
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsoliquiritigenin attenuates MiR-21 expression via induction of PIAS3 in breast cancer cells†
Shilong Ning‡
a,
Xiao Ma‡b,
Dongmei Zhuc,
Zhaoxia Shenc,
Jiao Liuc,
Yun Liuc,
Juan Chenc and
Zhong Li *c
*c
aDepartment of Clinical Nutrition, Jinhua Municipal Central Hospital, Jinhua, 321000, China
bDepartment of Health Education and Administration, Jinhua Municipal Central Hospital, Jinhua, 321000, China
cDepartment of Nutrition and Food Hygiene, School of Public Health, Nanjing Medical University, Nanjing, 211166, China. E-mail: lz-ny@njmu.edu.cn; Fax: +86-25-8652-7613; Tel: +86-25-8686-8451
First published on 24th March 2017
Abstract
We have previously documented that isoliquiritigenin (ISL), a dietary compound extracted from licorice, could exert anti-breast cancer effects via inhibition of miR-21, however, the intrinsic mechanisms remain obscure. Herein, we discovered that ISL down-regulated the primary miR-21 (pri-miR-21) and mature miR-21 expression in a dose-dependent manner, indicating transcriptional silencing of miR-21. The transcriptional factor signaling transducer and activator of transcription 3 (STAT3) could transcriptionally activate miR-21 expression. Our luciferase reporter and ChIP assay data demonstrated that ISL reduced the activity of STAT3 signaling by approximately 70% and the enrichment of activated STAT3 within miR-21 promoter in breast cancer cells. Next we found that ISL induced protein inhibition of activated STAT3 (PIAS3) a specific inhibitor of activated STAT3. Additionally, abrogation of PIAS3 via transfection of specific siRNA abolished the inhibitory effect of ISL on the activity of STAT3 signaling pathway and miR-21 expression. Taken together, our study documented for the first time that induction of PIAS3 mediated STAT3 signaling inhibition was responsible for the repression of miR-21 by ISL.
1 Introduction
Breast cancer is a serious public health problem worldwide, resulting in more than 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths and accounting for approximately 14.7 percent mortality of cancer-related deaths among females annually.1 The less satisfactory outcome from clinic is mainly due to cancer metastasis and much attention has been paid to unmasking the underling mechanisms consisting of dys-regulated molecules expression pattern.2,3
000 deaths and accounting for approximately 14.7 percent mortality of cancer-related deaths among females annually.1 The less satisfactory outcome from clinic is mainly due to cancer metastasis and much attention has been paid to unmasking the underling mechanisms consisting of dys-regulated molecules expression pattern.2,3
As currently known, microRNAs (miRNAs) located in RNA-induced silencing complex (RISC) are a cluster of naturally occurring non-coding RNA molecules, leading to repression of target gene expression via specific binding to the 3′-untranslated region (3′-UTR) within the target mRNA.4 It has been proved that dys-regulation of miRNAs expression profile is involved in breast cancer progression.5 And miRNAs have been selected as the ideal target for anti-tumor drug development.6 Some miRNAs called onco-miRNAs acted as oncogenes through targeting tumor suppressors, resulting in cancer initiation and progression.7 Among the widely known onco-miRNAs, miR-21 could promote malignant phenotype such as tumor cell invasion and metastasis via targeting several tumor suppressors including PDCD4, PTEN and RECK et al.8 Our recent research also confirmed the repressive effect of miR-21 on RECK expression via triggering mRNA translation repression in breast cancer cells.9 Besides, over-expression of miR-155, another oncogenic miRNA, might contribute to tumor progression via participating in oncogenic NF-κB signaling.10
Signal transducer and activator of transcription (STAT) family proteins, especially STAT3, play an essential role in initiating and maintaining pro-carcinogenic inflammatory microenvironment, leading to malignant transformation and cancer progression.11 STAT3 maintains monomer and quiescent status without stimulation. Upon stimulation such as the cytokine IL-6 treatment, STAT3 is activated by Janus kinase (Jak) via tyrosine residue phosphorylation modification. Furthermore, phosphorylation at tyrosine residue (Tyr705) is responsible for STAT3 activation, which contributes to its translocation to the nuclei as a dimer and subsequent activation of target genes via DNA binding to specific promoter sequence.11
Cytokine-induced STAT3 activation is transient in normal cells due to the existence of negative feedback loops consisted diverse proteins including protein inhibitor of activated STAT (PIAS) family, especially PIAS3. PIAS3, the negative STAT3 regulator, belongs to the PIAS protein family also including PIAS1, PIASγ, PIAS-xα and PIAS-xβ.12 Mechanistically, PIAS3 blocks the activity of STAT3 signaling via specifically interacted with the DNA binding domain of the activated STAT3 without affecting its phosphorylation course, leading to deficiency in target gene transcription.13–15 In fact, down-regulation of PIAS3 accompanied by abnormal activation of STAT3 signaling is a frequent event during cancer progression through analyzing tumor samples obtained from clinic.16 And the expression of PIAS3 in tumor positively related with the overall survival in patients with cancer.17,18
Growing evidence revealed the potential application of bioactive natural compounds isolated from dietary supplement in cancer prevention and therapy due to their ideal anti-tumor properties.19,20 Isoliquiritigenin (ISL), a dietary compound extracted from licorice, could exert anti-inflammation effect via inducing peroxisome proliferator-activated receptor-γ expression through activating extracellular regulated protein kinases 1/2 (ERK 1/2) pathway in intestinal epithelial cells.21 Additionally, ISL was also reported for its anti-tumor properties. ISL could prevent mammary carcinogenesis by inhibiting breast cancer stem cells through WIF1 demethylation.22 We recently demonstrated that induction of the tumor suppressor RECK by down-regulation of miR-21 was responsible for the inhibitory effect of ISL on breast cancer cellular invasiveness.9 In the present study, we aim to uncover the mechanism underling the attenuation of miR-21 by ISL in breast cancer cells.
2 Materials and methods
2.1 Cell culture and reagents
Human breast cancer cell lines Hs-578T and MDA-MB-231 were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured in accordance with the ATCC's recommendations. Briefly, MDA-MB-231 cells were maintained in L-15 medium; whereas Hs-578T cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Life Technologies/Gibco, Grand Island, NY, USA). Both of the medium contained 10% fetal bovine serum (FBS; Life Technologies/Gibco), 100 mg mL−1 streptomycin, and 100 U mL−1 penicillin (Life Technologies/Gibco). MDA-MB-231 cells were maintained in an incubator without CO2 at 37 °C; whereas, Hs-578T cells were grown in the presence of 5% CO2. Human umbilical vein endothelial cell line (HUVECs) was obtained from Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences. HUVECs were cultured in ECM medium (Invitrogen, Carlsbad, USA). The mediums were supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U mL−1 penicillin, 100 mg mL−1 streptomycin (Gibco), and 30 mg mL−1 endothelial cell growth supplement (for HUVECs, Sigma-Aldrich, MO, USA). ISL, dissolved in dimethyl sulfoxide (DMSO), was obtained from Sigma-Aldrich (St. Louis, MO, USA), with >98% purity and placed at −20 °C. All the other reagents used in our present study were of analytical grade or the highest grade available.2.2 Quantitative real-time polymerase chain reaction (qRT-PCR)
At the end of individual treatment, total cellular RNA was isolated using Trizol® (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's recommendations. For miR-21 detection, total RNA (1 μg) was reverse transcribed into cDNA using the TaqMan miRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) with miRNA-specific looped reverse primers. Forward and reverse primers were purchased from RiboBio (Guangzhou, China). The reverse transcription reaction conditions were as follows: 42 °C for 15 min and 85 °C for 5 s. The U6 snRNA was applied as the internal control to rule out the loading difference. The qRT-PCR assays were conducted with the Applied Biosystems 7300 Sequence Detection System (Applied Biosystems) for 40 cycles of 95 °C for 10 s, 60 °C for 20 s, and 70 °C for 30 s. For RECK, PIAS3, SOCS1 and miR-21 pri-cursor (pri-miR-21) detection, total RNA (2 μg) was reverse transcribed into cDNA with AMV Reverse Transcriptase (Promega, Madison, WI, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. The sequence of primers used in our study was listed in Table 1. The qRT-PCR assay was performed using the Applied Biosystems 7300 Sequence Detection System (Applied Biosystems) with cycling conditions as follows: 95 °C for 15 s and 60 °C for 1 min for 40 cycles. The two types of qRT-PCRs mentioned above were carried out with Power SYBR® Green master mix (Applied Biosystems). Fold changes in gene expression was calculated by a comparative threshold cycle (Ct) method with the formula 2-(ΔΔCt).| Sequences | ||
|---|---|---|
| RECK | Forward | 5′-AGCAACCGAGCCCGTATGT-3′ |
| Reverse | 5′-CCGAGTAGGCAGCACACACA-3′ | |
| PIAS3 | Forward | 5′-GCCGACATGGACGTGTCCTGTG-3′ |
| Reverse | 5′-TTCCCTCCTGGACTGCGCTGTAC-3′ | |
| pri-miR-21 | Forward | 5′-CATTGTGGGTTTTGAAAAGGTTA-3′ |
| Reverse | 5′-CCACGACTAGAGGCTGACTTAGA-3′ | |
| SOCS1 | Forward | 5′-CCCTGGTTGTTGTAGCAGCTT-3′ |
| Reverse | 5′-GGTTTGTGCAAAGATACTGGGTATATG-3′ | |
| GAPDH | Forward | 5′-GTCAGTGGTGGACCTGACCT-3′ |
| Reverse | 5′-AGGGGAGATTCAGTGTGGTG-3′ | |
2.3 Reverse-transcriptase polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated with Trizol® (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. RNA (2 μg) was transcribed into cDNA using AMV Reverse Transcriptase (Promega, Madison, WI, USA). Primers used are listed in Table 1. The reactions were determined by analyzing the PCR products on 2% (w/v) agarose gels. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) product was applied as the loading control.2.4 Western blotting
Cell lysis solution was obtained with RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China). The cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by transferring to polyvinylidene fluoride membranes (PVDF; Millipore, Billerica, MA, USA). The antigen–antibody complexes were detected with enhanced chemiluminescence. Highly specific antibodies used in our current study were anti-RECK, anti-PIAS3, anti-STAT3, anti-phosphorylated STAT3 (Cell Signaling Technology, Beverly, MA, USA) and anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA). And β-actin served as the internal control to eliminate the differences of protein loading.2.5 Invasion assay
Invasion assay was performed with growth factor-reduced Matrigel®-coated filters (8 mm pore size; BD Biosciences, Franklin Lakes, NJ, USA) in 24-well plates. The wells were pre-coated with matrigel at 4 °C for 12 h and placed in an incubator at 37 °C for another 1.5 h for solidification of matrigel. After individual treatments, breast cancer cells (2 × 104) were re-suspended in FBS-free medium and seeded onto the upper chamber of well plates. The lower chambers were filled with medium containing 100 ng mL−1 of epidermal growth factor (R&D Systems, Minneapolis, MN, USA). The chambers were incubated at 37 °C for 24 h in the absence or presence of CO2 for MDA-MB-231 and Hs-578T cells, respectively. At the end of the incubations, cells on the upper surface of the filter were removed gently with a cotton swab. Cells penetrated through the filter to the lower surface were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 10 min. Penetrated cells were photographed with a phase contrast microscope (Olympus, Tokyo, Japan) and counted in five randomly chosen fields.2.6 Chromatin immunoprecipitation (ChIP) assay
After individual treatment, cells were fixed by 1% formaldehyde for 10 min at 37 °C. Cells were washed twice with ice-cold phosphate buffer saline (PBS) containing 1 mM PMSF, the inhibitor of protease and then scraped. After centrifugation for 2 min at 4 °C, the cells population was resuspended in SDS lysis buffer containing 1 mM PMSF for 15 min and sonicated. The cell lysis sample was then centrifuged at 4 °C for 10 min at 12000 rpm. The supernatant was diluted in ChIP dilution buffer containing 1 mM PMSF. Phosphorylated STAT3 antibody and IgG was added and incubated at 4 °C overnight with gentle shaking. The immunocomplex was pulled down with protein A/G agarose and washed by low salt immune complex wash buffer, high salt immune complex wash buffer, LiCl immune complex wash buffer and TE buffer in order. The complex was eluted with elution buffer containing 1% SDS and 0.1 M NaHCO3 and the cross-links was reversed by heating at 65 °C for 4 h. The DNA was then purified by phenol/chloroform and applied to the subsequent PCR amplification. The sequence of promoter-specific primers for human miR-21 upstream region including activated STAT3 binding sites used in our study was as follows: forward: 5′-CCTCTGAGAAGAGGGGACAA-3′ and reverse: 5′-ACCGCTTCCAGCAA AAGAGT-3′. This pair of primers had been previously testified.23 Final product was approximately 235 bp. The amplified PCR products were resolved by on 2% (w/v) agarose gels and photographed by BioImage.
2.7 Cell transfection
Negative control (nc), and PIAS3 small interfering RNA (siRNA) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). These reagents were transiently transfected into breast cancer cells with Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions.2.8 Luciferase reporter assay
The STAT3 signaling pathway reporter plasmid was purchased from Shuntian Biology (Shanghai, China). And the construct containing only renilla luciferase was obtained from Promega and acted as the internal control to eliminate the difference in terms of the transfection efficiency and cytotoxicity of test chemicals. Briefly, the Hs-578T cells were seeded in a 24-well cell culture dish. The cells proliferated to approximately 80% confluence after 24 h of culture. The constructs were firstly transiently introduced into Hs-578T cells with application of Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. After an incubation period of 12 h, the transfection medium was replaced. Then, the Hs-578T cells were exposed to ISL at 0.0 or 10.0 μM for 24 h with normal medium. Following 24 h incubation, the cells were lysed with passive lysis buffer (Promega, Madison, WI, USA). Using a 96-well plate luminometer (Berthold Detection System, Pforzheim, Germany), the amounts of luciferase and renilla luciferase were measured with the Dual-Luciferase Reporter Assay System Kit (Promega, Madison, WI, USA) following the manufacturer's recommendations. The values of luciferase activity for each lysate were measured randomly in triplicate and normalized to the respective renilla luciferase activity. The relative activity was converted into fold induction compared with control group and presented as mean ± SD (n = 3).2.9 Statistical analysis
Values were presented as the means ± SD. The one-way analysis of variance (ANOVA) followed by Dunnett's t-test were applied to determined statistical differences between groups. P-values <0.05 were considered statistically significant.3 Results
3.1 ISL transcriptionally down-regulates miR-21 expression
Previously, we found that ISL did not exert cytotoxicity to breast cancer cells at the concentration of 10.0 and 20.0 μM for 24 h.9 Thus, we used the concentration of 10.0 and 20.0 μM ISL for further investigating the roles of ISL on invasion in breast cancer cells. Furthermore, no detectable cytotoxicity of ISL on non-cancer cells HUVECs was observed at these two concentrations indicating its safety of application (Fig. S1†).Next we examined the miR-21 expression pattern in breast cancer cells treated with ISL. As shown in Fig. 1A, the expression of mature miR-21 was attenuated in a dose-dependent manner. Moreover, the miR-21 pri-cursor (pri-miR-21) was also reduced in a dose-dependent manner (Fig. 1B). These results indicated the transcriptional repression of miR-21 expression by ISL at appropriate concentration.
 | ||
| Fig. 1 ISL transcriptionally down-regulates miR-21 expression. (A) Breast cancer MDA-MB-231 and Hs-578T cells were treated with ISL for 24 h at 0.0, 5.0, 10.0 and 20.0 μM. The expression of mature miR-21 was examined by qRT-PCR assay (mean ± SD, n = 3). (B) Breast cancer MDA-MB-231 and Hs-578T cells were treated with ISL for 24 h at 0.0, 5.0, 10.0 and 20.0 μM. The expression of miR-21 pri-cursor (pri-miR-21) was examined with qRT-PCR assay (mean ± SD, n = 3). *P < 0.05 and **P < 0.01 compared with the non-treated group. | ||
3.2 ISL reduces the DNA binding activity of activated STAT3
Research data from for other group suggested that the transcriptional factor STAT3 could translocate into nuclei and bind to specific sequence within the promoter of miR-21 gene, resulting in enhanced miR-21 expression at transcriptional level.24 Then, we examined the effect of ISL on STAT3 pathway. Surprisingly, we did not observe significant changes in the phosphorylation level of STAT3, confirmed with western blotting analysis (Fig. 2A). Similarly, immunofluorescence staining also demonstrated no obvious influence on expression or nuclear accumulation of phosphorylated STAT3 in cells treated with ISL (data not shown). As presented in Fig. 2B, however, we found the reduced signaling activity of STAT3 in Hs-578T cells exposed to ISL with STAT3 reporter plasmid. Furthermore, ISL significantly down-regulated the enrichment of phosphorylated STAT3 within miR-21 promoter in dose-dependent manner (Fig. 2C). These data indicated that ISL reduced the DNA binding activity of activated STAT3.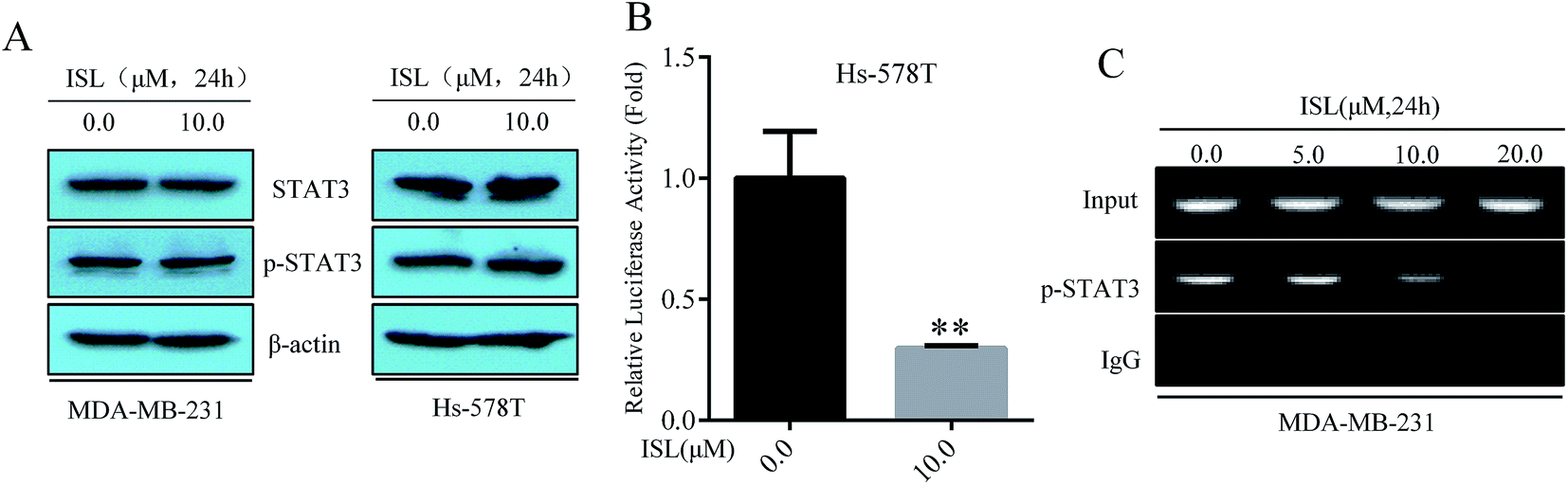 | ||
| Fig. 2 ISL reduces the DNA binding activity of activated STAT3. (A) MDA-MB-231 and Hs-578T cells were exposed to ISL at 0.0 or 10.0 μM for 24 h. Western blotting was carried out with specific antibodies. (B) Hs-578T cells were transiently transfected with the STAT3 reporter plasmid for 12 h followed by exposure to ISL at 0.0 or 10.0 μM for another 24 h. Then the luciferase activity was tested with dual-luciferase reporter assay system kit. The relative activity was converted into fold induction compared with control group and presented as mean ± SD (n = 3). **P < 0.01 compared with the non-treated group. (C) MDA-MB-231 cells were exposed to ISL at 0.0 or 10.0 μM for 24 h. The ChIP assay was performed with antibodies against p-STAT3. | ||
3.3 Induction of PIAS3 mediates the inhibition of the DNA binding activity of activated STAT3 by ISL
PIAS3 acts as a specific inhibitor of activated STAT3.13–15 Next, we attempted to elucidate the potential effect of ISL on the negative regulator of STAT3 signaling pathway namely PIAS3. As presented in Fig. 3A, PIAS3 mRNA was induced in dose-dependent manner in breast cancer cells treated with ISL. And the protein level of PIAS3 also increased due to ISL exposure (Fig. 3B). These results implied the involvement of PIAS3 in the repressive effect of ISL on STAT3 signaling. | ||
| Fig. 3 Induction of PIAS3 mediates the inhibition of the DNA binding activity of activated STAT3 by ISL. (A) Breast cancer MDA-MB-231 and Hs-578T cells were treated with ISL for 24 h at 0.0, 5.0, 10.0 and 20.0 μM. The expression of PIAS3 mRNA was measured with qRT-PCR assay (mean ± SD, n = 3). **P < 0.01 compared with the control group. (B) MDA-MB-231 and Hs-578T cells were exposed to ISL at 0.0 or 10.0 μM for 24 h. The expression of PIAS3 protein was examined with western blotting analysis. (C) MDA-MB-231 and Hs-578T cells were transiently transfected with specific siRNA for PIAS3. RT-PCR and western blotting analysis were performed simultaneously. MDA-MB-231 and Hs-578T cells were transiently transfected with si-nc or si-PIAS3, then exposed to ISL at 0.0 or 10.0 μM for 24 h. (D) The western blotting analysis and (E) the ChIP assay were then performed. | ||
Subsequently, the PIAS3 siRNA was transiently introduced into the breast cancer cells and data presented in Fig. 3C confirmed the high efficiency of the siRNA. As followed, these cells were transfected with si-nc or si-PIAS3 for 12 h, then treated with ISL at 0.0 or 10.0 μM for another 24 h. Herein, abrogation of PIAS3 expression by specific siRNA reversed the inhibitory effect of ISL on the enrichment of activated STAT3 in miR-21 promoter region (Fig. 3E). However, the phosphorylation level and nuclear location of activated STAT3 remained unchanged upon PIAS3 modulation (Fig. 3D). The results mentioned above certified that induction of PIAS3 mediated the inhibition of the DNA binding activity of activated STAT3 by ISL.
3.4 Induction of PIAS3 is crucial for the repression of miR-21 by ISL
As demonstrated in Fig. 4A, ISL attenuated the miR-21 expression, as determined by qRT-PCR assay; however, the repression of miR-21by ISL was abrogated by PIAS3 perturbance with siRNA. We previously confirmed the tumor suppressor RECK as a direct target of miR-21.9 Herein, the induction of RECK by ISL was also abolished with PIAS3 siRNA transfection (Fig. 4B). These results suggested that induction of PIAS3 was crucial for the repression of miR-21 by ISL in breast cancer cells.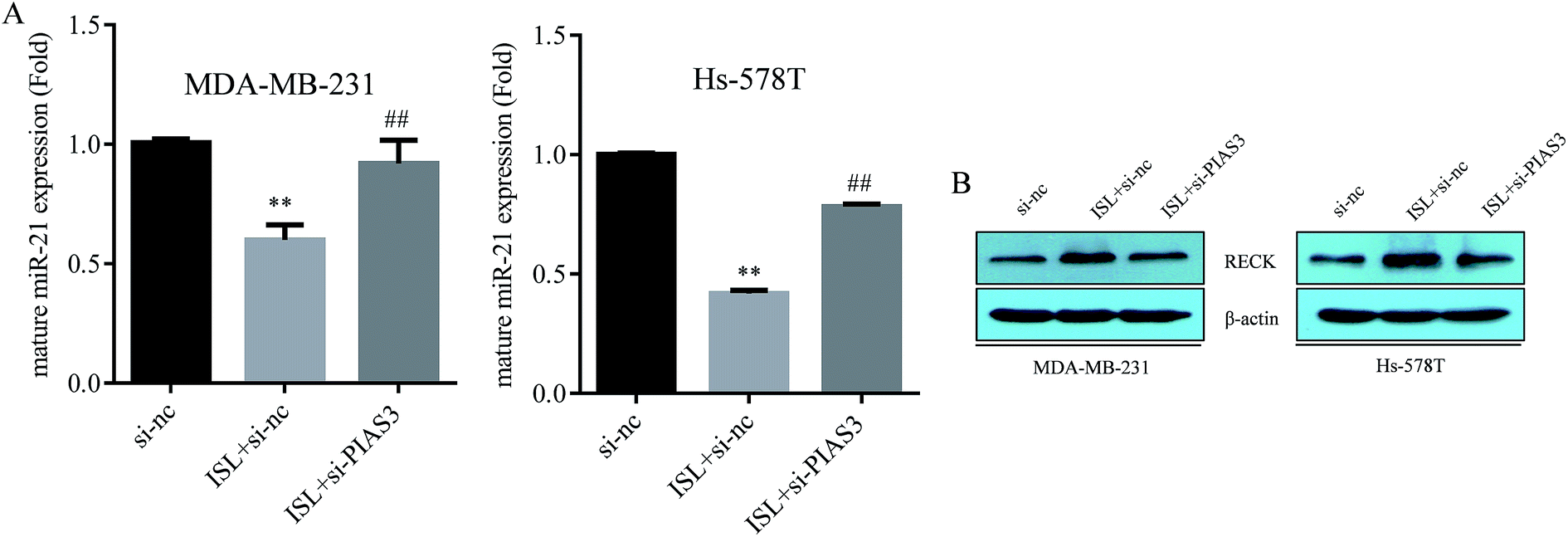 | ||
| Fig. 4 Induction of PIAS3 is crucial for the repression of miR-21 by ISL. MDA-MB-231 and Hs-578T cells were transiently transfected with si-nc or si-PIAS3, then exposed to ISL at 0.0 or 10.0 μM for 24 h. (A) qRT-PCR and (B) western blotting analysis were then performed to examine the expression of mature miR-21 and RECK, respectively. **P < 0.01 compared with the si-nc-treated group and ##P < 0.01 compared with the si-nc-treated group with ISL (10.0 μM) exposure. | ||
3.5 Induction of PIAS3 mediates the repressive effect of ISL on cellular invasiveness
Our recent study showed that repression of miR-21 mediated the inhibitory effect of ISL cellular invasiveness.9 In the present study, as demonstrated in Fig. 5, ISL attenuated the invasive capacity of breast cancer cells, as determined by invasion assay; however, perturbance of PIAS3 abrogated such effects. These results suggested that induction of PIAS3 mediated the repressive effect of ISL on cellular invasiveness. | ||
| Fig. 5 Induction of PIAS3 mediates the repressive effect of ISL on cellular invasiveness. MDA-MB-231 and Hs-578T cells were transiently transfected with si-nc or si-PIAS3, then exposed to ISL at 0.0 or 10.0 μM for 24 h. (A) Invasion assay was carried out. (B) Invaded cell number was determined (mean ± SD, n = 5). **P < 0.01 compared with non-treated cells and ##P < 0.01 compared with the si-nc-transfected cells treated with ISL at 10.0 μM. | ||
4 Discussion
Based on the evidence presented in our research, we supplied continued information on the anti-tumor mechanisms of ISL. Indeed, we firstly found that miR-21 was transcriptionally down-regulated by ISL treatment in breast cancer cells. Secondly, the inhibition of STAT3 signaling pathway was involved in the attenuation of miR-21 by ISL. Moreover, not the phosphorylation course but the DNA binding activity of STAT3 was responsible for the deficiency of STAT3 signaling caused by ISL. Lastly, further evidence showed that induction of PIAS3, an endogenous inhibitor of STAT3 pathway, was participated in the repressive effect of ISL on STAT3 DNA binding capacity. These results contributed to clear elucidation of the completed signaling caused by ISL in breast cancer cells.Quiescent STAT3 is activated via phosphorylation induced by appropriate stimulation and the STAT3 signaling could be modulated in many steps including phosphorylation by Jak and subsequent nuclear translocation even DNA binding. A number of endogenous inhibitors of STAT3 pathway has been discovered. In vitro studies, suppressor of cytokine signaling 1–3 (SOCS1–3) also known as STAT-induced STAT inhibitor (SSI1–3) could negatively regulate Jak-STAT3 pathway through direct interaction with activated Jak by binding to the activation loop in a phosphorylation-dependent manner, leading to deficiency in STAT3 phosphorylation.25–27 In addition, phosphatases responsible tyrosine dephosphorylation also play a critical role in suppression of IL-6 cytokine signaling via removal of phosphorylation group at specific site of STAT3.28 In our study, the data of luciferase reporter assay demonstrated that the activity of signaling drived by STAT3 in breast cancer cells was decreased due to ISL exposure. However, neither the phosphorylation status nor cellular location of STAT3 was affected by ISL treatment, demonstrated by western blotting and immunofluorescence staining, ruling out the involvement of SOCSs and tyrosine phosphatases. Moreover, our data presented in ESI Fig. 2† showed no obvious alteration on SOCS1 mRNA expression upon ISL treatment in breast cancer cells. Actually, neither phosphorylation nor translocation of STAT3 could represent the activation of STAT3 signaling because DNA binding was the last and vital step of STAT3 pathway. Thus, using plasmid reporter of STAT3 or examining the transcription of STAT3 target genes might be more intuitionistic and reasonable for monitoring the activity of STAT3 signaling pathway. In our study, we did not observe any alteration on phosphorylation or nuclear translocation of STAT3, however, our ChIP data showed that ISL down-regulated the enrichment of phosphorylated STAT3 at miR-21 promoter, indicating impaired DNA binding activity at post-phosphorylational level. PIAS3 acted as a negative regulator of STAT3 via inhibiting its DNA binding activity.13–15 Our subsequent results showed obvious restoration of PIAS3 in ISL-treated breast cancer cells. Additionally, PIAS3, the specific inhibitor of activated STAT3, served as the vital mediator during inhibition of the DNA binding activity of activated STAT3 and consequent miR-21 expression by ISL. Artificial restoration of PIAS3 in tumor-derived cell lines reversed STAT3-driven malignant phenotypes such as the highly invasive capacity and uncontrolled cell proliferation.29 Our data of invasion assay clearly showed that inhibition of cellular invasiveness by ISL was reversed upon PIAS3 perturbance with siRNA, which further verified the results of previous investigation focused on the function of PIAS3 on invasive nature of tumor cells.
In addition to STAT3, NF-κB, another critical transcriptional factor in tumor biology, could also bind to the promoter of miR-21, leading to transcriptional activation of miR-21.30 Moreover, previous evidence demonstrated that ISL could inactivate the DNA binding activity of NF-κB.31 Although initially identified as an inhibitor of STAT3 signaling, latter research established that PIAS3 could also block the signaling mediated by NF-κB via physical interaction with the P65/RelA subunit.32 It is rational to speculate that inhibition of NF-κB pathway by PIAS3 may also be involved in miR-21 repression by ISL, which needs further investigation.
In the present study, PIAS3 was certified as the upstream of miR-21. Recently, however, PIAS3 was identified as a direct target of miR-21 in multiple myeloma cells.33 Based on our and others' data, a negative regulation loop may exist between PIAS3 and miR-21.
In our study, we did not provide any evidence on the mechanisms underlying the up-regulation of PIAS3 by ISL. Former research suggested that exposure of lung cancer cells to 5-azacytidine, a highly effective inhibitor of DNA methylation, resulted in a significant increase in PIAS3 mRNA and protein expression, indicating the involvement of DNA methylation in PIAS3 deregulation in cancer cells.34 Reactive oxygen species (ROS)-induced oxidative stress could silence certain tumor suppressors via epigenetic mechanisms such as DNA methylation and histone modification, resulting in tumor progression.35 Besides, recruitment of DNA methyltransferases (DNMTs) to gene promoter plays a vital role in ROS-mediated DNA methylation.36 As previously confirmed, ISL could function as a naturally occurring anti-oxidant through eliminating intracellular ROS production.37 And demethylation was previously confirmed as the underling mechanism whereby ISL induced WIF1, an endogenous inhibitor of Wnt signaling.22 Based on these observations, promoter demethylation might be involved in the induction of PIAS3 by ISL through decreasing ROS level.
Taken together, our study showed transcriptional inhibition of miR-21 expression was found in MDA-MB-231 and Hs-578T breast cancer cells exposed to ISL. And PIAS3 was firstly identified as an important mediator in miR-21 intervention by ISL, leading to failure in tumor progression. By unmasking a novel mechanism by which ISL, a phytochemical with low cytotoxicity to normal cells, negatively regulates miR-21 via induction of PIAS3, our study not only expands understanding of the anti-tumor mechanisms of ISL, but also provides an alternative for the treatment of breast cancer.
Author contribution
Shilong Ning, Xiao Ma, and Zhong Li were involved in the study design. Shilong Ning, Xiao Ma, Dongmei Zhu and Zhaoxia Shen conducted the experiments and the statistical analysis. Shilong Ning and Xiao Ma wrote the first draft of the manuscript. Shilong Ning, Xiao Ma, Jiao Liu, Juan Chen and Zhong Li participated in manuscript editing. All authors contributed to the final version of the manuscript. Zhong Li supervised the study and contributed to the preparation of the manuscript.Conflict of interest statement
The authors do not have any competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81171987, 81673205), the Major Program of Natural Science Research of Jiangsu Higher Education Institutions (15KJA330001), the Research Fund for the Doctoral Program of Higher Education of China (20133234110007), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The funding agencies had no role in the study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.References
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- S. Valastyan and R. A. Weinberg, Cell, 2011, 147, 275–292 CrossRef CAS PubMed.
- A. J. Redig and S. S. McAllister, J. Intern. Med., 2013, 274, 113–126 CrossRef CAS PubMed.
- L. A. Yates, C. J. Norbury and R. J. Gilbert, Cell, 2013, 153, 516–519 CrossRef CAS PubMed.
- V. D. Haakensen, V. Nygaard, L. Greger, M. R. Aure, B. Fromm, I. R. Bukholm, T. Luders, S. F. Chin, A. Git, C. Caldas, V. N. Kristensen, A. Brazma, A. L. Borresen-Dale, E. Hovig and A. Helland, Int. J. Cancer, 2016, 139, 1117–1128 CrossRef CAS PubMed.
- A. M. McDermott, H. M. Heneghan, N. Miller and M. J. Kerin, Pharm. Res., 2011, 28, 3016–3029 CrossRef CAS PubMed.
- R. Gambari, E. Brognara, D. A. Spandidos and E. Fabbri, Int. J. Oncol., 2016, 49, 5–32 CAS.
- W. Wang, J. Li, W. Zhu, C. Gao, R. Jiang, W. Li, Q. Hu and B. Zhang, BMC Cancer, 2014, 14, 819 CrossRef PubMed.
- S. Ning, J. Mu, Z. Shen, D. Zhu, F. Jiang, X. Wang, Y. Li and Z. Li, RSC Adv., 2016, 6, 24719–24727 RSC.
- C. F. Chiu, Y. W. Chang, K. T. Kuo, Y. S. Shen, C. Y. Liu, Y. H. Yu, C. C. Cheng, K. Y. Lee, F. C. Chen, M. K. Hsu, T. C. Kuo, J. T. Ma and J. L. Su, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2526–E2535 CrossRef CAS PubMed.
- T. A. Zimmers, M. L. Fishel and A. Bonetto, Semin. Cell Dev. Biol., 2016, 54, 28–41 CrossRef CAS PubMed.
- K. Shuai, Cell Res., 2006, 16, 196–202 CrossRef CAS PubMed.
- C. D. Chung, J. Liao, B. Liu, X. Rao, P. Jay, P. Berta and K. Shuai, Science, 1997, 278, 1803–1805 CrossRef CAS PubMed.
- C. Levy, Y. N. Lee, H. Nechushtan, O. Schueler-Furman, A. Sonnenblick, S. Hacohen and E. Razin, Blood, 2006, 107, 2839–2845 CrossRef CAS PubMed.
- P. Starkel, C. De Saeger, I. Leclercq, A. Strain and Y. Horsmans, J. Hepatol., 2005, 43, 687–695 CrossRef PubMed.
- S. Dabir, A. Kluge, A. Kresak, M. Yang, P. Fu, B. Groner, G. Wildey and A. Dowlati, Clin. Cancer Res., 2014, 20, 5124–5132 CrossRef CAS PubMed.
- J. Li, J. Cui, J. Zhang, Y. Liu, L. Han, C. Jia, J. Deng and H. Liang, Int. J. Clin. Exp. Med., 2015, 8, 682–689 CAS.
- S. F. Yang, M. F. Hou, F. M. Chen, F. Ou-Yang, Y. C. Wu, C. Y. Chai and Y. T. Yeh, BMC Cancer, 2016, 16, 20 CrossRef PubMed.
- A. Bishayee and G. Sethi, Semin. Cancer Biol., 2016, 40–41, 1–3 CrossRef CAS PubMed.
- D. F. Romagnolo, K. D. Daniels, J. T. Grunwald, S. A. Ramos, C. R. Propper and O. I. Selmin, Mol. Nutr. Food Res., 2016, 60, 1310–1329 CAS.
- X. Y. Jin, D. H. Sohn and S. H. Lee, Arch. Pharmacal Res., 2016, 39, 1465–1471 CrossRef CAS PubMed.
- N. Wang, Z. Wang, Y. Wang, X. Xie, J. Shen, C. Peng, J. You, F. Peng, H. Tang, X. Guan and J. Chen, Oncotarget, 2015, 6, 9854–9876 CrossRef PubMed.
- L. Han, X. Yue, X. Zhou, F. M. Lan, G. You, W. Zhang, K. L. Zhang, C. Z. Zhang, J. Q. Cheng, S. Z. Yu, P. Y. Pu, T. Jiang and C. S. Kang, CNS Neurosci. Ther., 2012, 18, 573–583 CrossRef CAS PubMed.
- C. McClure, M. B. McPeak, D. Youssef, Z. Q. Yao, C. E. McCall and M. El Gazzar, Immunol. Cell Biol., 2017, 95, 42–55 CrossRef CAS PubMed.
- R. Starr, T. A. Willson, E. M. Viney, L. J. Murray, J. R. Rayner, B. J. Jenkins, T. J. Gonda, W. S. Alexander, D. Metcalf, N. A. Nicola and D. J. Hilton, Nature, 1997, 387, 917–921 CrossRef CAS PubMed.
- S. E. Nicholson, T. A. Willson, A. Farley, R. Starr, J. G. Zhang, M. Baca, W. S. Alexander, D. Metcalf, D. J. Hilton and N. A. Nicola, EMBO J., 1999, 18, 375–385 CrossRef CAS PubMed.
- J. U. Kazi, N. N. Kabir, A. Flores-Morales and L. Ronnstrand, Cell. Mol. Life Sci., 2014, 71, 3297–3310 CrossRef CAS PubMed.
- C. J. Greenhalgh and D. J. Hilton, J. Leukocyte Biol., 2001, 70, 348–356 CAS.
- X. Dai, K. S. Ahn, C. Kim, K. S. Siveen, T. H. Ong, M. K. Shanmugam, F. Li, J. Shi, A. P. Kumar, L. Z. Wang, B. C. Goh, J. Magae, K. M. Hui and G. Sethi, Mol. Oncol., 2015, 9, 818–833 CrossRef CAS PubMed.
- Y. J. Jeon, J. Middleton, T. Kim, A. Lagana, C. Piovan, P. Secchiero, G. J. Nuovo, R. Cui, P. Joshi, G. Romano, G. Di Leva, B. K. Lee, H. L. Sun, Y. Kim, P. Fadda, H. Alder, M. Garofalo and C. M. Croce, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3355–E3364 CrossRef CAS PubMed.
- K. L. Wang, S. M. Hsia, C. J. Chan, F. Y. Chang, C. Y. Huang, D. T. Bau and P. S. Wang, Expert Opin. Ther. Targets, 2013, 17, 337–349 CrossRef CAS PubMed.
- H. D. Jang, K. Yoon, Y. J. Shin, J. Kim and S. Y. Lee, J. Biol. Chem., 2004, 279, 24873–24880 CrossRef CAS PubMed.
- Q. Xiong, Q. Zhong, J. Zhang, M. Yang, C. Li, P. Zheng, L. J. Bi and F. Ge, J. Proteome Res., 2012, 11, 2078–2090 CrossRef CAS PubMed.
- A. Kluge, S. Dabir, I. Vlassenbroeck, R. Eisenberg and A. Dowlati, Mol. Oncol., 2011, 5, 256–264 CrossRef CAS PubMed.
- R. Zhang, K. A. Kang, K. C. Kim, S. Y. Na, W. Y. Chang, G. Y. Kim, H. S. Kim and J. W. Hyun, Gene, 2013, 524, 214–219 CrossRef CAS PubMed.
- Q. Wu and X. Ni, Curr. Drug Targets, 2015, 16, 13–19 CrossRef CAS PubMed.
- F. Peng, Q. Du, C. Peng, N. Wang, H. Tang, X. Xie, J. Shen and J. Chen, Phytother. Res., 2015, 29, 969–977 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25511f |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |