
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation and conversion of six temperature-dependent fluorescent ZnII-complexes containing two in situ formed N-rich heterocyclic ligands†
Qin Weia,
Mei-Juan Weia,
Yan-Jun Oua,
Ji-Yuan Zhangd,
Xia Huanga,
Yue-Peng Cai *abc and
Li-Ping Si*a
*abc and
Li-Ping Si*a
aSchool of Chemistry and Environment, South China Normal University, P. R. China. E-mail: caiyp@scnu.edu.cn
bGuangzhou Key Laboratory of Materials for Energy Conversion and Storage, Guangzhou 510006, P. R. China
cGuangdong Provincial Engineering Technology Research Center for Materials for Energy Conversion and Storage, Guangzhou 510080, P. R. China
dGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, P. R. China
First published on 20th January 2017
Abstract
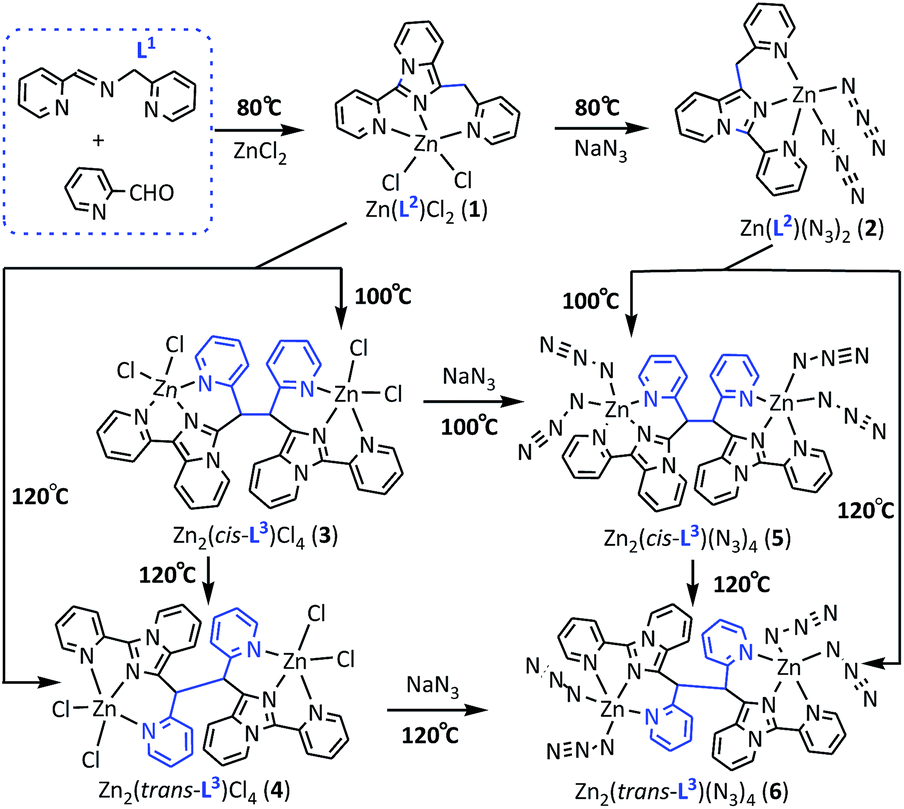
Six temperature-dependent Zn(II) complexes 1–6 based on the simple Schiff base L1 from condensation of equivalent 2-pyridine formaldehyde (2-Pfd) and 2-pyridylethylamine (2-Pea) were systematically studied for the first time. These six complexes are: two complexes of ZnL2X2 (1 and 2) involving in situ formed N-rich heterocyclic ligand L2 at 80 °C; two complexes of [Zn2(cis-L3)X4·S] (S = H2O for 3, S = 0 for 5) involving in situ formed azaheterocyclic ligand cis-L3 at 100 °C; and the rest two ones of [[Zn2(trans-L3)X4] (4 and 6) involving in situ formed azaheterocyclic ligand trans-L3 at 120 °C (where X = Cl (1, 3, 4) and N3 (2, 5, 6), L1 = N-(2-pyridylmethyl)-pyridine-2-carbaldimine, L2 = 1-pyridineimidazo-[1,5-a]pyridine, L3 = 1-(1,2-di(pyridin-2-yl)-2-(3-(pyridin-2-yl)H-imidazo-[1,5-a]-pyridin-1-yl)-ethyl)-3-(pyridin-2-yl)H-imidazo-[1,5-a]pyridine). Interestingly, three Cl-based complexes 1, 3, 4 under appropriate conditions can be irreversibly translated into the corresponding N3-based 2, 5, 6, respectively. The possible formation/conversion mechanism shows that the α-H activation in –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CH2– moiety of L1 coordinated to Zn2+ ion should be the original driving force for the intermolecular C–C/C–N coupling and ring formation reactions, meanwhile reaction temperature also plays a very important role during the formation/conversion of 1–6. Moreover, the results indicate that complexes 1–6 have good fluorescence properties as potential fluorescent materials.
N–CH2– moiety of L1 coordinated to Zn2+ ion should be the original driving force for the intermolecular C–C/C–N coupling and ring formation reactions, meanwhile reaction temperature also plays a very important role during the formation/conversion of 1–6. Moreover, the results indicate that complexes 1–6 have good fluorescence properties as potential fluorescent materials.
Introduction
The C–H bond in the α-position to an imino group –CHN–CH2– is activated after the imino nitrogen atom is coordinated to a metal center.1 Basic acceptors, such as pyridine, imidazole, etc., have the ability to deprotonate the imino carbon-bound hydrogen atom to form 1,3-dipole. According to the classsification of Huisgen, the 1,3-dipole of CN+–C− can be represented as XY+–Z− of allylic type.2 Schiff base N-(2-pyridylmethyl)-pyridine-2-carbaldimine (L1) with bipyridyl groups, condensed from equivalent 2-pyridine formaldehyde (2-Pfd) and 2-pyridylethylamine (2-Pea), may be led to various N-rich heterocyclic products under the guidance of the metal ion coordination induced effect, e.g. titanium(III), iron(II) and nickel(II) complexes containing 2,3,5,6-tetra-(pyridin-2-yl)piperazine/pyrrole3 as well as cadmium(II) complexes with 2,2′,2′′-(1-(pyridin-2-ylmethyl)imidazolidine-2,4,5-triyl)tripyridine4 are typical examples of such products.
Obviously, in situ formation of these N-rich heterocycles and their derivatives is very important in various fields: (i) azaheterocycles in organic syntheses can act as protective groups, since they are particularly easy to hydrolyze in acidic solutions and are stable in basic solutions. (ii) Some azaheterocycles can be used as intermediates in the biosynthesis of nucleotides, avoiding the shortcomings of traditional synthesis methods, and some of their metal complexes are found to be active as cytotoxic metallopharmaceuticals.5 (iii) They are important building blocks in biologically active compounds and carriers of pharmacologically active carbonyl compounds, and some of metal complexes are potential chemotherapeutic agents for DNA cleavage.6 (iv) Their transition-metal complexes have been also extensively studied for applications in OLEDs, luminescent materials,7 etc. Hence, it is very meaningful to explore the formation conditions and coordination behaviors of multi-substituted N-rich heterocycles by solvothermal in situ metal-induced reaction from simple Schiff base ligands. Herein we report the syntheses and their subsequent coordination chemistry of two imidazo[1,5-a]-pyridine-based azahetero-cycles involving 2-pyridine bis/tetra-substitutents(L2/L3), produced from zinc(II)-mediated inter-molecular ring-forming and C–C coupling reactions of L1 in the presence of another equimolar 2-Pfd under different reaction temperatures (Scheme 1), in which six new resulted ZnII coordination polymers, namely ZnL2Cl2 (1), ZnL2(N3)2 (2).
 | ||
| Scheme 1 The formation and conversion of six temperature-dependent Zn(II)-complexes containing in situ forming azaheterocycles L2/L3 from pyridine-type Schiff base L1 mediated by Zn2+ ion. | ||
Zn2(cis-L3)Cl4·H2O (3), Zn2(trans-L3)Cl4 (4), Zn2(cis-L3)(N3)4 (5) and Zn2(trans-L3)(N3)4 (6) (L1 = N-(2-pyridyl-methyl)pyridine-2-carbaldimine, L2 = 1-pyridine-imidazo-[1,5-a]pyridine, L3 = 1-(1,2-di-(pyridin-2-yl)-2-(3-(pyridin-2-yl)H-imidazo[1,5-a]-pyridin-1-yl)eth-yl)-3-(pyridin-2-yl)H-imidazo[1,5-a]pyridine), are involved. To the best of our knowledge, this is the first example that Zn2+-mediated C–C/C–N bond-forming strategy toward bis/tetra-substituted aza-heterocycles in situ from simple pyridine-type Schiff base.
Experimental section
Materials and methods
All reagent were grade obtained from commercial sources and used without further purification. Solvents were dried by the standard procedures. Elemental analyses for C, H, N were performed on a Perkin-Elmer 240C analytical instrument. IR spectra were recorded on a Nicolet FT-IR-170SX spectro-photometer in KBr pellets. Thermogravimetric analyses were performed on Perkin-Elmer TGA7 analyzer with a heating rate of 10 °C min−1 in flowing air atmosphere. The solid state luminescent spectra were recorded at room temperature on Hitachi F-2500 and Edinburgh-FLS-920 with a xenon arc lamp as the light source. In the measurements of emission and excitation spectra the pass width is 5.0 nm. X-ray powder diffraction patterns were measured on a Bruker D8 Advance diffractometer at 40 kV and 40 mA with a Cu target tube and a graphite monochromator. Nitrogen and hydrogen adsorption isotherms were taken on a Belsorp-Max surface area and pore size analyzer.Synthesis of L1
For complexes 1–6, they were prepared by a similar procedure except reaction temperature and auxiliary ligand NaN3. A mixture of N-(2-pyridylmethyl)pyridine-2-carbaldimine (L1) (0.0394 g, 0.2 mmol), ZnCl2 (0.0402 g, 0.2 mmol), 2-pyridine formaldehyde (0.022 g, 0.2 mmol), DMF (7 mL) and pyridine (3 mL) was sealed in a 15 mL Pyrex tube. The tube was heated for 3 days under autogenous pressure, and then slowly cooled the reaction solution to room temperature over 24 h and filtered to give pale yellow block single crystals for 1 at 80 °C, 3 at 100 °C and 4 at 120 °C. When NaN3 (0.130 g, 0.4 mmol) was added to the collected filtrate, three new complexes 2 (80 °C), 5 (100 °C) and 6 (120 °C) were obtained under the same reaction conditions, respectively. The crystals were collected by filtration, washed with Et2O (2 × 3 mL), and dried in air (Table 1).| a R1 = ∑‖Fo| − |Fc‖/∑|Fo|.b wR2 = {∑[w(Fo2 − Fc2)2]/∑(Fo2)2}1/2, where w = 1/(σ2(Fo2) + (aP)2 + bP), P = (Fo2 + 2Fc2)/3. | ||||||
|---|---|---|---|---|---|---|
| Complex | 1 | 2 | 3 | 4 | 5 | 6 |
| Empirical formula | C18H14Cl2N4Zn | C18H14N10Zn | C36H28Cl4N8OZn2 | C36H26Cl4N8Zn2 | C36H26N20Zn2 | C36H26N20Zn2 |
| Formula weight | 422.60 | 435.76 | 1246.63 | 843.19 | 869.51 | 869.51 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/n | C2/c | P21/c | P21/c | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 7.229(3) | 18.130(3) | 15.388(6) | 8.730(2) | 8.884(2) | 8.0218(8) |
| b (Å) | 14.023(6) | 18.615(3) | 12.271(5) | 13.047(4) | 19.730(5) | 10.0798(10) |
| c (Å) | 19.027(7) | 13.827(2) | 19.046(6) | 15.560(4) | 21.404(6) | 11.6154(12) |
| α (°) | 90 | 90 | 90 | 90 | 90 | 83.9840(10) |
| β (°) | 95.403(5) | 128.341(2) | 122.12(2) | 96.344(3) | 93.623(4) | 83.1740(10) |
| γ (°) | 90 | 90 | 90 | 90 | 90 | 76.6970(10) |
| V (Å3) | 1920.3(13) | 3660.1(10) | 3046.0(2) | 1761.5(8) | 3744.1(18) | 904.60(16) |
| Z | 4 | 8 | 2 | 2 | 4 | 1 |
| ρ(cald.) (mg m−3) | 1.462 | 1.582 | 1.359 | 1.590 | 1.543 | 1.596 |
| T (K) | 298(2) | 293(2) | 298(2) | 296(2) | 296(2) | 296(2) |
| μ (mm−1) | 1.564 | 1.371 | 0.688 | 1.705 | 1.340 | 1.386 |
| Rint | 0.0355 | 0.0395 | 0.0543 | 0.0298 | 0.0672 | 0.0168 |
| GOF | 1.027 | 1.021 | 1.040 | 1.052 | 1.050 | 1.032 |
| R1 [I > 2σ(I)]a | 0.0320 | 0.0455 | 0.0491 | 0.0395 | 0.0600 | 0.0483 |
| wR2 (all data)b | 0.0748 | 0.1137 | 0.1328 | 0.1007 | 0.1514 | 0.1387 |
Syntheses of complexes 1–6
Alternatively, two N3-based complexes 5, 6 can be also obtained via stirring reactions from NaN3 and the corresponding Cl-based complexes 1, 3 and 4 for 5 hours at 100 and 120 °C, respectively.
X-ray crystallographic studies
Complexes 1–6 were characterized by single crystal X-ray diffraction. Suitable single crystals were mounted on a glass fiber and the intensity data were collected on a Bruker APEX II diffractometer at 298 K using graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å). Absorption corrections were applied using the multi-scan program SADABS.8 Structural solutions and full-matrix least-squares refinements based on F2 were performed with the SHELXS-97 (ref. 9) and SHELXL-97 (ref. 10) program packages, respectively. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms on organic ligands were generated by the riding mode (C–H 0.96 Å). For 1 and 3, the SQUEEZE option in PLATON11 was used to remove the disordered solvent water molecules, and the actual water molecules in the unit cell are determined by elemental and thermo-gravimetric analyses (EA and TGA). A summary of the parameters for the data collection and refinements for nine complexes are given in Table S1.† Selected bond lengths and angles for complexes 1–6 are listed in Table S2.† Hydrogen bond lengths and angles for complex 3 are given in Table S3.† CCDC reference numbers for 1–6 are CCDC 1032239, 1508822, 1032241, 1508812, 1032243 and 1032244, respectively.Results and discussion
Syntheses and general characterization of complexes
Pale-yellow crystals of 1–6 (ca. 67–75% yield) were obtained from the in situ solvothermal treatment of N-(2-pyridylmethyl)-pyridine-2-carbaldimine (L1) with ZnCl2/ZnCl2 + NaN3 in the mixed solvents of DMF and pyridine (v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 80–120 °C for three days (see Scheme 1). The component diversification of the resulting discrete ZnII–L1–3 complexes 1–6 can be tuned simply by changing reaction temperature, in which in situ forming L2/L3 are the results of intermolecular C–C/C–N coupling and ring-forming reactions deriving from α-H activation at ortho-position of imine –CHN–CH2– moiety reduced by Zn(II) coordination (possible in situ forming mechanism in Scheme S1†). Elemental analysis and PXRD (Fig. S1 in the ESI†) confirmed the phase purity of the bulk materials. Compounds 1–6 are air-stable and insoluble in water and most organic solvents. Meanwhile, stirring reactions of NaN3 and the Cl-based complexes 1, 3 and 4 for 5 hours at 100 °C and 120 °C can irreversibly convert into corresponding N3-based complexes 5 and 6 in high yield, respectively. Or alternatively the direct reaction of L1 with ZnCl2 + NaN3 like the Cl-based complexes 1, 3 and 4 can also give the corresponding N3-based complexes 2, 5 and 6. However, they are not only low yield, but also difficult to isolate and purify. This probably stems from the diverse coordination modes of azide anion (such as η1-, μ(1,1)- and μ(1,3)-modes, etc.), especially under high temperature conditions. In contrast, azide substitution reactions based on Cl-based zinc complexes, due to a single final product, have obvious advantages.
:1) at 80–120 °C for three days (see Scheme 1). The component diversification of the resulting discrete ZnII–L1–3 complexes 1–6 can be tuned simply by changing reaction temperature, in which in situ forming L2/L3 are the results of intermolecular C–C/C–N coupling and ring-forming reactions deriving from α-H activation at ortho-position of imine –CHN–CH2– moiety reduced by Zn(II) coordination (possible in situ forming mechanism in Scheme S1†). Elemental analysis and PXRD (Fig. S1 in the ESI†) confirmed the phase purity of the bulk materials. Compounds 1–6 are air-stable and insoluble in water and most organic solvents. Meanwhile, stirring reactions of NaN3 and the Cl-based complexes 1, 3 and 4 for 5 hours at 100 °C and 120 °C can irreversibly convert into corresponding N3-based complexes 5 and 6 in high yield, respectively. Or alternatively the direct reaction of L1 with ZnCl2 + NaN3 like the Cl-based complexes 1, 3 and 4 can also give the corresponding N3-based complexes 2, 5 and 6. However, they are not only low yield, but also difficult to isolate and purify. This probably stems from the diverse coordination modes of azide anion (such as η1-, μ(1,1)- and μ(1,3)-modes, etc.), especially under high temperature conditions. In contrast, azide substitution reactions based on Cl-based zinc complexes, due to a single final product, have obvious advantages.
In the IR spectra of six compounds 1–6, the strong bands in the region of 3443–3567 cm−1 or so are due to a hydrogen bonding C–H⋯Cl(N) interactions. The strong CN bands occurring in the range of 1597–1635 cm−1 for these complexes are shifted considerably toward lower frequencies compared to that of the free Schiff base ligand L1 (1646 cm−1), showing that the azomethine nitrogen atom is coordinated to the metal.12 The weak bands in the region of 462–467 cm−1 for the six complexes can be assigned to ν(M–N/Cl),12 and provides further evidence for the coordination was through the terminal nitrogen/chlorine atoms. The position of the 2061–2063 cm−1 (νas-N3) falls within the absorption range of terminal azides in transition metal complexes,13 and the appearance of the νs-N3 band at 1341–1342 cm−1 indicates the asymmetric nature of the azide groups in three complexes 2, 5, 6.
Thermal stability of six complexes was investigated by the TGA technique (see Fig. S2†). The TG curve for 3 shows two steps of weight loss. The weight loss of the uncoordinated water molecules is observed from 83 to 112 °C for 3 (calc. 2.09% and exp. 1.96%). On further heating, the complexes start to be decomposed at 312 °C for 3. For the remaining five complexes 1, 2, 4, 5 and 6, their TGA curves are also similar and present one step of weight loss, namely decomposition of the complexes are observed at 205 °C for 1, 321 °C for 2, 307 °C for 4, 341 °C for 5 and 343 °C for 6, respectively. Although all six complexes are zero dimensional, their thermal stability is obviously different, probably due to the different dimensions of their supramolecular structures, in which the complex 3 has low dimensional (2D) supramolecular structure and shows the lowest thermal stability (see below for details).
Structural analysis and discussion
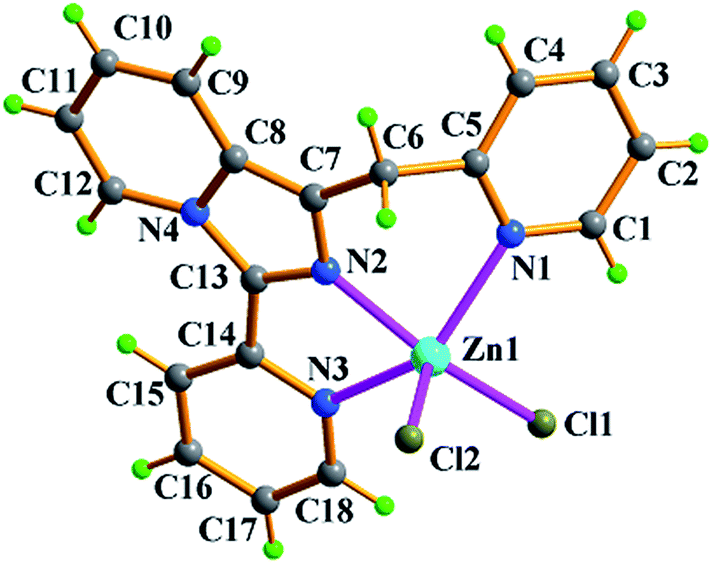 | ||
| Fig. 1 Molecular structure of compound ZnL2Cl2 (1) with atomic labels. | ||
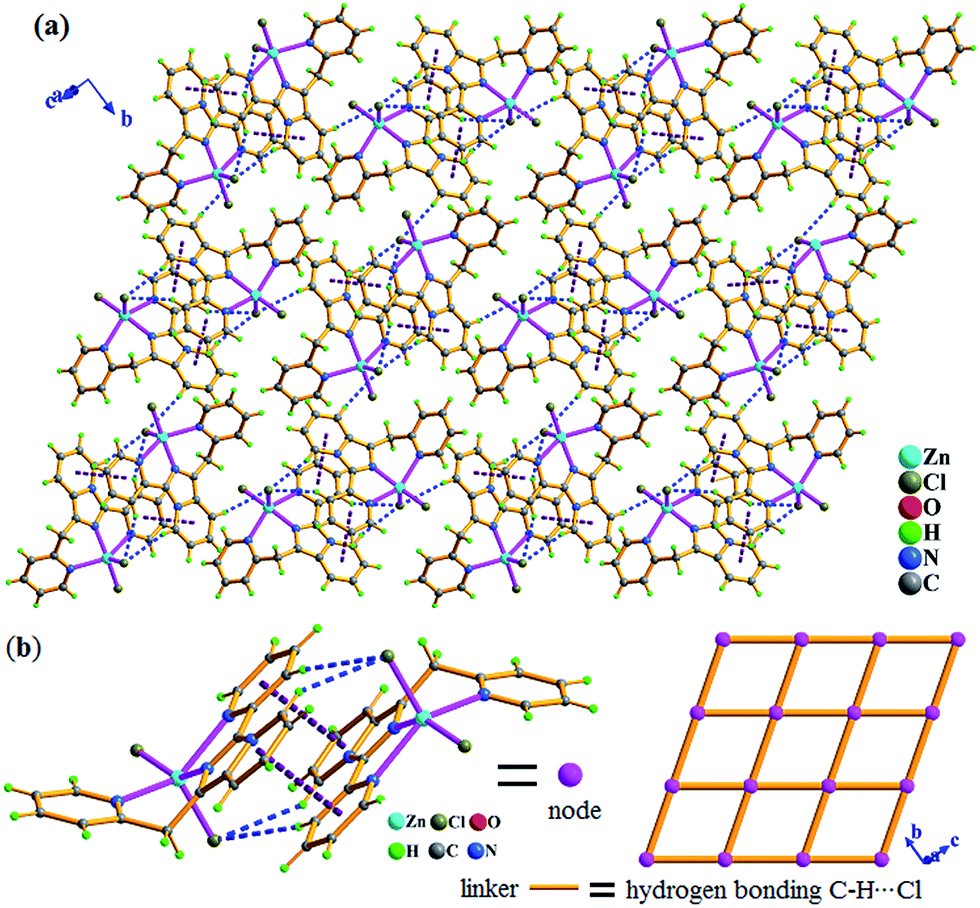 | ||
| Fig. 2 In 1, (a) 2-D supramolecular layer assembled by intermolecular hydrogen bonding C–H⋯Cl (blue color) and π⋯π packing (violet color) interactions in bc plane. (b) 44 topological supramolecular layer, in which the dimmer unit (ZnL2Cl2)2 as node constructed by intermolecular C–H⋯Cl and π⋯π packing interactions, interunit hydrogen bond C–H⋯Cl as linker. | ||
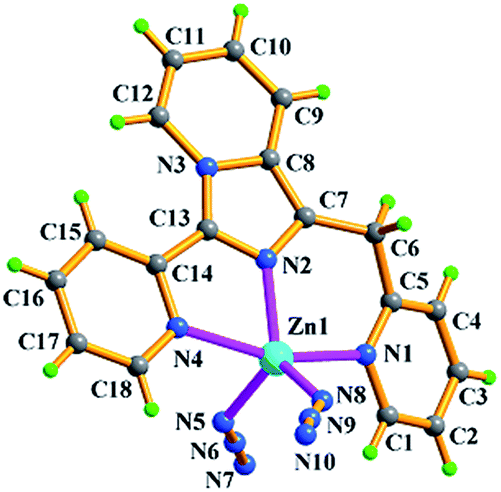 | ||
| Fig. 3 Molecular structure of ZnL2(N3)2 (2) with atomic labels. | ||
Zn2+ ion, one in situ formed L2 ligand, and two terminal monodentate η1-coordinated N3− ions. The coordination geometry around Zn1 is distorted trigonal bipyrimidal with five nitrogen N atoms (N1, N2, N4, N5, N8) from one L2 ligand and two azide anions, in which N2, N5, N8 atoms are the triangle plane, and N1 and N4 atoms at the apical positions. The ligand L2 in 2 also presents the same coordination mode: μ1-η1:η1:η1, namely, which as a terminal tridentate ligand using the three nitrogen atoms (N1, N2, N4) on the same side of L2 coordinates to one Zn2+ ion to form a 0-D discrete structure. The Zn–N bond distances are 1.950(4)–2.283(3) Å. And intermolecular hydrogen bonding C–H⋯Cl and π⋯π stacking interactions (Table S3†) resulting in 3-D supramolecular network with 36·413·56·63 topology further stabilize crystal structure (Fig. 4).
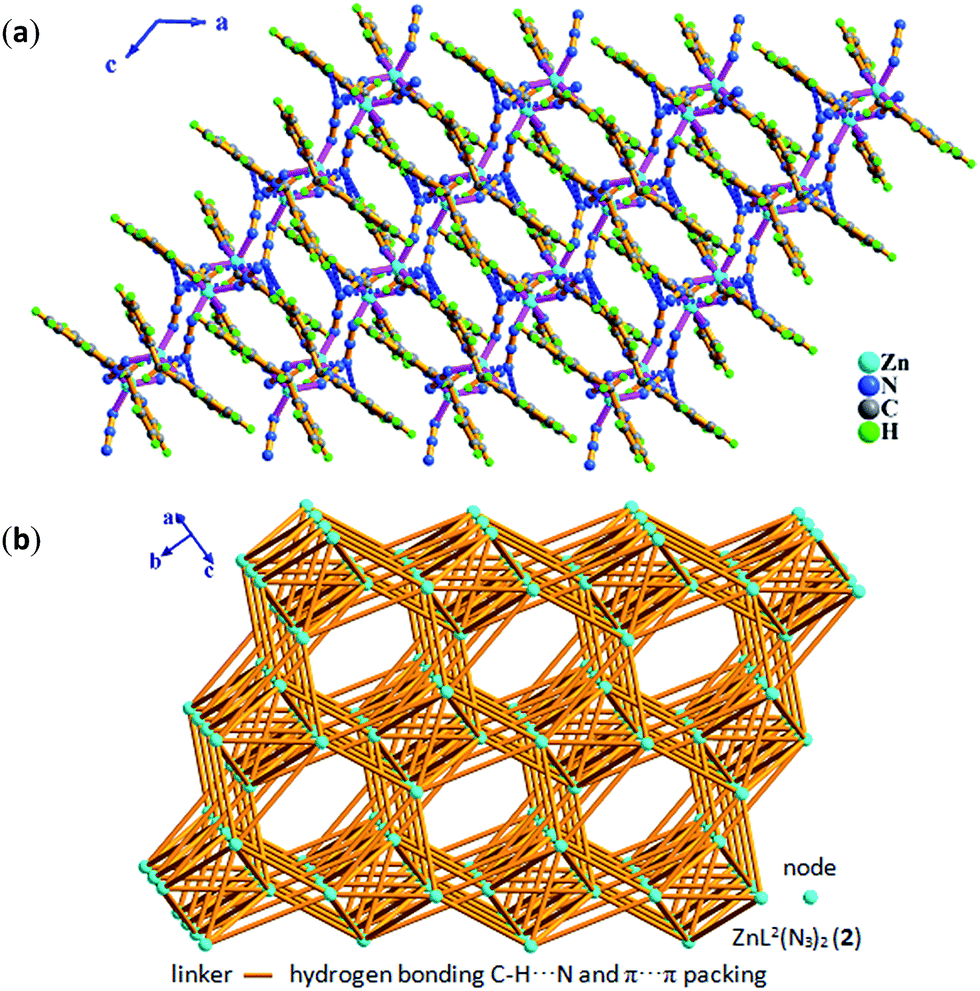 | ||
| Fig. 4 In 2, (a) 3-D supramolecular network assembled by inter-molecular hydrogen bonding C–H⋯N and π⋯π packing interactions in ac plane. (b) 3-D 36·413·56·63 topology. | ||
As shown in Fig. 5, the molecule of 3 with a C2 rotary axis contains two Zn(II) ions, one in situ formed ligand L3, four terminal Cl− ions and one lattice water molecule. Zn1 atom is penta-coordinated by three nitrogen atoms (N1, N2, N4) from the same ligand L3 and two terminal chloride atoms (Cl1, Cl2) and presents a slightly distorted trigonal bipyrimidal coordination geometry. The ligand L3, which adopts cis-conformation of four coordinated terminal groups referred to C6–C6a single bond and coordinates to two Zn2+ ions, shows μ2-η1:η1:η1:η1:η1:η1 coordination mode. The Zn–N bond length of (2.030(9)–2.323(11) Å), and Zn–Cl (2.220(4)–2.278(5) Å) bond distances are in the range expected for this coordination geometry with zinc(II). Meanwhile, the existence of intermolecular hydrogen bonding C–H⋯Cl and π⋯π packing interactions (Table S3†) makes dinuclear molecular unit Zn2(cis-L3)Cl4 to be linked into 1-D wave-like chain, and further assembled into 3-D network via inter-chain hydrogen bond C–H⋯O as shown in Fig. 6.
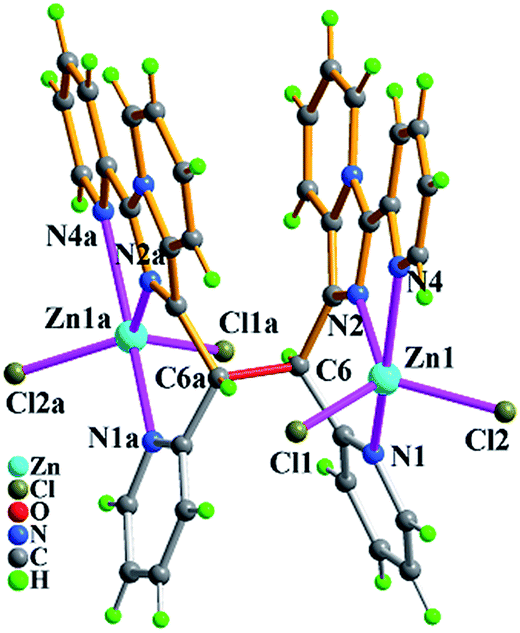 | ||
| Fig. 5 Molecular structure of compound Zn2(cis-L3)Cl4·H2O (3) with atomic labels, and one lattice water molecule was omitted for clarity. The symmetric code: (a) 2 − x, y, 0.5 − z. | ||
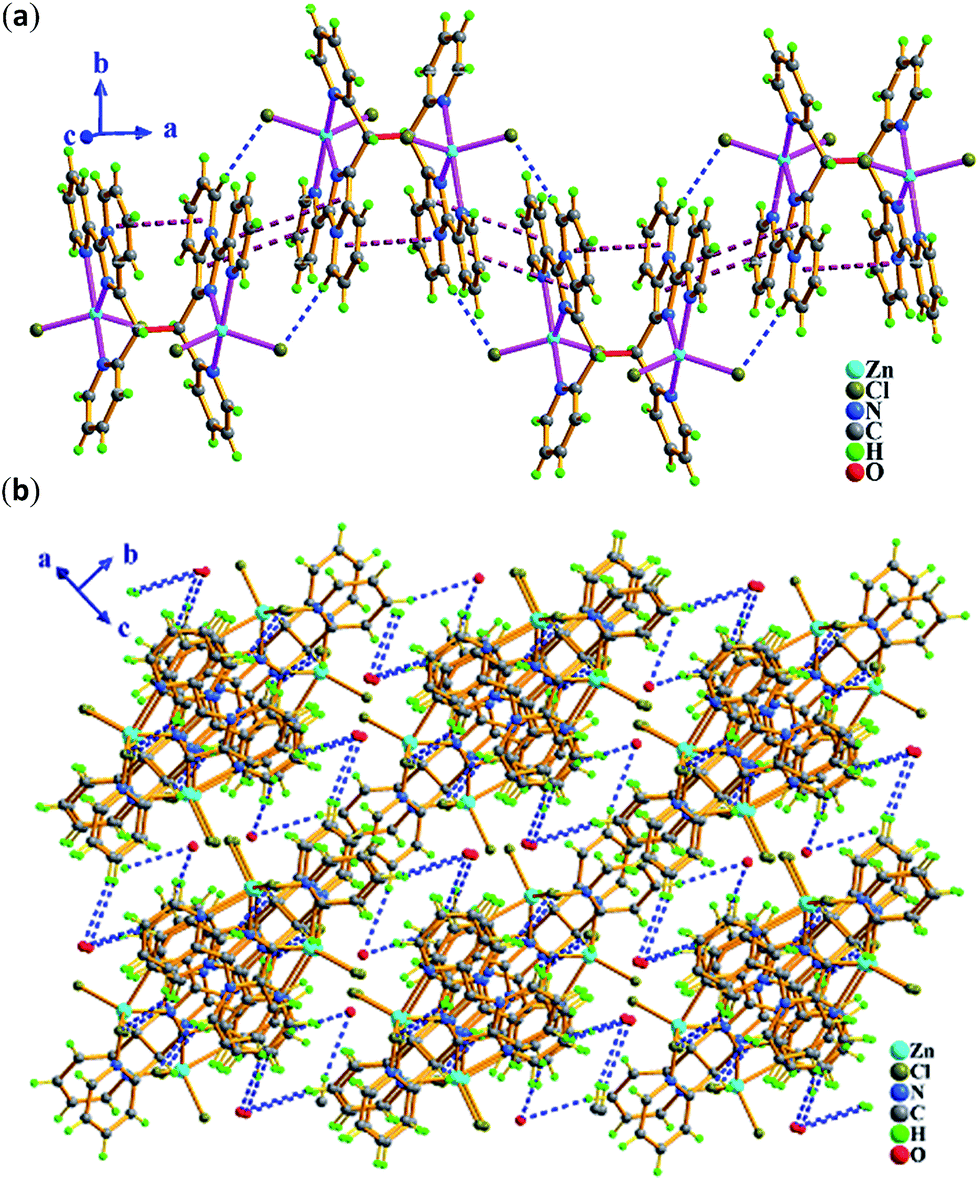 | ||
| Fig. 6 In 3, (a) 1-D wave-like chain constructed by inter/intra molecular hydrogen bonding C–H⋯Cl (blue dashed line) and π⋯π (plum dashed line) packing interactions along a axis. (b) 3-D supramolecular network assembled by interchain hydrogen bonding C–H⋯O (blue dashed line) interactions. | ||
Compared with Cl− ion, azide may have different spatial orientation, however compound 5 presents the same P2(1)/c space group. The molecule of 5 consists of two crystallographically independent Zn(II) ions showing a slightly distorted trigonal bipyrimidal coordination geometry, one in situ formed ligand L3 with μ2-η1:η1:η1:η1:η1:η1 coordination mode and four terminal η1-N3− ions as depicted in Fig. 7. The Zn–N bond length of 1.955(5)–2.2.344(5) Å is consistent with that of complex 3. Because of the hydrogen bonding C–H⋯N interactions from azide ion and pyridyl rings, each dinuclear molecular unit Zn2(cis-L3)(N3)4 links seven adjacent units to assemble into 3-D supramolecular network. If the dinuclear molecular unit Zn2(cis-L3)(N3)4 viewed as the node and the hydrogen bonding C–H⋯N interactions (Table S3†) as the linker, the resulting network can be simplified as 33·410·56·62 topology as displayed in Fig. 8.
 | ||
| Fig. 7 Molecular structure of compound Zn2(cis-L3)(N3)4 (5) with partial atomic labels. | ||
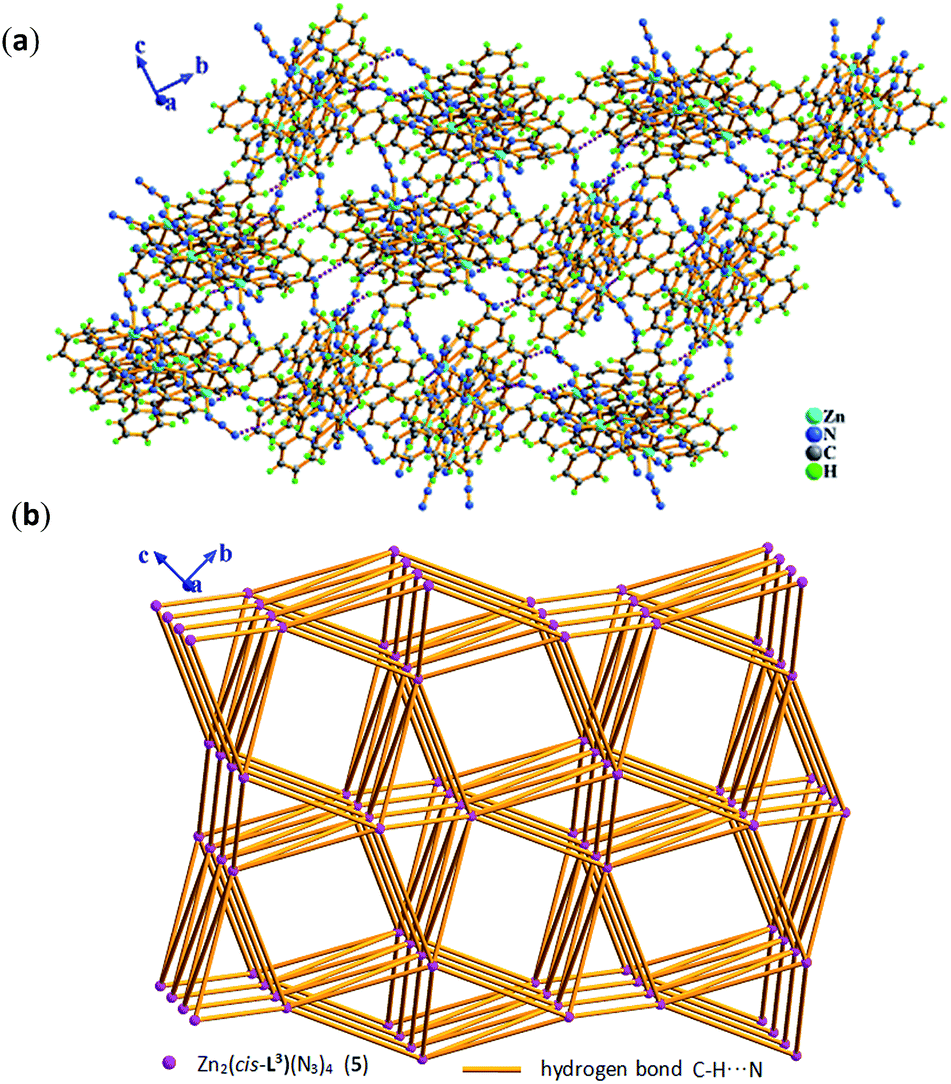 | ||
| Fig. 8 In 5, (a) 3-D supramolecular network assembled by inter-molecular hydrogen bonding C–H⋯N interactions in bc plane. (b) 3-D 33·410·56·62 topological network constructed by inter/intra molecular hydrogen bonding C–H⋯N interactions in bc plane, where pink sphere represents dinuclear molecular unit Zn2(cis-L3)(N3)4 (5) and light orange stick represents intermolecular hydrogen bonding C–H⋯N interactions. | ||
space group for 6, respectively.As shown in Fig. 9, the molecule of 4 possesses a crystallographically imposed inversion center located at the mid-point of C6–C6a single bond (blue color). Zn1 ion is penta-coordinated by three nitrogen atoms (N1, N2, N4) from the ligand L3 and two terminal chloride atoms (Cl1, Cl2), demonstrating trigonal bipyrimidal coordination geometry similar to that of its conformational isomer 3. One ligand L3, which is trans-conformation of four coordinated terminal groups referred to C6–C6a bond, coordinates to 2 equiv. of Zn2+ ions showing μ2-η1:η1:η1:η1:η1:η1 coordination mode. Meanwhile, the existence of intermolecular hydrogen bonding C–H⋯Cl interactions (Table S3†) makes dinuclear molecular [Zn2(trans-L3)Cl4] unit to be assembled into a 3-D supramolecular network with the topology of 35·418·65 as shown in Fig. 10 and 11.
 | ||
| Fig. 9 Molecular structure of compound Zn2(trans-L3)Cl4 (4) with partial atomic labels. The symmetric code: (a) 2 − x, −y, 2 − z. | ||
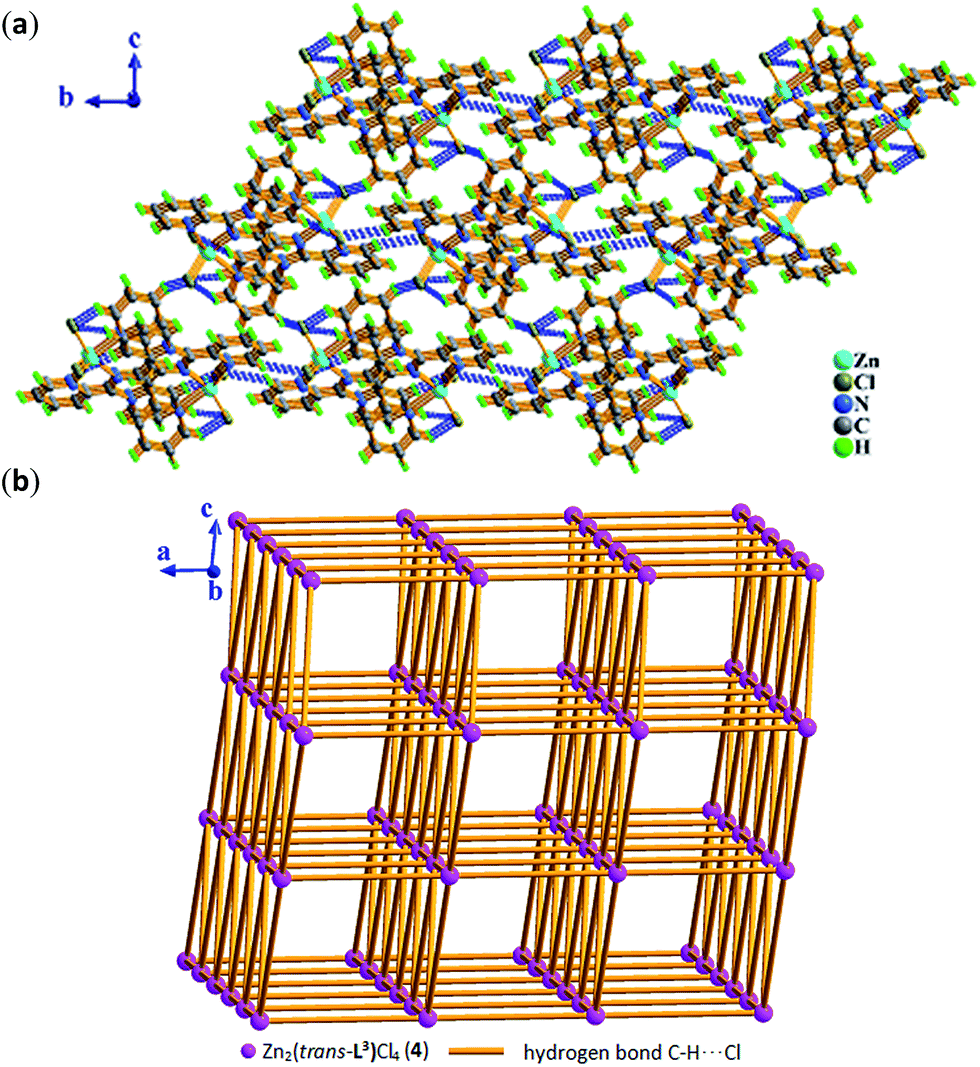 | ||
| Fig. 10 In 4, (a) 3-D supramolecular network assembled by inter-molecular hydrogen bonding C–H⋯Cl interactions in bc plane. (b) 3-D 35·418·65 topological network constructed by intermolecular hydrogen bonding C–H⋯Cl interactions in ac plane, where pink sphere represents dinuclear molecular unit Zn2(trans-L3)Cl4 (4) and light orange stick represents intermolecular hydrogen bonding C–H⋯Cl interactions. | ||
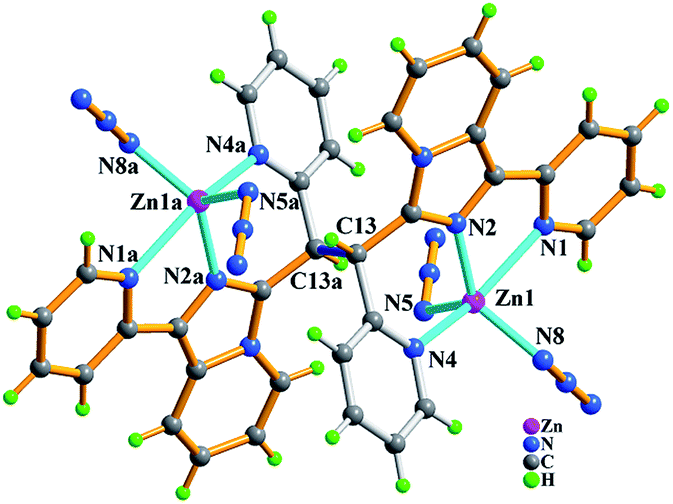 | ||
| Fig. 11 Molecular structure of compound Zn2(trans-L3)(N3)4 (6) with partial atomic labels. The symmetric code: (a) 2 − x, −y, −z. | ||
When Cl− ion in complex 4 was completely replaced by azide ion, its isologues 6 was obtained. From Fig. 12, two zinc ions (Zn1 and Zn1a) in 6 have the same coordination pattern of trigonal bipyrimidal coordination geometry as the zinc ions in 4. Meanwhile, replacement of the azide ions have no effect on trans-conformation of the in situ formed L2 ligand in 6. It is to be observed that the varied spatial orientations of azide ions make the supramolecular structure of complex 6 different from that of complex 4, exhibiting a 3-D supramolecular network with the topology of 34·44·620 through intermolecular hydrogen bonding C–H⋯N and π⋯π packing interactions (Table S3†) as displayed in Fig. 12.
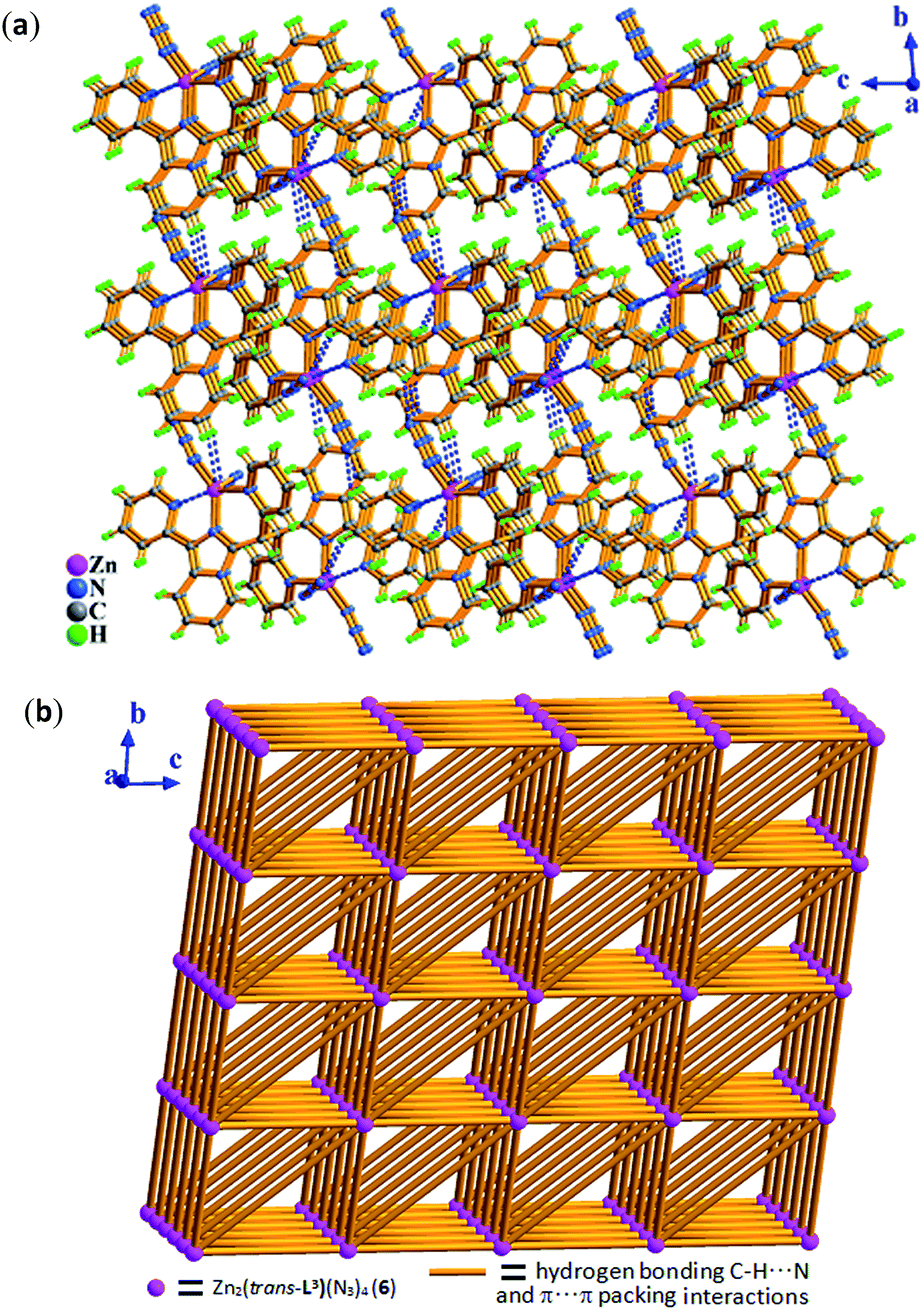 | ||
| Fig. 12 In 6, (a) 3-D supramolecular network assembled by inter-molecular hydrogen bonding C–H⋯N and π⋯π packing interactions in bc plane. (b) 3-D 34·44·620 topological network constructed by inter-molecular hydrogen bonding C–H⋯N and π⋯π packing interactions in bc plane, where pink sphere represents dinuclear molecular unit Zn2(trans-L3)(N3)4 (6) and light orange stick represents intermolecular hydrogen bonding C–H⋯N and π⋯π packing interactions. | ||
Structural in situ formation, transformation and temperature effect
In this contribution, the simple Schiff base ligand L1 with a donor set of N3 was firstly applied in the system of Zn-based aza-heterocyclic complexes. Initial reaction of ZnCl2 at 80 °C with L1 together with 2-pyridine formaldehyde afforded Zn-mono-nuclear compound ZnL2Cl2 (1) containing one in situ formed N-rich heterocycle L2 with a donor set of N3 via intermolecular C–C/C–N coupling and ring forming reactions (Scheme S1†). And then two terminal coordinated Cl− ions in 1 could be easily replaced by two N3− anions and produced the corresponding mononuclear zinc(II) compound ZnL2(N3)2 (2). Increasing the reaction temperature to 100 °C, Zn2+-induced intermolecular C–C coupling reaction between two L2 molecules resulted in the formation of cis-conformation L3 and the corresponding Cl-based compound [Zn2(cis-L3)Cl4·H2O (3) as well as N3-based substitute [Zn2(cis-L3)(N3)4 (5). Continuing to increase the reaction temperature to 120 °C, in situ formed cis-L3 can be lightly converted to the trans-L3 and the corresponding Cl-based compound Zn2(trans-L3)Cl4 (4) as well as N3-based substitute [Zn2(trans-L3)(N3)4] (6) with high thermal stability via the rotation of the C–C single bond derived by reaction temperature.A careful inspection of experimental condition reveals that the reaction temperature has a significant contribution to final structures of two aza-heterocycles of L2–3 as well as six corresponding Zn-based compounds 1–6, which might mainly depends on the influence of reaction temperature as reaction driving force. When reaction temperature is higher than 80 °C, the ortho-hydrogen atom of an imino group –CHN–CH– in ligand L1 can be markedly activated by the coordination induction of Zn2+ ion,1,2 and pyridine as basic acceptor has the ability to deprotonate the imino carbon-bound α-position hydrogen atom and form 1,3-dipolar, leading to the occurrence of inter-molecular C–C/C–N coupling, [3+2] cycloaddition, transformation of cis/trans-isomers caused by C–C single rotation and formation of final Zn-imidazo[1,5-a]pyridine complexes L2/L3–Zn (Scheme S1†).
Fluorescence/UV absorption spectra and titration of L1 + 2-Pfd
The luminescence properties of complexes 1–6 have been studied in the solid state at room temperature as depicted in Fig. S3.† Observably, 1–6 are blue emission with peak wavelengths at the range from 428 to 450 nm. These emissions could be tentatively assigned as resulting from the ligand-to-metal charge transfer (LMCT).5 Meanwhile, the fluorescence response of receptor L1 towards Zn2+ was studied in 0.10 mM mixture solution of DMF and pyridine (volume ratio of 2:1) at the different temperatures. Upon excitation at 355 nm, L1 and 2-pyridine formaldehyde (2-Pfd) (molar ratio of 1:1) in DMF and pyridine at 80–120 °C presented dark blue emission at 469 nm which could be attributed to the intra-ligand π–π* transition.14 As stepwise addition of ZnCl2 solution in DMF at 80 °C with concentration ranging from 0.1 equiv. to 2.0 equiv., fluorescent emission of L1 + 2-Pfd gradually red-shifted to 498 nm with their fluorescent intensity at 498 nm enhanced linearly with the Zn2+ concentration and saturated when [Zn2+] reached to 1.00 equiv. (Fig. 13). Interaction between Zn2+ and L1 + 2-Pfd was also studied by UV absorption spectra (Fig. S4†). Concomitant addition of ZnCl2 into L1 + 2-Pfd in DMF + pyridine causes a new absorption peak at 385 nm with intensity increased while that at 225, 261, 338, 430 nm decreased with four isosbestic points at 247, 272, 305, 348 nm. The binding ratio between Zn2+ and ligand was estimated to be 1:1 as confirmed by its job plot (Fig. S5†) and the related crystal structures.
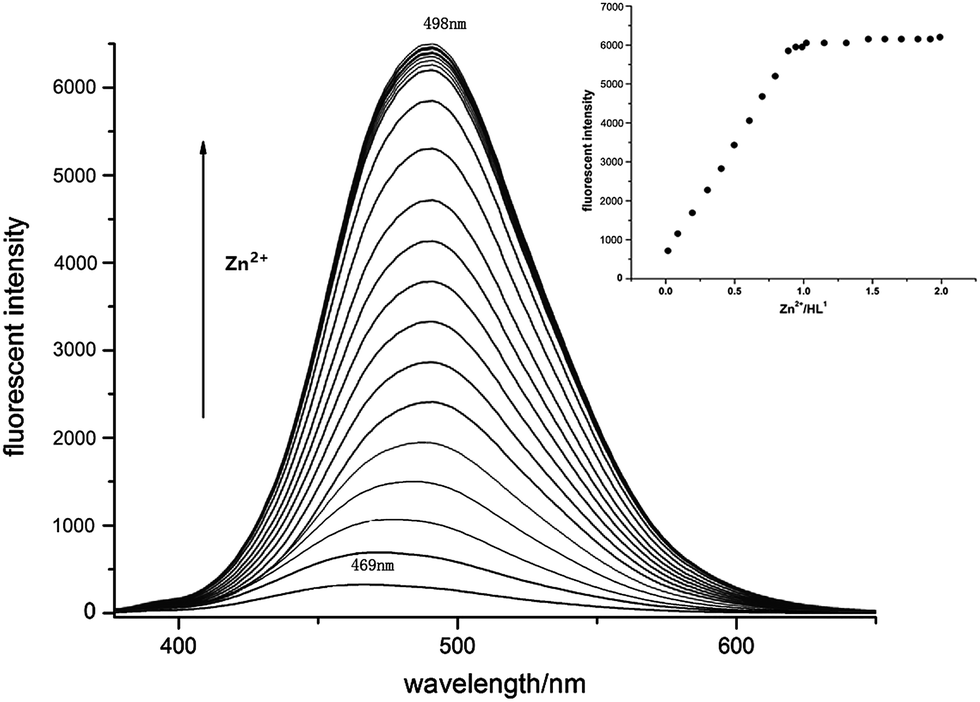 | ||
| Fig. 13 Fluorescence emission spectra of L1 + 2-Pfd upon addition of ZnCl2 in DMF + pyridine. λex = 355 nm at 80 °C ([L1 + 2-Pfd] = 0.10 mM); [Zn2+] = 0, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.131, 0.132, 0.133, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20 mM. (Inset: corresponding ZnCl2 titration profile according to the fluorescence intensity, indicating 1:1 stoichiometry for Zn2+/L1 + 2-Pfd.) | ||
As the temperature of the mixture [ZnCl2 + L1 + 2-Pfd] increased from 80 °C to 100 °C and 120 °C, the emission peak wavelength of the mixture ZnCl2 + L1 + 2-Pfd has an obvious red-shift to 525 nm for 3 (at 100 °C) and 531 nm for 4 (at 120 °C), respectively. The fluorescent intensity for 3 (Fig. S6†) and 4 (Fig. S7†) were both enhanced, but the peak shape were unchanged when compared to that at 80 °C (for 1) due to the formation of 2D/3D dimensional supramolecular network assembled by a large number of intramolecular and intermolecular hydrogen bonding C–H⋯Cl and/or π⋯π stacking interactions.
The azide anion substitution effect of Cl-based Zn-compounds 1, 3 and 4 was examined by fluorescence titration of 1/3/4 + NaN3 at 80 °C, 100 °C and 120 °C, respectively (Fig. 14 and S8 and S9†). The fluorescence intensity of 1/3/4 + NaN3 was, to some extent, enhanced with almost unchanged peak shape and position compared to the corresponding Cl-based Zn-compounds, in which the further enhanced fluorescence intensity of N3-based Zn-compounds could be attributed to the stronger intermolecular/intramolecular hydrogen bonding interactions supported by more stable and higher dimensional supramolecular structures in the solid state.
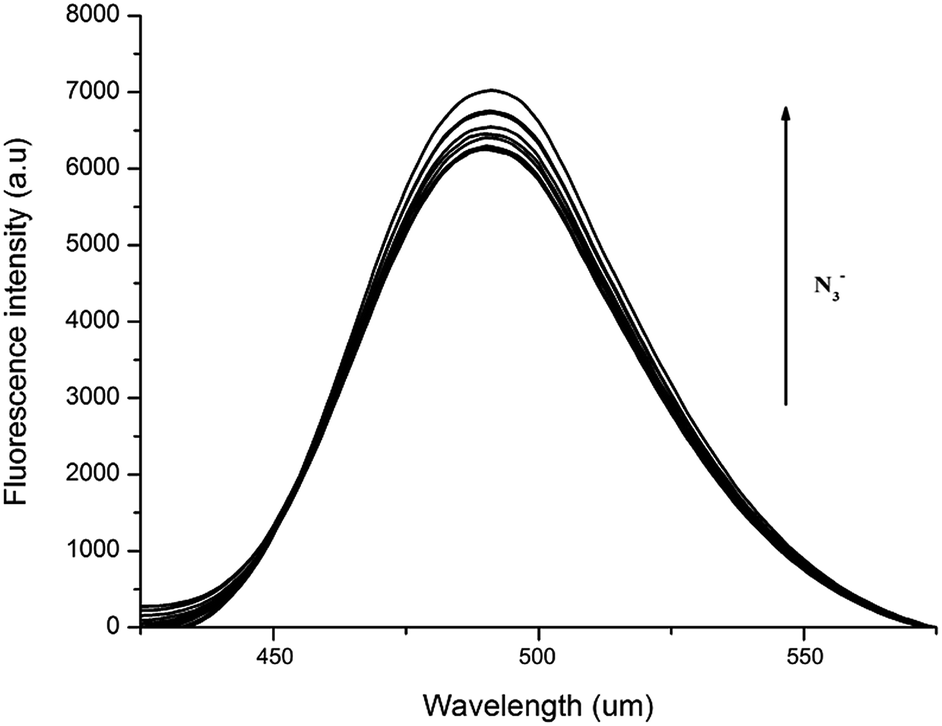 | ||
| Fig. 14 Fluorescence emission spectra of 1 upon addition of NaN3 in DMF + pyridine. λex = 355 nm at 80 °C ([1] = 0.10 mM); [N3−] = 0, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.10, 0.11, 0.12, 0.13, 0.131, 0.132, 0.133, 0.14, 0.15, 0.16, 0.17, 0.18, 0.19, 0.20 mM. | ||
In conclusion, this work describes the first example of efficient syntheses and complexation of two asymmetric N-rich heterocyclic ligands (L2–3) containing imidazo-[1,5-a]-pyridine core involving solvothermal in situ ligand generation from simple tridentate N3-set pyridine-type Schiff base ligand (L1) mediated by Zn2+. Further studies on in situ formation and conversion of ligands L1–3 and six corresponding Zn-complexes 1–6 show that temperature effect is the key factor rather than anion effect, which can tune their supramolecular connections and topologies of resulting zinc complexes. Obviously, this synthesis strategy may bring a broad interest in the construction of novel nitrogen-rich multidentate asymmetric heterocycles relating to metal ion induced α-H activation chemistry on imine bond –CHN–, and relevant metal–organic frameworks as well as inorganic–organic hybrid materials with advanced luminescent and biological functions.
Acknowledgements
The authors are grateful for financial aid from the National Natural Science Foundation of P. R. China (Grant No. 21471061, 21671071 and 911220008), the Doctoral Program of Higher Education of China (Grant No. 20124407110007), Science and Technology Planning Project of Guangdong Province, Guangzhou, China (Grant No. 2013B010403024 and 2015B010135009) and the N. S. F. of Guangdong Province (Grant No. 2014A030311001 and C86186).References
- (a) S. M. Chiu, T. W. Wong, W. L. Man, W. T. Wong, S. M. Peng and T. C. Lau, J. Am. Chem. Soc., 2001, 123, 12720 CrossRef CAS PubMed; (b) J. Xiang, W.-L. Man, S.-M. Yiu, S.-M. Peng and T.-C. Lau, Chem.–Eur. J., 2011, 17, 13044 CrossRef CAS PubMed; (c) J. Xiang, W.-L. Man, J.-F. Guo, S.-M. Yiu, G.-H. Lee, S.-M. Peng, G.-C. Xu, S. Gao and T.-C. Lau, Chem. Commun., 2010, 46, 6102 RSC; (d) V. M. Aryuzina and M. N. Shchukina, Khim. Geterotsikl. Soedin., 1968, 3, 506 Search PubMed; (e) M. P. Kochergin and V. A. Lifanov, Khim. Geterotsikl. Soedin., 1994, 4, 490 Search PubMed; (f) M. E. Bluhm, M. Ciesielski, H. Görls, O. Walter and M. Döring, Inorg. Chem., 2003, 42, 8878 CrossRef CAS PubMed.
- (a) R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, John Wiley & Sons, New York, 1984, vol. 1 Search PubMed; (b) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187 CrossRef CAS PubMed.
- (a) B. A. Frazier, P. T. Wolczanski, I. Keresztes, S. DeBeer, E. B. Lobkovsky, A. W. Pierpont and T. R. Cundari, Inorg. Chem., 2012, 51, 8177 CrossRef CAS PubMed; (b) B. A. Frazier, V. A. Williams, P. T. Wolczanski, S. C. Bart, K. Meyer, T. R. Cundari and B. Lobkovsky, Inorg. Chem., 2013, 52, 3295 CrossRef CAS PubMed; (c) B. A. Frazier, P. T. Wolczanski, E. B. Lobkovsky and T. R. Cundari, J. Am. Chem. Soc., 2009, 131, 3428 CrossRef CAS PubMed.
- Y.-J. Ou, Z.-P. Zheng, X.-J. Hong, L.-T. Wan, L.-M. Wei, X.-M. Lin and Y.-P. Cai, Cryst. Growth Des., 2014, 14, 5339 CAS.
- (a) H. A. Staab, Angew. Chem., Int. Ed. Engl., 1962, 7, 351 CrossRef; (b) J. Chang-Fong, K. Benamour, B. Szymonski, F. Thomasson, J.-M. Morand and M. Cussac, Chem. Pharm. Bull., 2000, 48, 729 CrossRef CAS PubMed; (c) C. R. Sage, M. D. Michelitsch, T. J. Stout, D. Biermann, R. Nissen, J. Finer-Moore and R. M. Stroud, Biochemistry, 1998, 37, 13893 CrossRef CAS PubMed; (d) V. C. Sharma, L. Crankshaw and D. Piwnica-Worms, J. Med. Chem., 1996, 39, 3483 CrossRef CAS PubMed; (e) H. Sakuta and K. Okamoto, Eur. J. Pharmacol., 1994, 259, 223 CrossRef CAS PubMed; (f) G. J. Kant, J. L. Meyerhoff and R. H. Lenox, Biochem. Pharmacol., 1980, 29, 369 CrossRef CAS PubMed.
- (a) A. R. Katritzky, K. Suzuki and H.-Y. He, J. Org. Chem., 2002, 67, 3109 CrossRef CAS PubMed; (b) M. Roy, B. V. S. K. Chakravarthi, C. Jayabaskaran, A. A. Karande and A. R. Chakravarty, Dalton Trans., 2011, 40, 4855 RSC.
- (a) Y.-M. Chen, L. Li, Z. Chen, Y.-L. Liu, H.-L. Hu, W. Q. Chen, W. Liu, Y.-H. Li, T. Lei, Y.-Y. Cao, Z.-H. Kang, M.-S. Lin and W. Li, Inorg. Chem., 2012, 51, 9705 CrossRef CAS PubMed; (b) C. Garino, T. Ruiu, L. Salassa, A. Albertino, G. Volpi, C. Nervi, R. Gobetto and K. I. Hardcastle, Eur. J. Inorg. Chem., 2008, 3587 CrossRef CAS.
- G. M. Sheldrick, SADABS, Version 2.05, University of Göttingen, Göttingen, Germany Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Determination, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, John Wiley & Sons, New York, 1984, vol. 1 Search PubMed.
- (a) M. A. S. Goher, Acta Chim. Hung., 1984, 117, 215 CAS; (b) Z. Dori and R. F. Ziolo, Chem. Rev., 1973, 73, 247 CrossRef CAS; (c) J. L. Manson, A. M. Arif and J. S. Miller, Chem. Commun., 1999, 1479 RSC; (d) J. Ribas, M. Monfort, B. K. Ghosh and X. Solans, Angew. Chem., Int. Ed. Engl., 1994, 33, 2087 CrossRef; (e) X. Hao, Y. Wei and S.-W. Zhung, Chem. Commun., 2000, 2271 RSC.
- B. Bosnich, J. Am. Chem. Soc., 1968, 90, 627 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The TGA curves, liquid-state emission as well as the related UV absorption spectra at 100 °C and 120 °C, XRD patterns of compounds 1–6; and Tables S1–S3 of selected bond lengths, bond angles, hydrogen bonds, crystal data and structure refinement, as well as X-ray crystallographic files in CIF format for six compounds 1–6 are available in ESI. CCDC 1032239, 1508822, 1032241, 1508812, 1032243 and 1032244 for 1–6, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25678c |
| This journal is © The Royal Society of Chemistry 2017 |