all the FTIR spectra (Kacurakova et al., 2000; Nejatzadeh-Barandozi & Enferadi, 2012). The broad absorption peaks at around 2930 cm−1 are due to the characteristic stretching vibration of CH2. The peak at 1387 cm−1 is due to the presence of OH deformations in the C–OH groups and at 1656 cm−1 is due to the presence of sp2 bond of graphite in GO. The band at 1060 cm−1 is assigned to C–O (epoxy) group which is consistent with XPS studies while the band at 1100 cm−1 is usually contributed to alkoxy group.43–45 | ||
| Fig. 7 FTIR spectra of ZrO2 NM and ZrO2/rGO NC. | ||
The absence of peaks at 1060, 1210 and 1656 cm−1 in the ZrO2/rGO confirms that GO has been reduced to rGO. The peak at 470 cm−1 can be assigned to the Zr–O vibration. These observations may signify that most oxygen functionalities in the GO were removed and ZrO2/rGO has been formed. The intensity of all moieties containing oxygen present in GO has been reduced suggesting efficient and successful reduction of GO to rGO.
3.6 Optical characterization
The difference between the valence band (EVB) and the conduction band (ECB) gives energy band gap (Eg), from which the electronic structure of ZrO2/rGO NC was characterized. In the photocatalytic mechanism, reactive oxygen species has been induced by metal oxide nanoparticles. The production of these reactive oxygen species such as ˙OH and O2˙ by ZrO2/rGO were related to the energy band gap and also to redox potential of different reactive species.46–48 So calculation of energy band gap for the synthesized NC is very important and it is determined by the Wood–Tauc relationship. As shown in Fig. 8, an extrapolation of the linear region of a plot of F(R)2 on the Y-axis versus energy (eV) on the X-axis gives the value of the Eg. The Eg value of ZrO2 NM and ZrO2/rGO NC was found to be 4.6 and 3.25 eV respectively. This Eg value for ZrO2/rGO was very close to the Eg value reported by Braj Raj Singh and his co-workers.49 The very less value of ZrO2/rGO NC compared to ZrO2 NM clearly depicts that the NC has good electron transport and hence can act as a very good photocatalyst with enhanced photocatalytic activity under sunlight irradiation.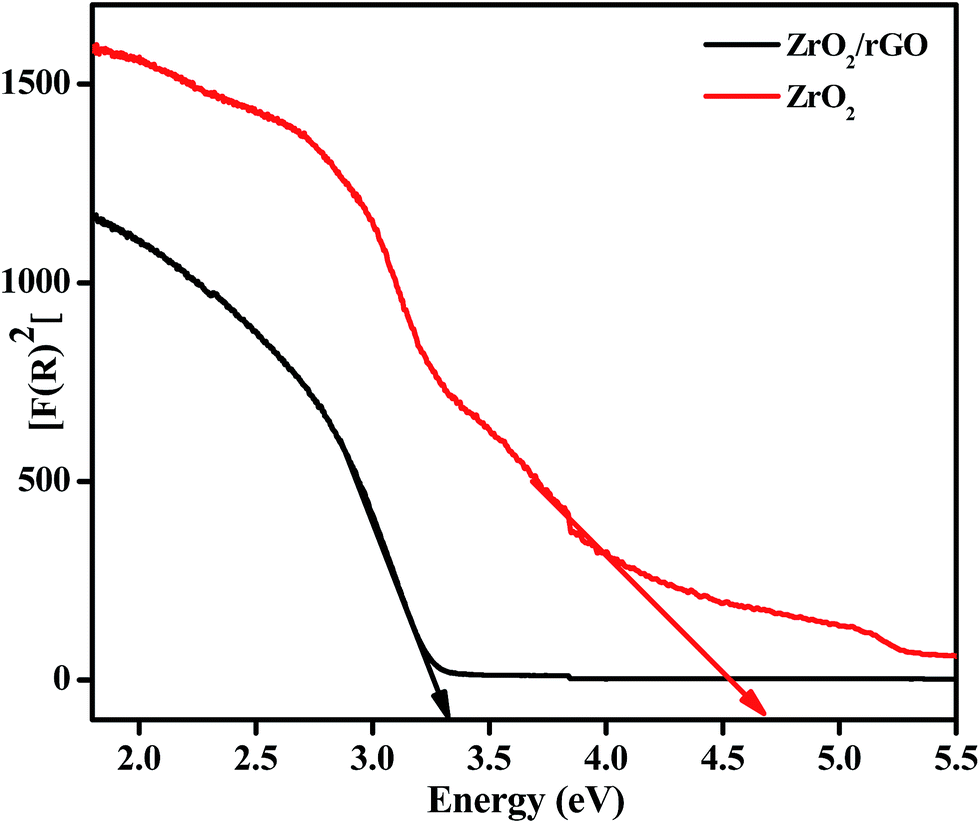 | ||
| Fig. 8 Plots of F(R)2 vs. energy (eV) for ZrO2 NM and ZrO2/rGO NC to find energy band gap. | ||
The decrease in the Eg value might be due to high crystallinity or less oxygen vacancies of ZrO2/rGO NC as confirmed by PL. The narrowing of band gap could be ascribed to chemical bonding between ZrO2 and specific sites of carbon during the preparation, which is similar to the case of carbon nanotube–TiO2 composite materials.50,51 The value of Eg is decreased to 3.25 eV after anchoring of ZrO2 onto the enhanced rGO sheet, signifying that the ZrO2/rGO can absorb the visible light irradiation efficiently. The narrowing in the band gap can be attributed to the formation of Zr–C and Zr–O–C bond between ZrO2 and rGO.52 Compared to ZrO2 NM, ZrO2/rGO NC has favorable energy band gap which agrees with the black appearance of the NC consistent with SEM analysis. These results suggest that ZrO2/rGO NC could exhibit an enhanced photocatalytic activity.
3.7 Photoluminescence studies
Fig. 9a shows the excitation spectrum of ZrO2 NM by monitoring the emission wavelength at 482 nm. The excitation spectrum shows two sharp emission peaks centred at 308 and 428 nm and a less intense peak at 234 nm. These peaks results from electron transfer from valence band to conduction band of the ZrO2 host. Due to trapping of electrons on the surface states, broad emission peaks arise at 482 nm. This may be also due to the interstitial oxygen lattice defects and oxygen vacancies of the ZrO2 to valence band of ZrO2 nanoparticles. The other low intense emission peak centered at 587 nm is owing to the band edge emission of ZrO2 NM. Compared to ZrO2 NM, the PL emission peak of ZrO2/rGO NC for both broad emission and low intense peak was found to be quenched. Moreover, the broad peak at 482 nm has been slightly shifted towards lower wavelength (467 nm) in ZrO2/rGO NC. The decrease in PL intensity of the composite stems from the band alignment at the interface of ZrO2 and rGO takes place as shown in inset of Fig. 9b.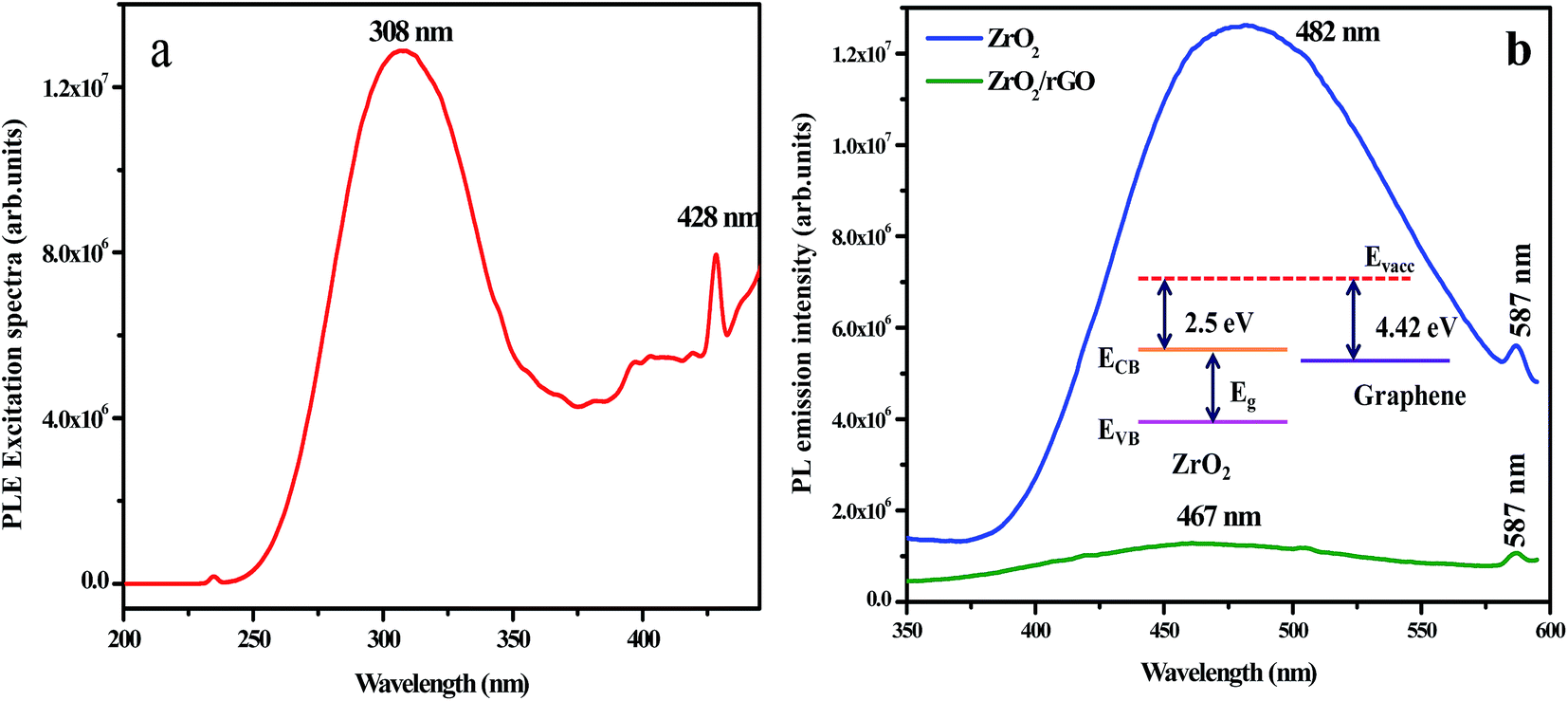 | ||
| Fig. 9 PL excitation spectrum of ZrO2 NM excited at 308 nm (b) PL emission spectrum of ZrO2 NM and ZrO2/rGO NC (inset: decrease in PL intensity of the composite stems from the band alignment at the interface of ZrO2 and rGO). | ||
Fig. 9b depicts PL emission spectra of ZrO2 NM and ZrO2/rGO NC excited at 308 nm. When ZrO2 NM are attached to the rGO, it is clear from the figure that the electron affinity for ZrO2 is 2.5 eV (ref. 53) and the Fermi level of the graphene is known to be 4.42 eV.54 This is due to the direct electron transfer from the conduction band of photo excited ZrO2 NM to the rGO which is energetically constructive, resulting in the formation of interfaces between the ZrO2 and rGO. This results in the efficient charge separation of the photo-excited charge carriers leading to inhibition of the recombination of photo-excited electron–hole pairs.55–58 Hence due to the interaction of ZrO2 surface with rGO sheets, PL quenching appears with decrease in intensity.57 Further, there is considerable PL quenching observed in ZrO2–rGO compared to ZrO2 NM, suggesting additional pathways for the disappearance of charge carriers. Obviously, ZrO2 NM shows higher PL intensity than ZrO2/rGO NC, indicating ZrO2 has the highest optical combination rate that deteriorated the photodegradation.
PL spectra mainly depend on the size of the particle, defects and impurities found in the materials. The PL properties of ZrO2 nanomaterial have been widely investigated by many researchers59–61 however, the role of particle size and the crystal phases of ZrO2 nanomaterial has not yet been analyzed in a systematical manner. Harrison et al.62 proved that the luminescent centers constituted by Zr4+ ions in an asymmetric site bounded by the oxygen ions contribute to the emission peak. The presence of defects and impurities in the systems could be the major reason for the luminescent properties of ZrO2 nanomaterials. The broad emission band arises due to the singly ionized oxygen vacancies defects in ZrO2 nanomaterials. The radiative recombination of a photo generated hole with an electron occupying the oxygen vacancy results in an emission band. The electron–hole pairs were obtained when the excitation energy exceeds to the band gap of ZrO2 nanomaterials. The electrons were trapped rapidly by the oxygen vacancies (VO) thereby producing F centers. The Zr4+ ions adjacent to the bulk oxygen vacancies had retained the electrons leading to the formation of Zr3+ ions.63,64 The holes recombine with the F centers forming the excited states of the emitter. These excited emitters undergo radiative transitions to the ground state. The recombination phenomenon could be summarized by the following reactions:65
| ZrO2 + hν → h+ + e− | (3) |
| Vo + e− → F center | (4) |
| F center + h+ → (Vo)* → Vo + hν | (5) |
The mechanism possibly involved in the emission phenomenon of the sample ZrO2 has been demonstrated with the help of schematic diagram as shown in Fig. 10. The major source of defects centers in ZrO2 sample was oxygen vacancies/interstitial, Zr vacancies/interstitials (Zr3+ interstitials).
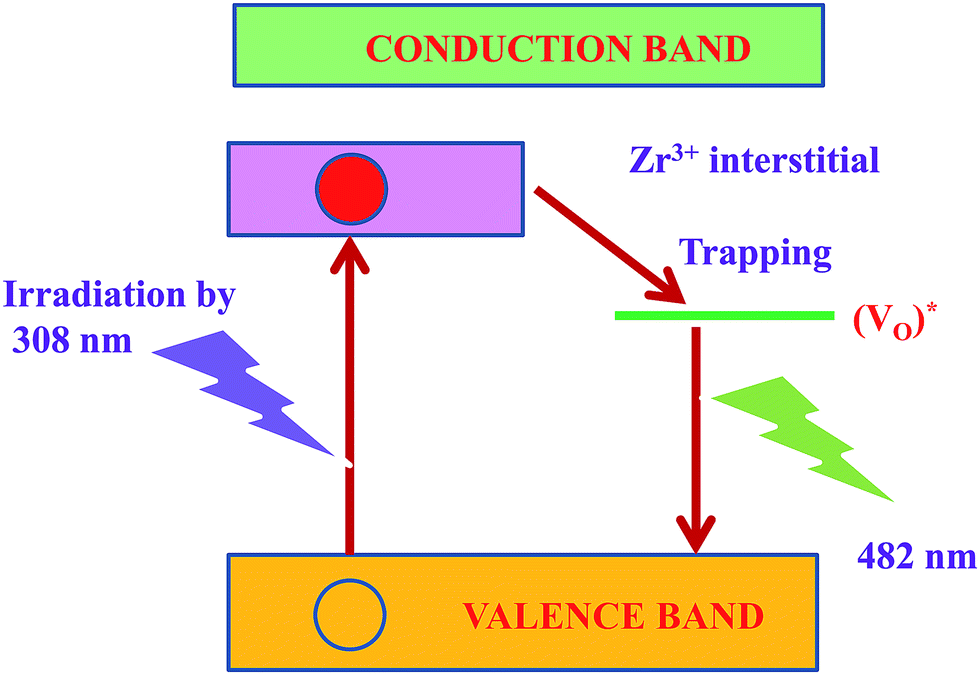 | ||
| Fig. 10 Energy level diagram for emission mechanism in ZrO2 nanophosphor. | ||
Moreover, the broad peak at 482 nm has been slightly shifted towards lower wavelength (467 nm) in ZrO2/rGO NC. The decrease in PL intensity of the composite stems from the band alignment at the interface of ZrO2 and rGO takes place as shown in inset Fig. 9b. This blue shifting of the emission band could have originated from the modification of stress/strain in the samples (Table 1) is mainly because of lattice distortions. The decrease in PL intensity is due to the decrease in density of oxygen vacancies. The increase in the particle size could be the possible reason for the decrease in the density of oxygen vacancies.
3.8 Electrochemical properties
The Cyclic Voltammetry (CV) data were detected within controlled potential region (−1.2 to 1.2 V) to set a collection of the reproducible current–potential dependencies in 0.1 M sodium nitrate (NaNO3) solution using three electrode cell arrangement. Fig. 11a shows the capacitance variations of the ZrO2/rGO electrodes at various scanning speed from 10 to 50 mV s−1. The capacitance increases with increase in the scanning speeds and predominantly became more obvious at the high scanning speed. For comparison purpose, CV plot of ZrO2 NM and ZrO2/rGO NC were run at 30 mV s−1 and depicted in Fig. 11b. It is seen from the figure that ZrO2 NM found to have a very broad semi-rectangular plot while ZrO2/rGO NC has less broad semi rectangular shape. This indicated that the capacitance value for ZrO2 NM is more compared to ZrO2/rGO.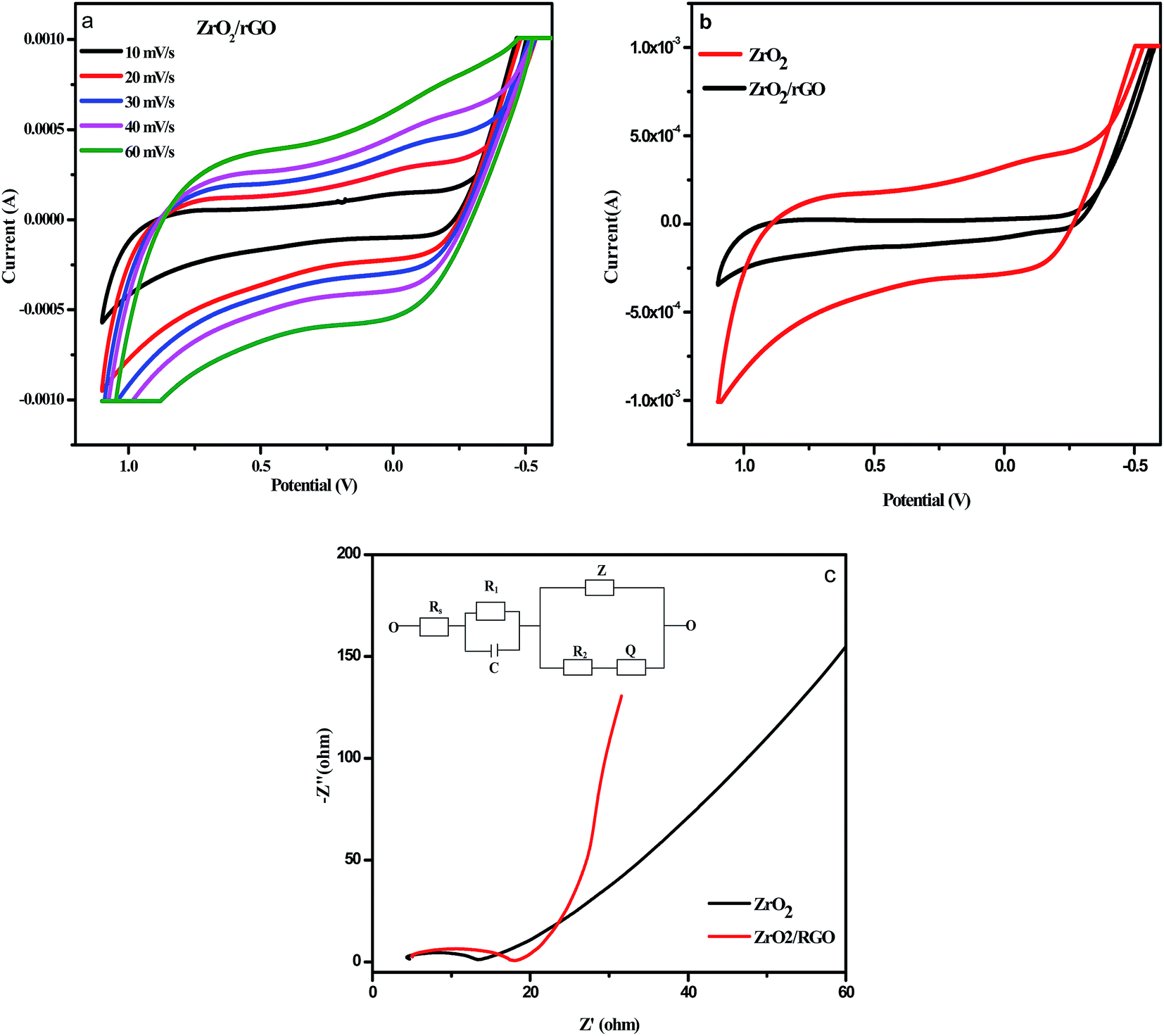 | ||
| Fig. 11 (a) Comparison of results of cyclic voltammetry (CV) test on ZrO2/rGO NC at different scan rates tested in 0.1 M NaNO3 (b) comparison of results of cyclic voltammetry (CV) test on ZrO2 NM and ZrO2/rGO NC at different scan rates tested in 0.1 M NaNO3 with scan rate of 30 mV s−1 (c) impedance spectroscopy plot for ZrO2 NMs and ZrO2/rGO NC (inset: circuit fitting program for ZrO2/rGO NC). | ||
Electrochemical impedance spectroscopy (EIS) were recorded by the application of an AC voltage with 10 mV amplitude in the frequency range of 105 to 10−2 Hz in 0.1 M sodium nitrate (NaNO3) using three electrode cell that was connected to an electrochemical station, where the prepared samples served as the working electrode, a platinum foil as the counter electrode and the silver as the reference electrode. As an extensively used electrochemical method, EIS is very effective to demonstrate the properties of the electron-transfer phenomenon across the ZrO2–electrolyte interfaces.66,67 Fig. 11c illustrates the Nyquist plots of the electrodes with X-axis as real part Z′ of impedance and Y-axis as the inverse of imaginary part −Z′′. This semicircular curve in high frequency region and inclined straight lines in low frequency region represents a typical AC impedance curve for super capacitor.
The diameter of the semicircle in high frequency region represents the charge transfer resistance (Rct) value in terms of ohms, which generally associated with the pore structure of electrode materials and faraday pseudo capacitive reaction.68 The less impedance arc radius in Nyquist plots indicates the faster electron transfer rate.69 The diameter for ZrO2 NM and ZrO2/rGO NC were found to be 14 and 8 Ω respectively. The diameter for ZrO2/rGO was found to be less indicating less charge transfer resistance and hence enhanced photocatalytic activity and decrease in photoluminescence intensity.
Inset Fig. 11c represents the circuit used for fitting the experimental data for impedance plot. Electrolyte resistance for 0.1 M NaNO3 solution is depicted as element Rs. Element R1 corresponds to electrolyte resistance present in porosity of the material, while element R2 corresponds to charge transfer resistance present in phase interface and is inversely proportional to surface area. The element C is the double layer capacitance that formed when ions from the solution move towards the electrode surface. Z is Warburg impedance that characterized by having identical real and imaginary contributions, resulting in a phase angle of 45°, which generates a line in the impedance plot. This double layer capacitance (C), Warburg impedance (Z) and Q constitutes the Constant Phase Element (CPE). Q is included in the constant phase element to avoid the lack of homogeneity in the electrode.
3.9 Photocatalytic experiment
For evaluating the optical properties of photocatalyst, the performance of UV-Vis light absorption is an important factor.70 UV-Vis absorption spectra of the ZrO2 NM and ZrO2/rGO NC are shown in Fig. 12a. In the present work, photocatalytic activity is studied for ZrO2 NM and ZrO2/rGO NC for degradation of Reactive Blue 4 (RB 4) dye under UV and sunlight irradiation for 120 min. The enhanced photocatalytic activity was observed in ZrO2/rGO NC than ZrO2 NM due to high absorption of sunlight and because of quenching in recombination of electron–hole pair in metal oxides with the existence of reduced graphene oxide. After achieving the complete adsorption of dye molecules on the composite material, the photocatalytic experiment was conducted.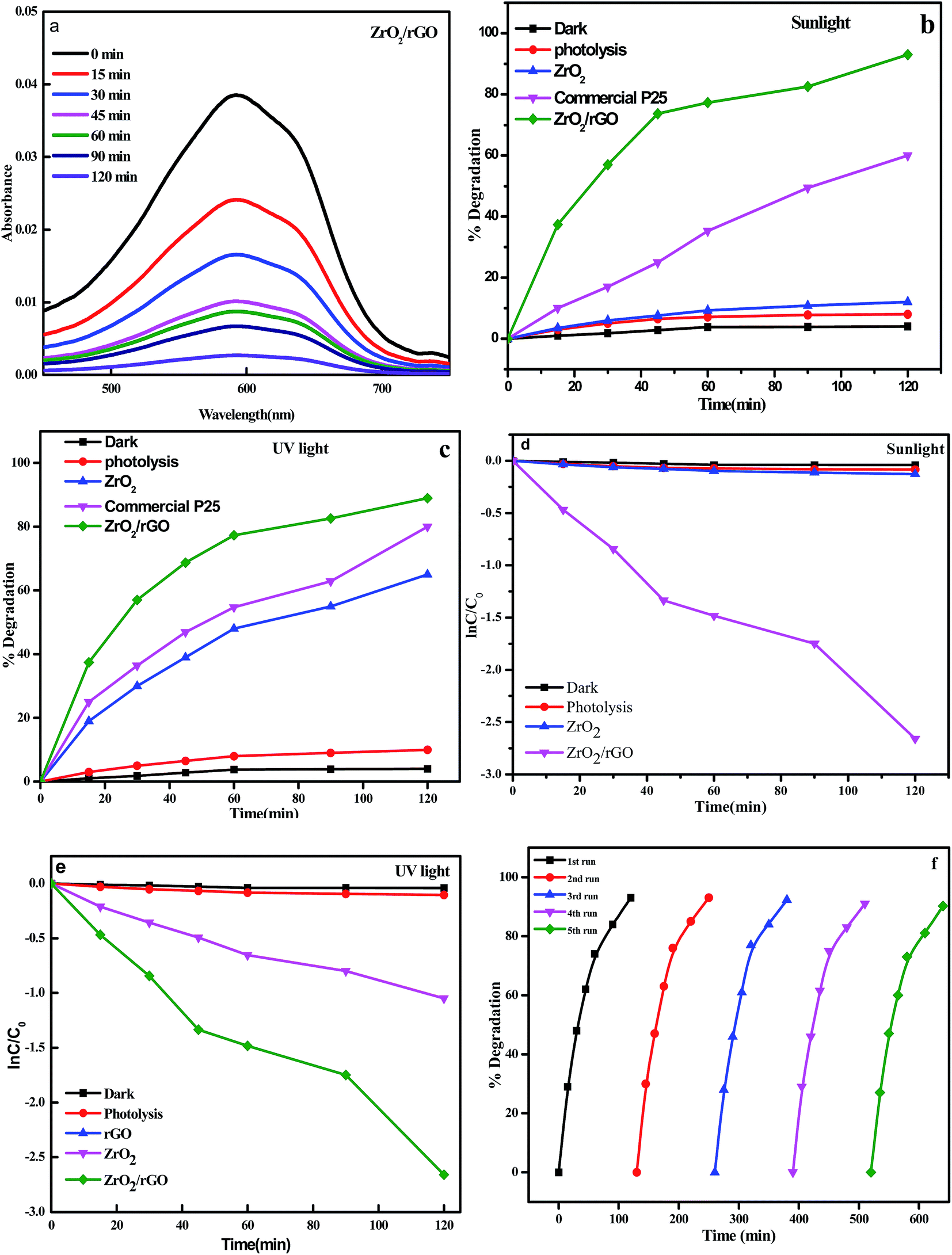 | ||
| Fig. 12 (a) Spectral absorbance changes with ZrO2/rGO photocatalyst; (b) plot of % decomposition of RB 4 under sunlight irradiation; (c) plot of % decomposition versus irradiation time for the degradation of RB 4 under UV irradiation; (d) plot of ln(C/C0) versus irradiation time for the degradation of RB 4 under sunlight irradiation; (e) plot of ln(C/C0) versus irradiation time for the degradation of RB 4 under UV light irradiation; (f) reusability of the ZrO2/rGO photocatalyst for five consecutive recycle runs. | ||
It was noted that there was very low decomposition of dye under dark condition. But under UV and sunlight irradiation, in the absence of the photocatalyst, RB 4 was very stable. After 120 min of UV and sunlight irradiation, the photolysis efficiency of the dye was negligible. The degradation efficiencies of RB 4 by ZrO2 NM and ZrO2/rGO NC under sunlight irradiation have been measured to be of 12% and 93% respectively as depicted in Fig. 12b. The photocatalytic decomposition of RB 4 under UV light irradiation for ZrO2 NM and ZrO2/rGO NC has been carried out for comparison purpose and found to be 65% and 89% (Fig. 12c). Commercial P25 was tested for the photocatalytic activity under the same experimental condition and the degradation percentage was found to be 80% for sunlight and 65% for UV light irradiation. However, it is seen that ZrO2/rGO NC has got higher photocatalytic activity compared to commercial P25. It was evident that the absorbent capacity of ZrO2/rGO NC towards RB 4 was stronger than that of ZrO2 NM, which was because rGO presented large surface area to bind dye molecules through the π–π conjugation with offset face-to-face direction. This degradation is due to the stepwise energy level structure present in the NC. As per the earlier reported data, the order for the conduction band offset is ZrO2 (−1.16) > rGO (−4.4), that shows conduction band for ZrO2 NM is less negative than compared to rGO.71 Hence under the sunlight irradiation, system absorbs sufficient amount of energy from irradiated light.
As a result of lower Fermi level and greater electrical conductivity of rGO, the excited electrons will migrate from the CBs of ZrO2 to rGO, leaving h+ behind in the ZrO2. The rGO acts as an e− reservoir with ZrO2 in this system by accepting the induced-photo electrons. This results in the disconnection of generated electron and hole pairs and increases life span of holes and photoelectrons, thus improving the activity of the photocatalyst dramatically. The obtained results were in consistent with photoluminescence and electrochemical impedance spectroscopic studies. Plots of lnC/C0 vs. time for degradation of RB 4 under sunlight and UV light irradiation is represented in Fig. 12d and e. The determined rate constant for ZrO2 NM and ZrO2/rGO NC under sunlight irradiation was found to be 0.0014 and 0.0203 min−1 and under UV light irradiation was found to be 0.0083 and 0.018 min−1 respectively. The obtained results portrayed excellent photocatalytic performance in the presence of sunlight.
Recycling experiments were performed in order to study the photostability of the photocatalysts under sunlight (Fig. 12f). The recycling experiment is carried out for ZrO2/rGO NC for five cycles by choosing the same sample and loading a fresh set of dye for degradation in each cyclic runs. The samples were washed for each cycle and dried before starting the next cycle. For four recycles, the percentage of degradation was found to be same and after the fourth cycle, the degradation was decreased which may be due to loss in the sample while washing. Hence it is confirmed that the ZrO2/rGO NC shows a very good recycling efficiency and it is also stable.
Fig. 13 depicts the time dependency TOC data for degradation of RB 4 under sunlight irradiation. The degradation of RB 4 was characterized by choosing TOC concentration as mineralization index. As the reaction time increases, the TOC removal concentration decreased gradually. Most of the TOC was mineralized at 120 min of irradiation of RB 4 indicating the maximum removal at 78% which played a vital role for practical applications in order to avoid secondary pollution.
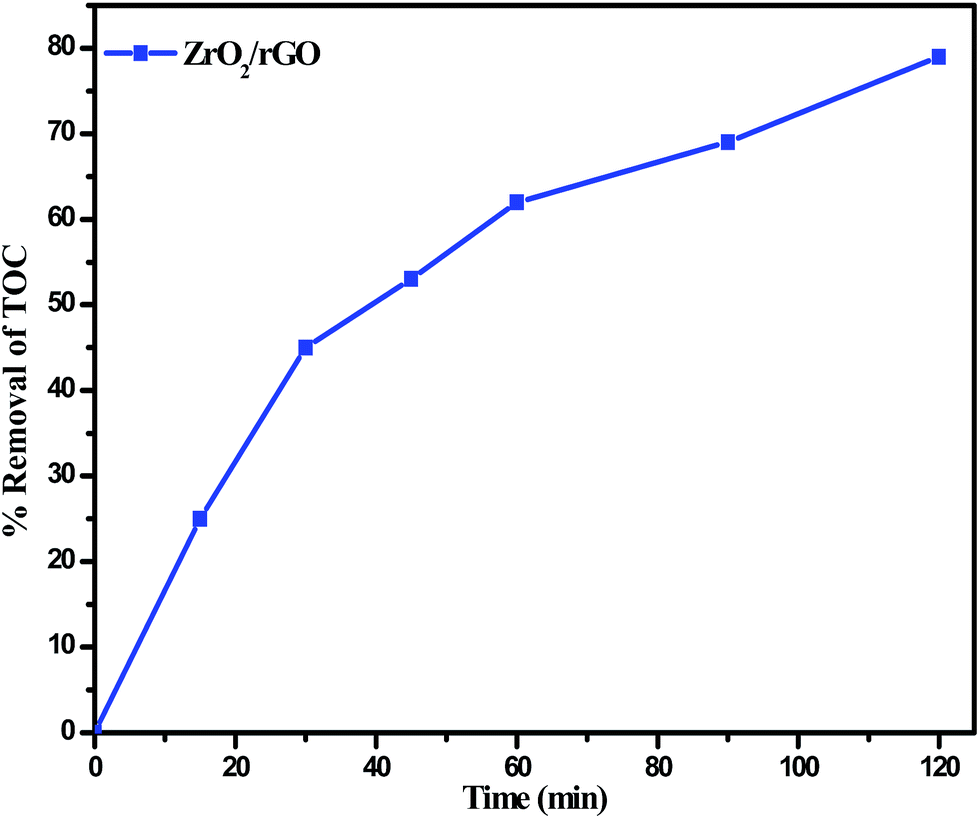 | ||
| Fig. 13 Percentage TOC removal for the degradation of RB 4 under sunlight in presence of ZrO2/rGO photo catalyst. | ||
When the NC is exposed to sunlight, surface adsorbed oxygen molecules and hydroxyl ions of ZrO2/rGO NC are the vital active species for the mechanism that reacts with excited electron and holes to convert it into ˙O2 and ˙OH radicals respectively (Fig. 14). These reactive oxygen species degrade the dye by breaking it into simple organics and furthermore into carbon dioxide and water. The main steps involved in the reaction process of photocatalytic degradation under sunlight irradiation is as follows:
| ZrO2/rGO + hν → e− + h+ | (6) |
| ZrO2 (h+) + OH− → ZrO2 + OH− | (7) |
| rGO (e−) + O2 → rGO + ˙O2− | (8) |
| Dye + ˙O2− + ˙OH → CO2 + H2O | (9) |
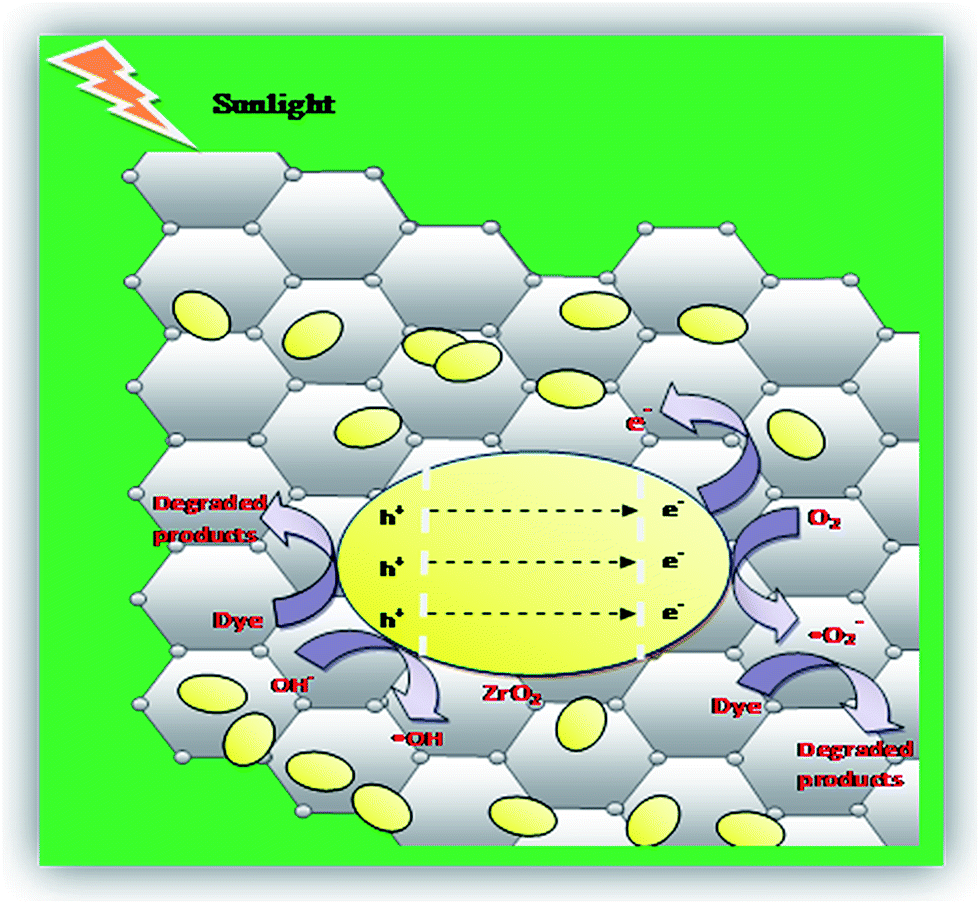 | ||
| Fig. 14 Mechanism for the photocatalytic degradation of RB 4 over ZrO2/rGO photocatalyst. | ||
4. Conclusions
The present work demonstrates an eco-friendly, green route based and simple approach for the rGO synthesis using Cinnamon extract. X-ray diffraction analysis ensured that the ZrO2 NM has a monoclinic and tetragonal mixed phase. The increase in intensity of ZrO2/rGO peak in PXRD confirms the incorporation of ZrO2 into rGO sheets and also clearly evidenced from SEM and TEM morphological analysis. Shift of peaks in the Raman spectrum and formation of C–C bond and C–O bond in deconvoluted peak for C 1s XPS spectrum confirms the presence of ZrO2 and RGO in ZrO2/rGO NC. The interaction between the surface of the ZrO2 NM and rGO sheets leads to PL quenching and therefore the intensity of PL gets decreased. The ZrO2/rGO NC serves as a very good photocatalyst for the decomposition of RB 4 under sunlight. The NC shows enhanced photocatalytic activity compared to commercial P25 with 93% degradation under sunlight. The photocatalyst was found to be stable even after five cyclic runs indicating its high reusability for several practical applications.Acknowledgements
The authors S. C. Sharma and K. S. Anantharaju thank VGST, Govt. of Karnataka, India for sanctioning the research project (VGST/CESEM/2015-16).References
- A. Umar, M. S. Akhtar, A. Al-Hajry, M. S. Al-Assiri, G. N. Dar and M. Saif Islam, Chem. Eng. J., 2015, 262, 588 CrossRef CAS.
- B. Liu, J. Hu and J. S. Foord, Electrochem. Solid-State Lett., 2011, 14, D22 CrossRef.
- A. T. Ezhil Vilian, S.-M. Chen, M. Ajmal Alib and F. M. A. Al-Hemaidb, RSC Adv., 2014, 4, 30358 RSC.
- K. Samson, M. S. liwa, R. P. Socha, K. Gora-Marek, D. Mucha, D. Rutkowska-Zbik, J. F. Paul, M. Ruggiero-Mikołajczyk, R. Grabowski and J. Słoczynski, ACS Catal., 2014, 4, 3730 CrossRef CAS.
- Y. L. Yu, P. Zhang, Y. J. Kuang, Y. H. Ding, J. H. Yao, J. J. Xu and Y. A. Cao, J. Phys. Chem. C, 2014, 118, 20982 CAS.
- C. Wu, X. Zhao, Y. Ren, Y. Yue, W. Hua, Y. Cao, Y. Tang and Z. Gao, J. Mol. Catal. A: Chem., 2005, 229, 233 CrossRef CAS.
- H. Zheng, K. Liu, H. Cao and X. Zhang, J. Phys. Chem. C, 2009, 113, 18259 CAS.
- M. Alvarez, T. Lopez, J. A. Odriozola, M. A. Centeno, M. I. Domínguez, M. Montes, P. Quintana, D. H. Aguilar and R. D. Gonzalez, Appl. Catal., B, 2007, 73, 34 CrossRef CAS.
- B. Neppolian, H. Choi, S. Sakthivel, V. M. B. Arabindoo, B. Neppolian and V. Murugesan, J. Hazard. Mater., 2002, 89, 303 CrossRef CAS PubMed.
- B. M. Reddy and A. Khan, Catal. Rev., 2007, 47, 257 Search PubMed.
- A. Burri, N. Jiang and S. E. Park, Catal. Sci. Technol., 2012, 2, 514 CAS.
- B. Neppolian, Q. Wang, H. Yamashita and H. Choi, Appl. Catal., A, 2007, 333, 264 CrossRef CAS.
- A. Kambur, G. S. Pozan and I. Boz, Appl. Catal., B, 2012, 115–116, 149 CrossRef CAS.
- L. L. Tan, S. P. Chai and A. R. Mohamed, ChemSusChem, 2012, 5(10), 1868 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41(2), 782 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Nanoscale, 2011, 3(9), 3670 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438(7065), 197 CrossRef CAS PubMed.
- G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2(7), 1487 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4(1), 380 CrossRef PubMed.
- J. Shen, B. Yan, M. Shi, H. Ma, N. Li and M. Ye, J. Mater. Chem., 2011, 21(10), 3415 RSC.
- K. Zhou, Y. Zhu, X. Yang, X. Jiang and C. Li, New J. Chem., 2011, 35(2), 353 RSC.
- W. Fan, Q. Lai, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2011, 115(21), 10694 CAS.
- X. Yang, J. Qin, Y. Li, R. Zhang and H. Tang, J. Hazard. Mater., 2013, 261, 342 CrossRef CAS PubMed.
- K. Gurushantha, K. S. Anantharaju, S. C. Sharma, H. P. Nagaswarupa, S. C. Prashantha, K. R. Vishnu Mahesh, L. Renuka, Y. S. Vidya and H. Nagabhushana, J. Alloys Compd., 2016, 685, 761 CrossRef CAS.
- L. Renuka, K. S. Anantharaju, S. C. Sharma, H. P. Nagaswarupa, S. C. Prashantha, H. Nagabhushana and Y. S. Vidya, J. Alloys Compd., 2016, 672, 622 CrossRef.
- P. Mishra, Y. P. Singh, H. P. Nagaswarupa, S. C. Sharma, Y. S. Vidya, S. C. Prashantha, H. Nagabhushana, K. S. Anantharaju, S. Sharma and L. Renuka, J. Alloys Compd., 2016, 685, 656 CrossRef CAS.
- P. Ranasinghe, S. Pigera, G. A. S. Premakumara, P. Galappaththy, G. R. Constantine and P. Katulanda, BMC Complementary Altern. Med., 2013, 13, 275 CrossRef PubMed.
- K. Sambaiah and K. Srinivasan, Nahrung, 1991, 35, 47 CrossRef CAS PubMed.
- G. Singh, S. Maurya, M. P. DeLampasona and C. A. Catala, Food Chem. Toxicol., 2007, 45, 1650 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Gurunathan, J. W. Han, V. Eppakayala and J. Kim, Colloids Surf., B, 2013, 102, 772 CrossRef CAS PubMed.
- D. Suresh, Udayabhanu, H. Nagabhushana and S. C. Sharma, Mat. Lett., 2015, 142, 4 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- S. Calderon, R. E. Galindo, N. Benito, C. Palico, A. Cavaleiro and S. Carvalho, J. Phys. D: Appl. Phys., 2013, 46, 325303 CrossRef.
- Y. Liu, RSC Adv., 2014, 4, 36040 RSC.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS PubMed.
- J. Shen, T. Li, Y. Long, M. Shi, N. Li and M. Ye, Carbon, 2012, 50, 2134–2140 CrossRef CAS.
- D. C. Luo, G. X. Zhang, J. F. Liu and X. M. Sun, J. Phys. Chem. C, 2011, 115, 11327–11335 CAS.
- H. Teymourian, A. Salimi, S. Firoozi, A. Korani and S. Soltanian, Electrochim. Acta, 2014, 143, 196 CrossRef CAS.
- H. L. Wang, J. T. Robinson, X. L. Li and H. J. Dai, J. Am. Chem. Soc., 2009, 131, 9910–9911 CrossRef CAS PubMed.
- P. Wanga, J. Wanga, X. Wanga, H. Yua, J. Yub, M. Lei and Y. Wang, Appl. Catal., B, 2013, 132–133, 452–459 CrossRef.
- S. Kumar, J. G. Sharma, S. Maji and B. D. Malhotra, Biosens. Bioelectron., 2016, 15, 497 Search PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806 CrossRef CAS PubMed.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS PubMed.
- X. Li, Q. Wang, Y. Zhao, W. Wu, J. C. Feng and H. Meng, J. Colloid Interface Sci., 2013, 411, 69 CrossRef CAS PubMed.
- M. Shoeb, B. R. Singh, J. A. Khan, W. Khan, B. N. Singh, H. B. Singh and A. H. Naqvi, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2013, 4, 035015 CrossRef.
- J. A. Khan, M. Qasim, B. R. Singh, S. Singh, M. Shoeb, W. Khan, D. Das and A. H. Naqvi, Spectrochim. Acta, Part A, 2013, 109, 313 CrossRef CAS PubMed.
- C. D. Vecitis, K. R. Zodrow, S. Kang and M. Elimelech, ACS Nano, 2010, 4, 5471 CrossRef CAS PubMed.
- B. Raj Singh, M. Shoeb, W. Khan and A. H. Naqvi, J. Alloys Compd., 2015, 651, 598 CrossRef.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233 CrossRef CAS.
- Y. Yu, J. C. Yu, C. Y. Chan, Y. K. Che, J. C. Zhao, L. Ding, W. K. Ge and P. K. Wong, Appl. Catal., B, 2005, 61, 1 CrossRef CAS.
- Y. L. Min, K. Zhang, W. Zhao, F. C. Zheng, Y. C. Chen and Y. G. Zhang, Chem. Eng. J., 2012, 193–194, 203 CrossRef CAS.
- W. Zheng, K. H. Bowen, J. Li, I. Dabkowska and M. Gutowski, J. Phys. Chem. A, 2005, 109, 11521 CrossRef CAS PubMed.
- Y.-C. Yeo, T.-J. King and C. Hu, J. Appl. Phys., 2002, 92, 7266 CrossRef CAS.
- Y. B. Shahar, F. Scotognella, N. Waiskopf, I. Kriegel, S. D. Conte, G. Cerullo and U. Banin, Small, 2015, 11, 462 CrossRef PubMed.
- W. T. Chen, T. T. Yang and Y. J. Hsu, Chem. Mater., 2008, 20, 7204 CrossRef CAS.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Adv. Funct. Mater., 2013, 23, 5444 CrossRef CAS.
- D. Zhang, B. Hua, D. Guan and Z. Luo, Catal. Commun., 2016, 76, 7 CrossRef CAS.
- C. Lin, C. Zhang and J. Lin, J. Phys. Chem. C, 2007, 111, 3300 CAS.
- A. Mondal, A. Zachariah, P. Nayak and B. B. Nayak, J. Am. Ceram. Soc., 2010, 93, 387 CrossRef CAS.
- H. Cao, X. Qiu, B. Luo, Y. Liang, Y. Zhang, R. Tan, M. Zhao and Q. Zhu, Adv. Funct. Mater., 2004, 14, 243 CrossRef CAS.
- D. E. Harrison, N. T. Melamed and E. C. Subbarao, J. Electrochem. Soc., 1963, 110, 23 CrossRef CAS.
- H. Liu, L. Feng, X. Zhang and Q. Xue, J. Phys. Chem., 1995, 99, 332 CrossRef CAS.
- S. Kumar and A. K. Ojha, J. Alloys Compd., 2015, 644, 654 CrossRef CAS.
- N. G. Petrik, D. P. Taylor and T. M. Orlando, J. Appl. Phys., 1999, 85, 6770 CrossRef CAS.
- A. Zaban, A. Meier and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 7985 CrossRef CAS.
- F. F. Santiago, G. G. Belmonte, J. Bisquert, A. Zaban and P. Salvador, J. Phys. Chem. B, 2002, 106, 334 CrossRef.
- M. Toupina, D. Belanger, I. R. Hill and D. Quinn, J. Power Sources, 2005, 140, 203 CrossRef.
- H. Liu, S. Cheng, M. Wu, H. Wu, J. Zhang, W. Li and C. Cao, J. Phys. Chem. A, 2000, 104, 7016 CrossRef CAS.
- L. S. Wang, M. W. Xiao, X. J. Huang and Y. D. Wu, J. Hazard. Mater., 2009, 161, 49 CrossRef CAS PubMed.
- F. T. Johra and W. G. Jung, Appl. Catal., A, 2015, 491, 52 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |