
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of the surface properties of resorcinol–formaldehyde resin/carbon nanocomposites and their carbonization products on the solid-phase extraction of explosives†
Viktor M. Bogatyrov a,
Mariia V. Galaburdaa,
Waldemar Tomaszewski*b and
Jadwiga Skubiszewska-Ziębac
a,
Mariia V. Galaburdaa,
Waldemar Tomaszewski*b and
Jadwiga Skubiszewska-Ziębac
aChuiko Institute of Surface Chemistry, 17 General Naumov Street, Kiev 03164, Ukraine
bFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: wtomaszewski@ch.pw.edu.pl
cFaculty of Chemistry, Maria Curie-Skłodowska University, Maria Curie Sklodowska Sq 3, 20-031 Lublin, Poland
First published on 20th January 2017
Abstract
Two polymer–nanocarbon composites, containing carbon nanotubes or exfoliated graphite, were prepared using resorcinol–formaldehyde resin. By carbonization of the composites, two additional carbon-based nanomaterials were obtained. The resulting materials were investigated with respect to their textural and morphological characteristics. The prepared composites were employed as sorbents for the separation of explosives (nitramines, nitrate esters and nitroaromatics) from aqueous samples by solid-phase extraction (SPE). The SPE results, i.e., the adsorption and desorption rates, are separately discussed. Both polymeric and carbonized composites (with nanotubes as a filler) gave high recovery rates, which reached 90%, and proved to be very effective for the extraction of explosives. The observed differences in the SPE performance of the carbonized composites were explained in terms of their porous structure, which depended on the route followed in the carbonization process. It was suggested that this process was predominantly influenced by the properties of the nanofillers employed, e.g., their high thermal conductivity, tendency to agglomerate and interactions with resin components.
1 Introduction
One of the most frequently used methods of sample preparation in the areas of science and industry as environmental, biological and forensic investigations for subsequent chromatographic analyses is solid-phase extraction.1 Since the time of the pioneering publication by Junk et al.,2 novel trapping materials and a variety of bed formats and instrumentation have been introduced. Particular emphasis has been placed on methods for development of novel adsorbents that feature increasingly more selective properties, of which carbon adsorbents provide outstanding examples.3,4 This is due to their superior properties in comparison with silica or polymeric adsorbents, i.e., a higher sample capacity and breakthrough volume, satisfactory recovery of polar and water-soluble analytes, and a wide pH stability range. For this reason, it seems natural that since the advent of a new generation of carbon materials, more specifically, carbon nanomaterials, some attempts have also been made to employ them in SPE, which has been discussed in a few reviews.5,6 In accordance with the reports just mentioned, the most frequently used carbon nanomaterials include single and multiwall nanotubes, graphene and graphene oxide. The major advantages of these adsorbents, in comparison with the most commonly used graphitized adsorbents of the GCB (graphitized carbon black) and PGC (porous graphitic carbon) types,3 are a large specific surface area to volume ratio, higher affinity for the analyzed substances and a wide spectrum of chemical modifications. However, from the viewpoint of the fundamental requirements related to adsorbents in SPE,7 nanocarbons exhibit certain drawbacks which make their direct use in “classic” syringe- or cartridge-type SPE columns difficult. Mention may also be made of their particle size, where one dimension (diameter/thickness) is on the nm scale, whereas the remaining dimensions are as large as several dozen μm. The significant polydispersity of these dimensions is also worthy of note.8 A bed formed from such materials may exhibit low permeability or even be entirely impermeable to the samples under analysis. In addition, small adsorbent particles may pass through the bottom filter that supports the bed and contaminate the extracts. Bed settling connected with dislocation of the smallest particles of the material most commonly results in channeling and considerable variability in recovery. Another disadvantage of carbon nanomaterials is the tendency of their particles to aggregate, which may restrict their sorptive capacity.8 A recommended solution of the abovementioned problems has proved to be the preparation of graphene-based beds or beds based on nanotubes immobilized on a solid support or introduced into a sample matrix. In the former case use was made, for instance, of stir-bar sorptive extraction,9 SPME,10 membranes,11 polymeric composites12 or inorganic composites.13 On the other hand, the dispersion of adsorbents into the sample matrix was accomplished by a number of methods, i.e., by direct addition of carbon adsorbents to solid samples, followed by extraction with solvents, in the method referred to as matrix solid-phase extraction (MSPD),14 by addition to liquid samples with subsequent mechanical shaking, which is known as dispersive SPE (dSPE),15 and in an analogous way using magnetic adsorbent particles, which is known as magnetic SPE (MSPE),16 in which the stirring, separation and extraction of the bed were made easier by the magnetic properties of the adsorbent particles.Despite the rather unavoidable drawbacks of conventional cylindrical beds formed from nanocarbons, studies can be found in which such a solution has continued to be used. By way of examples, a glass column packed with 20 mg graphene was employed for the extraction of chlorophenols from aqueous samples,17 and a column containing 30 mg carbon nanotubes for the determination of pesticides in virgin olive oil.18 Attention should be called to the fact that the classical SPE column provides one of the simplest and at the same time most easily repeatable procedures for the extraction of liquid samples: sample loading, bed drying and desorption are simple manual operations. Thus, in this work we have decided to employ this classical SPE format with the use of beds containing carbon nanotubes or exfoliated graphite, yet in an immobilized form in a polymer matrix. The composites that were prepared, which contained 2.5% nanofiller, were based on resorcinol–formaldehyde resin. The initial carbon–polymer composites were also subjected to further carbonization to obtain, as a result, two carbon/carbon nanotubes and carbon/exfoliated graphite composites. The adsorbents obtained were used in the extraction of explosives that belonged to a variety of groups, i.e., nitramines, nitric acid esters, and nitroaromatics, from aqueous samples. One of the reasons underlying the decision to launch studies of this kind is the fact that currently no reference can be found to the employment of nanocarbons in the SPE of explosives. Furthermore, the search for selective adsorbents for trapping explosives continues to be a significant analytical issue as, for example, imprinted polymers have still not seen more uses than the determination of certain nitroaromatics, e.g., trinitrotoluene.19 It is also a matter of common knowledge that the contamination of the natural environment with carcinogenic explosives in the areas affected by numerous military conflicts is currently growing at an immense rate.20
2 Experimental
2.1 Materials
The following materials were used: resorcinol (Farm, China), formaldehyde solution (37 wt% aqueous solution) and anhydrous sodium carbonate (Sigma-Aldrich), distilled water, multiwalled carbon nanotubes (CNT) (Development Department of Chuiko Institute of Surface Chemistry of NAS of Ukraine) and exfoliated graphite (EG) (Termografenit-E, Ukraine).2.2 Preparation of nanocomposites
The general synthesis scheme of the resin (RF) composites consisted of the following steps: the estimated amount of resorcinol was added to a solution of sodium carbonate (0.02 M) under stirring and, after the complete dissolution of all components, formaldehyde was added at room temperature. Then, either CNT or EG was added using mechanical stirring, with subsequent ultrasonic treatment in order to obtain a homogeneous suspension. This mixture was hermetically sealed, placed in a thermostatic oven, and treated at 85 °C for 20 h. A polymerization reaction took place with the formation of a dark brown solid polymer composite, which was then dried at 90 °C for 10 h, milled and sieved in order to obtain a 0.25–0.50 mm fraction. Two initial composites were obtained, i.e., RFR-43 (with CNT) and RFR-44 (EG). Then, the composites were placed in quartz tubes and set in a tubular furnace in a vertical position. The polymers were carbonized under an argon atmosphere from room temperature to 800 °C at a heating rate of 5 °C min−1 and were kept at the maximum temperature for 2 h. The inflow of argon and outflow of the volatile reaction products took place through the tube at the top of the furnace. This structure ensured that the atmosphere in the reaction zone contained not only argon but also the volatile products of pyrolysis. Two carbonized composites were obtained, i.e., RFC-43 (CNT) and RFC-44 (EG). The initial compositions of the reaction mixtures are shown in Table 1.| Sample | Filler content (wt%) | Components ratio | ||
|---|---|---|---|---|
| R/W (g g−1) | R/F (mol mol−1) | R/Na (mol mol−1) | ||
| a R/W, R/F and R/Na are the weight ratio of resorcinol to water and the mole ratios of resorcinol to formaldehyde and the Na2CO3 catalyst, respectively. The filler content is the content by weight in the composite vs. resorcinol. | ||||
| RFR-43 | CNT, 2.5 | 1/4.44 | 1/2 | 130/1 |
| RFR-44 | EG, 2.5 | 1/4.44 | 1/2 | 130/1 |
2.3 Characterization methods
The surface topography of the prepared samples was studied using Quanta 3D FEG (FEI, USA) and JEOL JSM 6700F (USA) high-resolution scanning electron microscopes.Raman spectra were recorded using an inVia Reflex Raman microscope (DMLM, Leica Research grade, Renishaw, UK). Excitation was achieved using a 514 nm laser.
Thermal analysis, including thermogravimetry (TG), differential thermogravimetry (DTG) and differential thermal analysis (DTA), was carried out using a Derivatograph C (MOM, Hungary). A ceramic crucible was used with a sample with a weight of 25–28 mg.
To analyze the textural characteristics, low-temperature (77.4 K) nitrogen adsorption–desorption isotherms were recorded using a Micromeritics ASAP 2405N adsorption analyzer.
The specific surface area (SBET) was calculated according to the standard BET method.21 The total pore volume Vp was determined from the adsorption of nitrogen at p/p0 = 0.98–0.99 (p and p0 denote the equilibrium and saturation pressures of nitrogen at 77.4 K, respectively). Non-local density functional theory (NLDFT) with models of slit/cylindrical pores in carbons was used to calculate the differential pore size distribution (PSD) (dV/dR).22 Furthermore, the surface areas related to micropores were calculated by the t-plot method.
2.4 Solid-phase extraction
SPE columns containing 100 mg of an adsorbent were prepared using 3 mL polyethylene tubes with Teflon frits (Baker). The columns were rinsed with 5 mL acetonitrile (ACN) and conditioned with 5 mL water. Then, 10 mL aqueous solutions of samples (containing 5 vol% ACN to ensure solubility of the explosives) were applied and the effluents were collected. Then, 5 mL ACN extracts collected in 10 mL flasks were made up with water. The initial concentration of the explosives in the samples was 0.005 mg mL−1. The following three groups of explosives were used: nitrate esters, i.e., diethyleneglycol dinitrate (DNTG), trinitroglycerin (NG), and pentaerythritol tetranitrate (PETN); nitroaromatics, i.e., 2,4-dinitrotoluene (DNT) and 2,4,6-trinitrotoluene (TNT); and nitramines, i.e., RDX (hexogen), HMX (octogen), CL-20 (2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaaza-isowurtzitane) and TEX (4,10-dinitro-2,6,8,12-tetraoxa-4,10-diaza-isowurtzitane). NG was obtained from SPP Pionki (Poland). DNTG and nitramines were synthesized at the Faculty of Chemistry, Warsaw University of Technology. PETN and nitroaromatics were obtained from different sources. The purity of the explosives was determined by chromatographic and thermal methods. Quantification by HPLC was carried out as previously reported.23 Using the concentrations of the explosives determined in the effluent Ceff and in the extract Cext, as well as the initial concentration in the samples Cref = 0.005 mg mL−1, the following parameters that described the SPE results were calculated: the adsorption rate Rads, desorption rate Rdes, and recovery rate Rrec. These parameters, expressed as percentage values, were calculated according to the following expressions:
 | (1) |
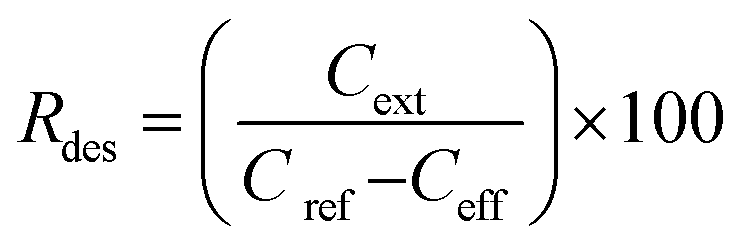 | (2) |
 | (3) |
3 Results and discussion
3.1 Characteristics of the composites
 | ||
Fig. 1 SEM images: RFR-43 (a) and RFC-43 (b), magnification ×80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; RFR-44 (c) and RFC-44 (d–f); magnifications: (c and d) ×10000, (e) ×12000, (f) ×35000. 000; RFR-44 (c) and RFC-44 (d–f); magnifications: (c and d) ×10000, (e) ×12000, (f) ×35000. | ||
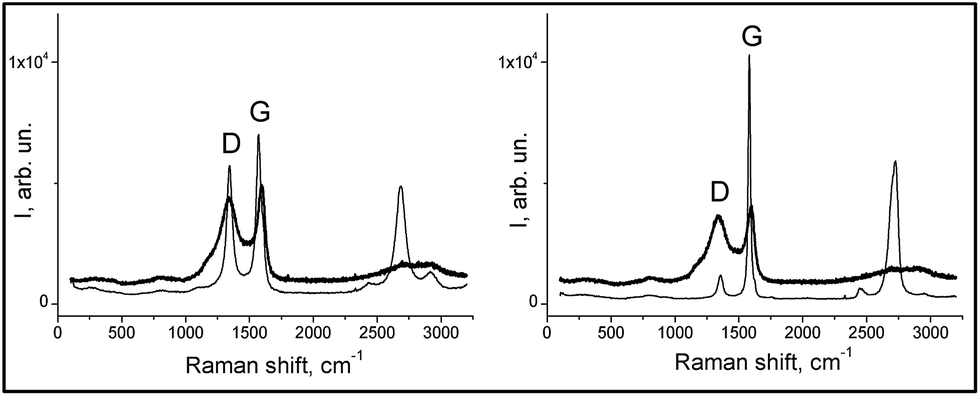 | ||
| Fig. 2 Raman spectra of the composites (thick lines): RFC-43 left-hand graph, RFC-44 right-hand graph, and fillers (thin lines): CNT on the left-hand side and EG on the right-hand side. | ||
| Sample | D1 (cm−1) | D2 (cm−1) | G1 (cm−1) | G2 (cm−1) | AD2/AG2 |
|---|---|---|---|---|---|
| CNT | 1183 | 1344 | — | 1574 | 0.82 |
| EG | — | 1356 | — | 1582 | 0.22 |
| RFC-43 | 1190 | 1340 | 1520 | 1595 | 2.66 |
| RFC-44 | 1182 | 1340 | 1524 | 1595 | 2.93 |
These results indicate that the surface of the RFC-43 sample, despite the presence of a filler with a lower degree of crystallinity, has a more ordered structure in comparison to RFC-44. These conclusions represent a good complement to the conclusions that can be drawn from the SEM observation: in fact, they even suggest a coincidence with the latter conclusions. At this point, it may be advisable to recall the conclusions from SEM observations, which indicate that the RFC-43 composite is formed from rather regularly spaced, even though randomly arranged, CNTs. In the case of RFC-44, a manifest disintegration of EG agglomerates has taken place, and its surface consists largely of planes formed from amorphous resinous carbonizate. For RFC-43, parts of the CNTs seem, on the basis of the changed appearance of their surface, to be covered with amorphous carbon. The higher degree of order of the RFC-43 surface may also result from the fact that CNT inclusions, on account of their exceptionally high thermal conductivity, may bring about, as in the case of nanodiamonds,32 localized graphitization.
| Sample | SBET (m2 g−1) | Smicro (m2 g−1) | Smeso (m2 g−1) | Vp (cm3 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Vmicro/Vp (%) |
|---|---|---|---|---|---|---|---|
| a SBET is the specific surface area; Smicro is the specific surface area of micropores determined by the t-plot method; Smeso is the specific surface area of mesopores estimated by subtracting Smicro from SBET; Vp is the total pore volume; Vmicro is the volume of micropores; and Vmeso is the volume of mesopores calculated by subtracting Vmicro from Vp. | |||||||
| RFR-43 | 378 | 254 | 124 | 0.296 | 0.136 | 0.160 | 46 |
| RFR-44 | 392 | 284 | 108 | 0.298 | 0.153 | 0.145 | 51 |
| RFC-43 | 515 | 420 | 95 | 0.299 | 0.211 | 0.088 | 70 |
| RFC-44 | 510 | 431 | 79 | 0.270 | 0.216 | 0.054 | 80 |
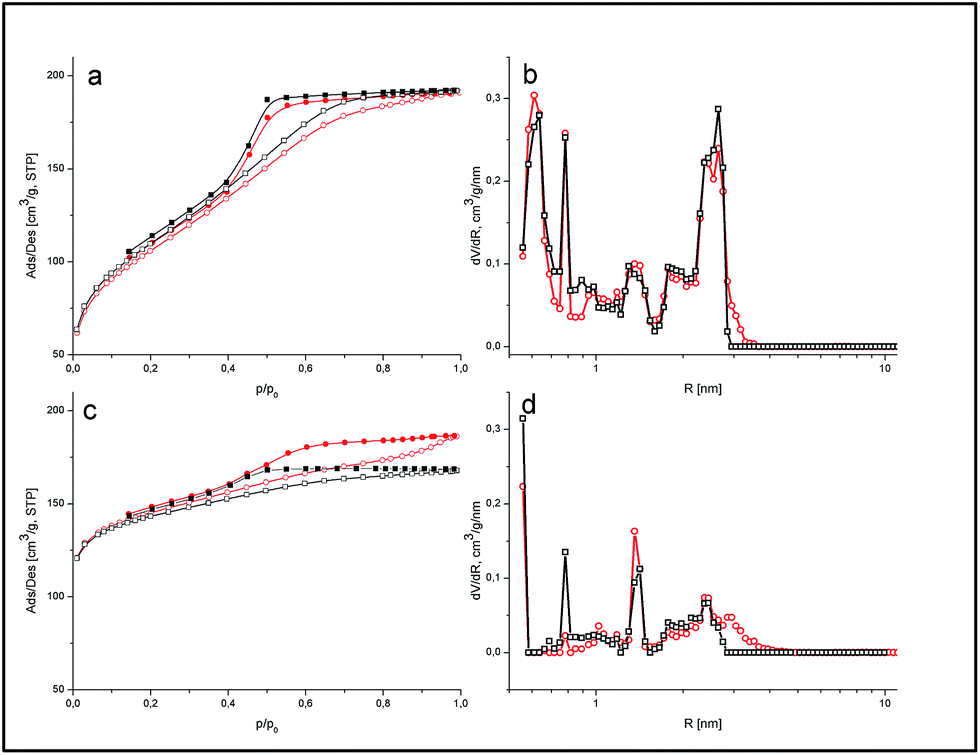 | ||
| Fig. 3 Nitrogen adsorption/desorption isotherms (a and c) and pore size distributions (b and d) of the polymeric (a and b) and carbonized (c and d) composites. For both isotherms and PSDs, circles represent RFR-43 and RFC-43, whereas squares represent RFR-44 and RFC-44. For isotherms open symbols represent adsorption, whereas full symbols represent desorption. | ||
 | ||
| Fig. 4 TG, DTG and DTA curves for RFC-43 (solid lines) and RFC-44 (dashed lines) composites. | ||
3.2 SPE results
To facilitate the comparison of the average Rads, Rdes and Rrec values for individual substances on two analogous adsorbents, i.e., the two RFR composites and two RFC carbonizates, a statistical test was applied, i.e., the analysis of variance or the F-Snedecor test for the statistical comparison of intra- and intergroup variation.34 For a level of significance of p = 0.05 and for a number of degrees of freedom of f = 1 = 2 − 1 (two adsorbents) and f = 4 = 6 − 2 (6 measurements, i.e., three for each adsorbent), the value of the F statistic was determined and was then compared with the critical value of F0.05,1,4, which was equal to 7.71 (Table ESI 1, ESI†). In the case when F < F0.05,1,4, it was assumed that the observed rates (adsorption and desorption) did not differ from one another; in the contrary case, it was assumed that there existed statistically significant differences between these results.SPE experiments were also performed with two additional adsorbents without nanofillers, namely, a resin and its carbonization product, which were obtained under the same conditions as the RFR and RFC composites. It was found that the retention of the studied explosives on such adsorbents was negligible, which means that the adsorption rates observed were lower than 5%. Taking into consideration the uncertainty in the SPE results, which reached ca. 4–6%, it was assumed that the polymeric or carbonaceous matrix had no significant impact on the SPE results.
 | ||
| Fig. 5 SPE results: adsorption rate (upper graphs), desorption rate (middle graphs) and recovery rate (lower graphs). The left-hand graphs represent resins, whereas the right-hand graphs represent carbonized samples. In each graph white bars indicate the results for CNT composites, grey bars the results for EG composites. In the individual graphs the pairs of bars successively represent RDX, HMX, TEX, CL-20, NG, DNTG, PETN, TNT, and DNT. | ||
It follows from the SEM images (Fig. 1a) that either the nanotube fragments or one or both tube ends are covered with a polymer layer. For the EG-containing composite, considerable fragments of the filler aggregate remain outside the polymer matrix. The surface oxygen functionalities in the case of both CNTs and EG are known to be primarily located (disregarding the presence of surface defects) at the edges of the graphene planes. In nanotubes, they are located at their ends, whereas in graphite, they are located at the edges of graphene surfaces. Taking into consideration the distribution of carbon fillers in the polymer matrix, it can be assumed that the number of surface oxygen groups available on the surface of the RFR-44 composite is significantly greater than that on RFR-43. This conclusion is in good agreement with the results of XPS measurements, which enabled the surface concentration of the major oxygen functionalities to be quantified (Table ESI 2, ESI†). The results demonstrate that the surface concentration of polar carbonyl groups was higher on the RFC-44 composite in comparison to the RFC-43 composite. Moreover, it was demonstrated in earlier studies35 that nitrate esters are weakly adsorbed on graphitized carbons, whereas they effectively become concentrated on oxidized carbons. Considering these two facts together, the weaker adsorption of nitrate esters on the RFR-43 composites in comparison with the RFR-44 composites appears understandable.
The desorption rates presented in the middle graph attained, with the exception of TEX, high values in the range of 90 to 100%. The low Rdes value for TEX in contrast to the other explosives was most probably due to the small size of this molecule.36 The Dmin value, which is equal to 0.49 nm, is comparable to the size of the smallest molecules of recognized explosives. The Dmax value, however, which barely equals 0.78 nm, is already the lowest among the remainder of these substances. Therefore, TEX molecules may become adsorbed in small pores into which the penetration of other explosives is rendered more difficult. A smaller pore size results in stronger interactions of TEX particles with the pore walls, which results in a decline in their desorption by acetonitrile.
On the basis of the F statistics, it may be assumed that the Rdes values for nitramines and nitroaromatics on the RFR-43 and RFR-44 adsorbents do not differ from one another. As regards the aliphatic nitrates (NG, DNTG, and PETN), the Rdes values differ from one another and are greater by 12 to 14% for the RFR-43 composite. The more effective desorption of these esters correlates with their weaker adsorption, as discussed above.
The final parameter that describes the efficiency of the SPE method is the recovery rate Rrec, which depends on both the adsorption and the desorption rate (see eqn (3)). It determines the effectiveness of the isolation of an analyte from a sample and thus the selectivity of the SPE method employed. The recovery rates on RFR-43 and RFR-44 (Fig. 5, lower left-hand graph) show that the highest values (over 90%) were for TNT and DNT. The Rrec values for the majority of esters and nitramines fell within the range of 60 to 80%. For both esters and nitramines, the lower Rrec values stemmed from their weaker adsorption in comparison with the aromatic explosives. Such recovery rates in excess of 50% are an excellent starting point for further optimization of the method.37
4 Conclusions
Four novel composites containing nanocarbons, i.e., carbon nanotubes and exfoliated graphite immobilized in polymeric and carbon matrices, were prepared. The initial two composites were obtained using resorcinol–formaldehyde resin and the two additional composites by their subsequent carbonization. Columns packed with the composites proved to be useful for SPE experiments. The beds were stable, the particles of adsorbents did not settle, and excellent permeability for the solvents used (water and acetonitrile) was observed. It must be remembered that neither the initial resin nor the applied nanocarbons can be used separately as adsorbents for solid-phase extraction. However, the derived composites, which contained only 2.5 wt% of the immobilized filler, proved to be a promising material for the solid-phase extraction of aliphatic, cyclic and aromatic explosive compounds from an aqueous matrix. Both the polymeric composites and the carbonized composite with nanotubes as a filler gave high recovery rates for explosives, which reached 90%. Although similar SPE results were obtained on the polymeric composites, the recoveries on the carbonized samples were quite different. This shows that the course of the carbonization process was strongly dependent on the presence of nanofillers. The differences in the adsorption and desorption rates for explosives were discussed on the basis of the surface properties (graphitization rate) and porous structures (micro- and mesoporosity) of the composites and the properties of the explosive molecules themselves. As an example, the polymeric composites are mesoporous materials, whereas the carbonized composites, to a large extent, are microporous. However, it should be emphasized that in the carbonized sample containing nanotubes mesoporosity is still demonstrated. The reasons for this are probably the homogeneous dispersion of nanotubes, their strong interactions with the resin matrix, and the stabilization of the porous structure. Therefore, the mesoporosity of the composite was partially preserved during carbonization. In addition, the carbonized sample containing exfoliated graphite is primarily microporous, and the adsorption of aliphatic explosives (nitrate esters and nitramines) in small pores is strongly hindered by water clusters.References
- B. Buszewski and M. Szultka, Crit. Rev. Anal. Chem., 2012, 42, 198–213 CrossRef CAS.
- G. A. Junk, J. J. Richard, M. D. Grieser, D. Witiak, J. L. Witiak, M. D. Arguello, R. Vick, H. J. Svec, J. S. Fritz and G. V. Calder, J. Chromatogr. A, 1974, 99, 745–762 CrossRef CAS PubMed.
- M. C. Hennion, J. Chromatogr. A, 2000, 885, 73–95 CrossRef CAS PubMed.
- E. Matisová and S. Skrabáková, J. Chromatogr. A, 1995, 707, 145–179 CrossRef.
- K. Pyrzyńska, Sep. Purif. Rev., 2008, 37, 372–389 CrossRef.
- N. Ye and P. Shi, Sep. Purif. Rev., 2015, 44, 183–198 CrossRef CAS.
- M. Henry, SPE technology – principles, and practical consequence, in Solid-phase extraction, principles, techniques, and applications, ed. N. J. K. Simpson, Marcel Dekker, New York, 2000, pp. 149–172 Search PubMed.
- Handbook of Carbon Nano Materials, Graphene-Fundamental Properties, ed. F. D'Souza and K. M. Kadish, World Scientific, Singapore, 2014, vol. 5, p. 159 Search PubMed.
- Z. Ayazi and P. Rafighi, Anal. Methods, 2015, 7, 3200–3210 RSC.
- Q. H. Wu, C. Feng, G. Y. Zhao, C. Wang and Z. Wang, J. Sep. Sci., 2012, 35, 193–199 CrossRef CAS PubMed.
- Z. Wang, Q. Han, J. Xia, L. Xia, M. Ding and J. Tang, J. Sep. Sci., 2013, 36, 1834–1842 CrossRef CAS PubMed.
- M. R. Nabida, R. Sedghia, A. Bagheria, M. Behbahania, M. Taghizadeha, H. A. Oskooieb and M. M. Heravib, J. Hazard. Mater., 2012, 203–204, 93–100 CrossRef PubMed.
- J. L. Zhang, R. M. Cheng, S. S. Tong, X. W. Gu, X. J. Quan, Y. L. Liu, Q. Jia and J. B. Jia, Talanta, 2011, 86, 114–120 CrossRef CAS PubMed.
- Q. Liu, J. Shi, J. Sun, T. Wang, L. Zeng, N. Zhu and G. Jiang, Anal. Chim. Acta, 2011, 708, 61–68 CrossRef CAS PubMed.
- R. Su, X. Xu, X. Wang, D. Li, X. Li, H. Zhang and A. Yu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3423–3428 CrossRef CAS PubMed.
- Y. N. Jiao, L. Ding, S. L. Fu, S. H. Zhu, H. Li and L. B. Wang, Anal. Methods, 2012, 4, 291–298 RSC.
- Q. Liu, J. B. Shi, L. X. Zeng, T. Wang, Y. Q. Cai and G. B. Jiang, J. Chromatogr. A, 2011, 1218, 197–204 CrossRef CAS PubMed.
- S. López-Feria, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2009, 1216, 7346–7350 CrossRef PubMed.
- W. J. Cheong, S. H. Yang and F. Ali, J. Sep. Sci., 2013, 36, 609–628 CrossRef CAS PubMed.
- http://www.thepeninsulaqatar.com/views/political-views/371557/syria-after-the-war-a-polluted-desert.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, 2. Auflage, Academic Press, London, 1982, pp. 41–109 Search PubMed.
- A. V. Neimark and P. I. Ravikovitch, Microporous Mesoporous Mater., 2001, 44/45, 697–707 CrossRef.
- W. Tomaszewski and V. M. Gun'ko, J. Sep. Sci., 2015, 38, 2488–2495 CrossRef CAS PubMed.
- M. C. Gutiérrez, F. Rubio and F. del Monte, Chem. Mater., 2010, 22, 2711–2719 CrossRef.
- T. L. Wu, T. S. Lo and W. S. Kuo, Polym. Compos., 2010, 31, 292–298 CAS.
- H. Peng and X. Sun, Chem. Phys. Lett., 2009, 471, 103–105 CrossRef CAS.
- A. Yasmin, J. J. Luo and I. M. Daniel, Compos. Sci. Technol., 2006, 66, 1179–1186 Search PubMed.
- H. G. Chae, Y. H. Choi, M. L. Minus and S. Kumar, Compos. Sci. Technol., 2009, 69, 406–413 CrossRef CAS.
- F. Tuinstra, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- C. S. Sharma, D. K. Upadhyay and A. Sharma, Ind. Eng. Chem. Res., 2009, 48, 8030–8036 CrossRef CAS.
- T. H. Ko, W. S. Kuo and Y. H. Chang, Polym. Compos., 2000, 21, 745–750 CrossRef CAS.
- E. Duffy, X. He, E. P. Nesterenko, D. Brabazon, A. Dey, S. Krishnamurthy, P. N. Nesterenko and B. Paull, RSC Adv., 2015, 5, 22906–22915 RSC.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CrossRef CAS.
- P. C. Meier and R. E. Zund, Statistical Methods in Analytical Chemistry, J.Wiley & Sons, New York, 2nd edn, 2000, pp. 69–72 Search PubMed.
- R. Leboda, J. Skubiszewska-Zięba, V. M. Gun'ko, W. Tomaszewski and B. J. Trznadel, J. Colloid Interface Sci., 2001, 239, 489–500 CrossRef CAS PubMed.
- W. Tomaszewski, V. M. Gun'ko, J. Skubiszewska-Zięba, B. Charmas and R. Leboda, Colloids Surf., A, 2015, 468, 76–86 CrossRef CAS.
- Optimizing solid phase extraction methods, in Forensic and Clinical Applications of Solid Phase Extraction, ed. M. J. Telepchak, T. F. August and G. Chaney, Humana Press Inc., Totowa, 2004, pp. 109–128 Search PubMed.
- V. M. Gun'ko, V. M. Bogatyrov, V. V. Turov, R. Leboda, J. Skubiszewska-Zięba and I. V. Urubkov, Appl. Surf. Sci., 2013, 283, 683–693 CrossRef.
- B. Kaur, Porous graphitic carbon, a new material for high performance liquid chromatography, Ph.D. thesis, University of Edinburgh, 1986.
Footnote |
| † Electronic supplementary information (ESI) available: Table SI 1. The results of the analysis of variance (F-Snedecor's test) for the adsorption and desorption rates. See DOI: 10.1039/c6ra25822k |
| This journal is © The Royal Society of Chemistry 2017 |