DOI:
10.1039/C6RA25501A
(Paper)
RSC Adv., 2017,
7, 5649-5659
Theoretical prediction of the synthesis of 2,3-dihydropyridines through Ir(III)-catalysed reaction of unsaturated oximes with alkenes†
Received
19th October 2016
, Accepted 5th January 2017
First published on 17th January 2017
Abstract
In spite of their widespread use as catalysts, 1,2,3,4,5-pentamethylcyclopentadienyl (Cp*) iridium complexes have been rarely employed in the synthesis of pyridine derivatives. Herein, we used density functional theory (DFT) calculations to predict the [Cp*Ir(OAc)]+-catalysed synthesis of 2,3-dihydropyridines, which are important starting materials for pharmaceuticals, from α,β-unsaturated oxime pivalates and alkenes. The corresponding Cp*Rh complex-catalysed processes were discussed in comparison. The simulated catalytic cycle consists of several elementary reactions, such as reversible acetate-assisted metalation–deprotonation, migratory insertion of the alkene, pivaloyl transfer, and reductive elimination. The migratory insertion of the alkene is identified as the rate-determining step, and the reductive elimination to furnish the product-ligated species makes the reaction irreversible (exergonic by about 48 kcal mol−1). The stabilities of the intermediates and the energy barrier of migratory insertion of the alkene can be affected by introducing substituent groups with different electronic characteristics into Cp* and the 2-position of α,β-unsaturated oxime pivalates, as well as by using polarised alkenes. The apparent activation energy of the reaction can be increased by increasing the electron-donating ability of the substituent group on Cp*, and by introducing electron-withdrawing groups into the terminus of alkenes. When a strong electron-donating group such as the amido group is introduced into the 2-position of α,β-unsaturated oxime pivalates, the apparent activation energy is greatly reduced so that the reaction can occur at room temperature. In contrast, changing phenyl into the highly electron-deficient p-CF3-phenyl makes the reaction more difficult. Diastereoselectivity of the reaction was examined using cyclohexylethylene as a substrate, and a method for enhancing diastereocontrol was suggested.
Introduction
Iridium and rhodium-catalysed reactions, such as (de)hydrogenation, olefin functionalisation, cyclisation, and C–H activation have attracted much attention, due to their high efficiency, broad substrate tolerance, and high stereoselectivity.1–5 Li et al. have pointed out that Ir and Rh complexes as efficient catalysts have complementary substrate scopes in the alkynylation of arenes.6 Ozawa et al. described the different reactivities of Ir- and Rh-complexes toward ammonia due to thermodynamic reasons.7 In carbine carbonylation reactions, Ir- and Rh-complexes also show distinct catalytic performance.8 Various coordination models for Ir and Rh metals with pincer ligands have been reported.9 In particular, 1,2,3,4,5-pentamethylcyclopentadienyl M(III) (Cp*M(III), M = Ir, Rh) complexes are important catalysts that deliver excellent performance for transfer (de)hydrogenation and C–H activation with high catalytic reactivity.1d,e,6,10–15 Davies et al. discovered that owing to electronic effects, Cp*Ir could catalyse the cyclometallation of benzylamine and alkyl and aryl imines at room temperature, while the Cp*Rh complex only works for imimes.16 Jin et al. reported that when using Cp*Ir, the insertion of internal and terminal alkynes into the Ir–C bond of the cycloirridation intermediate induced another regiospecific C–H activation, and that Cp*Rh catalysed the oxidative coupling of aromatic imines with an internal alkyne to give an indenone imine product.17 Singh et al. pointed out that the acetate-assisted cyclometallation with Cp*Rh is more sensitive to steric effects compared to that with Cp*Ir, and also thermodynamically less favoured.18 Among the substrates tested in these studies, oxime, triazole, imidazole, and amine work well with Cp*Ir but not with Cp*Rh, indicating the higher reactivity of the former for such reactions. In addition, Cp*Ir and Cp*Rh complexes have been used as supramolecular building blocks.1e,19,20 Thus, iridium-based complexes not only efficiently emulate the known catalytic capabilities of rhodium-based ones, but also exhibit additional unique activities. These findings urge us to explore the possibility of replacing Rh- with Ir-complexes in the catalytic reactions.
Nitrogen-containing heterocycles, such as pyridine and dihydropyridine derivatives, are the most prevalent structural components in both natural products and pharmaceuticals.21,22 Thus, their syntheses have been extensively explored, especially through transition metal (Co, Ni, Cu, Ru, Rh, Pd, and Au)-catalysed C–H activation pathways with external or internal oxidants.23–26 Owing to their high efficiency, broad substrate scope, and high functional tolerance, Rh complexes with pivaloyl oximes as internal oxidants have been frequently applied to the catalytic synthesis of pyridines and dihydropyridines.27–29 Although 2,3-dihydropyridines are not found in bioactive molecules due to their kinetic instability, they are regarded as versatile intermediates in biosynthetic methods. They can also be reduced to piperidines, which are important building blocks for numerous pharmaceuticals.30–33 Recently, Rovis and co-workers reported the synthesis of 2,3-dihydropyridines through the Cp*Rh complex-catalysed reaction of unsaturated oximes with alkenes, which features a broad range of substrates and high diastereoselectivity but requires a temperature of 50–60 °C.34 The cation [Cp*RhOAc]+ is the true active species for this reaction,35 which has been suggested to consist of concerted metalation–deprotonation (CMD), migratory insertion of alkene into the Rh–C bond, pivaloyl transfer to the Rh centre, and reductive elimination, among other steps.34,36 In 2008, Jones et al. reported the synthesis of polycyclic isoquinoline salt through C–H activation with Cp*M (M = Ir, Rh) complexes.37 However, compared to Cp*Rh, there are few examples of synthesising pyridines and dihydropyridines through Cp*Ir complex-catalysed C–H activation. In order to further explore the application of Cp*Ir complexes in the synthesis of nitrogen heterocyclic compounds, herein we theoretically study the feasibility of synthesising 2,3-dihydropyridines through the reaction of unsaturated oxime pivalates with alkenes catalysed by [Cp*IrOAc]+ in place of [Cp*RhOAc]+.
Our simulated catalytic cycle is shown in Scheme 1. The unsaturated oxime pivalate a1 coordinates to the active species followed by a CMD process, releasing the formed acetic acid molecule to produce the five-membered iridacycle 4. This is followed by the coordination and migratory insertion of alkene b1 into the Ir–C bond. The resultant seven-membered metallacycle 6d undergoes a conformational change, pivaloyl transfer to the Ir centre, and the subsequent reductive elimination, eventually furnishing 2,3-dihydropyridines and regenerating the active species. In order to reveal the effects of substitutions on the catalyst and substrates and investigate the possible diastereoselectivity, the following substituted species were studied: catalysts bearing CF3, CH3, NH2, N(CH3)2, tertiary butyl (tBu) groups on Cp*; α,β-unsaturated oxime pivalates bearing CH3, NH2, phenyl, and p-CF3-phenyl groups at the 2-position; and alkenes bearing NH2, Cl, CO2Et, and cyclohexyl (Ch) groups at the terminus.
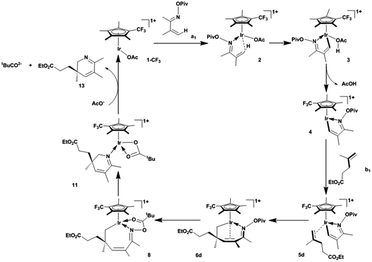 |
| | Scheme 1 Simulated catalytic cycle. | |
Computational details
All calculations were performed using the G09 program package,38 and influences of hydrogen bonds were not taken into account. The molecular structures were optimised by density functional theory (DFT) calculation using the B3LYP/BS1 method.38a,39,40 In the G09 program, there are no physical parameters pertaining to hexafluoroisopropanol (HFIP),38 which is used experimentally for the Cp*Rh complex-catalysed reaction.34 With 2-propanol instead of HFIP, the calculated results for this reaction are in good agreement with the experimental observations.36 Therefore, in order to compare to the catalytic performance of the Cp*Rh complex in this work, 2-propanol was also used to simulate the solvent effects, by invoking the PCM solvation model.41 The basis set (BS1) was defined as follows. Ir atoms are described with the LANL2DZ basis sets with ECP, which are modified by Couty and Hall.42 C and N atoms directly connected to Ir (including those involved in the conjugated π bonds of α,β-unsaturated oxime pivalates and alkenes), H atoms connected to the unsaturated π bonds, C atoms of the carbonyls of oxime pivalates, and the O atoms are described with the 6-311+G(d,p) basis sets.43 The other atoms are represented by the 6-31G basis sets, except for F, N, and Cl atoms in the substituent groups which are described with the 6-31+G(d) basis sets. To validate our results, we calculated the Cp*Ir complex M using the same method (see the ESI†), and the obtained main bond parameters are in good agreement with X-ray data.44a The method described here has also been used in similar studies.44b The frequency calculations were carried out using the optimised structures, in order to confirm that the obtained species are either energy minima (no imaginary frequency) or transition states (only one imaginary frequency), and to provide thermodynamic corrections at 1 atm and 298.15 K. In addition, intrinsic reaction coordinate (IRC) calculations were performed to ensure that the obtained transition states are correctly connected to the intermediates.45 Finally, more accurate energies were calculated using the 6-311G(d,p) in place of 6-31G basis sets (BS2). In order to take the dispersion effects into account, the B3LYP-D3/BS2//B3LYP/BS1 calculations were carried out at the same time.1f,38b,46 Natural bond orbital (NBO) analysis was performed at the stationary points, and the natural charges and Wiberg bond indices (WBIs) were computed by the NBO program as implemented in the G09 package.38a Corrected Gibbs free energies were used to describe the energetic profiles of the reaction. For the cationic system (the counter anion is ignored in this study), the dispersion effects were much overestimated. Hence, we only discuss these effects on the main energy barriers with great care, and hope to obtain some insights into how the dispersion effects influence the elementary reaction steps.
Results and discussion
Energetic profiles of the reaction catalysed by 1-CF3
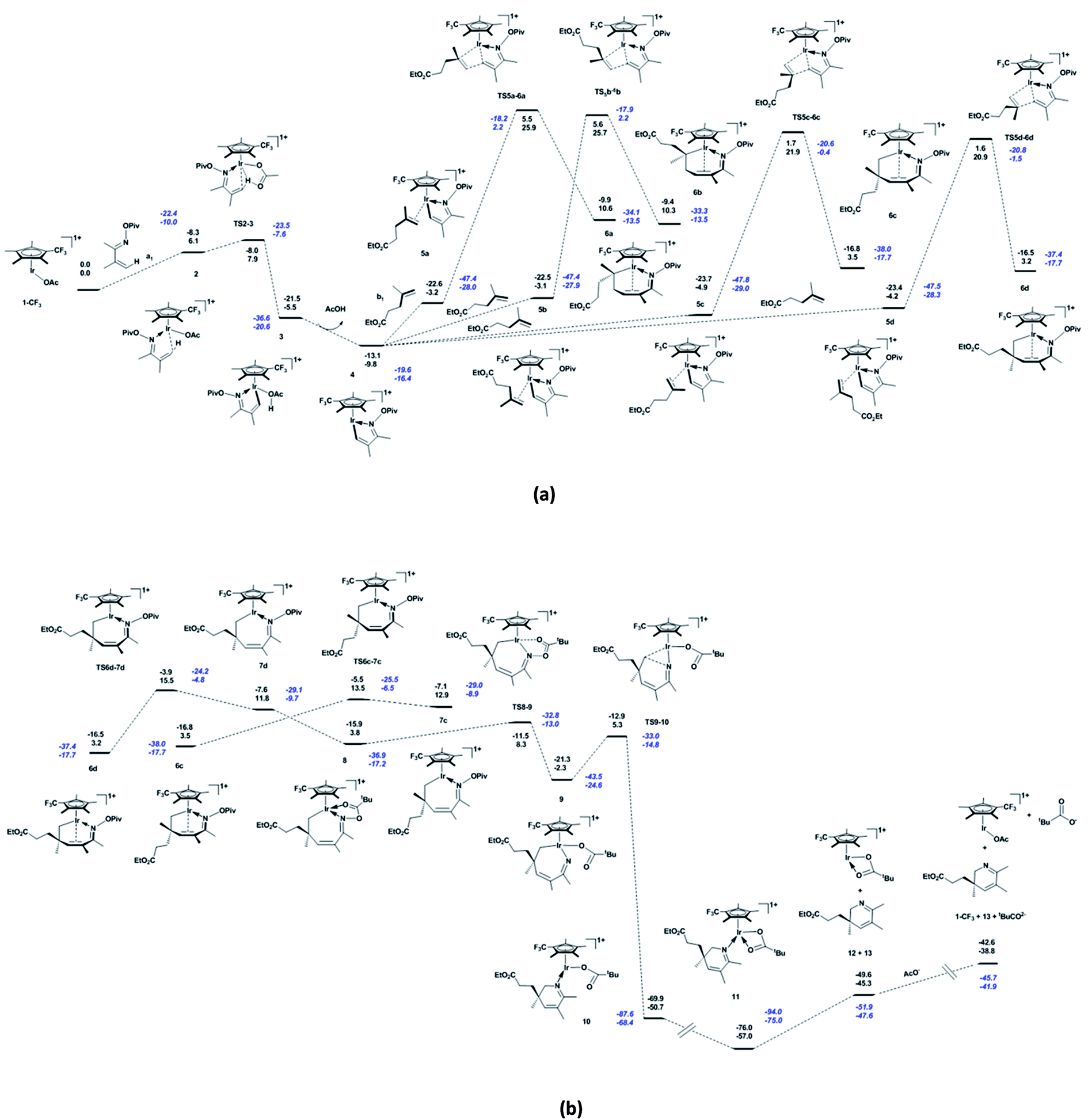
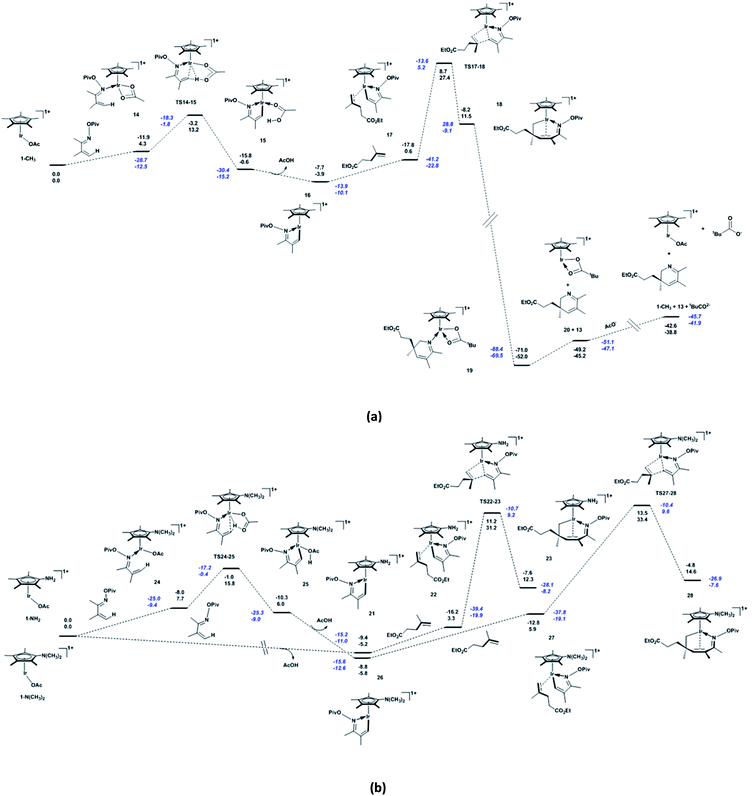
Based on the catalytic cycle shown in Scheme 1, we calculated the reaction of unsaturated oxime a1 (bearing CH3 at the 2- and 3-positions) with alkene b1 (bearing the ester group CO2Et at the terminus) catalysed by 1-CF3 (bearing a CF3 substitution on Cp*). As shown in Fig. 1a, the unsaturated oxime pivalate a1 coordinates to the active species 1-CF3 with the endergonicity of 6.1 kcal mol−1, the system entropy is reduced by 47.7 cal mol−1 K−1. In 2, there is an agostic interaction between the Ir atom and the C1–H1 bond, as evidenced by the length of the Ir–H1 bond (2.18 Å, see Fig. S1a in ESI†), the calculated interaction energy between the C1–H1 bonding orbital and the valence p orbital on Ir is 30.2 kcal mol−1, and the WBI of the Ir–N bond is calculated to be 0.50. Then, C–H activation occurs through an acetate-assisted CMD process. The transition state TS2-3 (Ir–C1 = 2.18 Å, Ir–H1 = 2.21 Å, C1–H1 = 1.27 Å, and O4–H1 = 1.43 Å) is slightly higher in energy (by 1.8 kcal mol−1) than 2. The dispersion-corrected energy barrier is 2.4 kcal mol−1, which is larger than the uncorrected value of 1.8 kcal mol−1. Therefore, the dispersion effects increase the energy barrier of the acetate-assisted CMD process. The length of Ir–H1 in TS2-3 indicates that the Ir atom mediates the C–H activation. The coordination of the substrate and C–H activation result in an exergonicity of 5.5 kcal mol−1. The calculated energy barrier (1.8 kcal mol−1) and exergonicity (5.5 kcal mol−1) are comparable with those reported for the acetate-assisted C–H activation of dimethylbenzylamine (DMBA-H) catalysed by Cp*Ir complex (0.9 and 4.8 kcal mol−1, respectively).5a For further comparison, the corresponding theoretical values for the acetate-assisted C–H activation for imidazolium salt with Ir(COD)(OAc) (COD = cyclooctadiene) as catalyst are 2.2 and 4.5 kcal mol−1, respectively.5i The release of the formed acetic acid molecule is exergonic by 4.3 kcal mol−1, and this step furnishes the five-membered iridacycle 4 with a vacant site (Ir–C1 = 2.12 Å and Ir–H1 = 2.03 Å), followed by the coordination of substrate b1. The increase in entropy (42.8 cal mol−1 K−1) is responsible for the exergonicity. The formation of iridacycle 4 requires the additional energy of 7.9 kcal mol−1, and the resulting system exergonicity of 9.8 kcal mol−1 indicates the reversibility of this process. For the CF3-substituted Cp*Rh complex, the corresponding process requires 11.9 kcal mol−1 and releases 5.5 kcal mol−1 in energy.36 In the case of CF3-substituted Cp*Rh, the reversibility of the reaction is known to be supported by a primary kinetic isotopic effect (KIE = 1.41).34 These data clearly show that 1-CF3 is a more robust catalyst than the CF3-substituted Cp*Rh complex for the CMD process. There are four pathways for the alkene b1 to approach to form the four complexes labelled 5a–5d in Scheme 2. The system is endergonic by 4.9–6.7 kcal mol−1 and the entropy is reduced by 53.2 to 55.8 cal mol−1 K−1. The complexes 5a–5d are slightly different in energy (within 1.8 kcal mol−1 from each other), which is ascribed to a marginal clash between the substituent groups on Cp* and alkenes (in 5d, Ir–C5 = 2.21 Å and C1–C6 = 2.90 Å). The clash becomes stronger in the subsequent migratory insertion of alkene b1 into Ir–C1 in 5a and 5b, making the transition states TS5a-6a and TS5b-6b higher in energy (25.9 and 25.7 kcal mol−1, respectively) than TS5c-6c and TS5d-6d (21.9 and 20.9 kcal mol−1, respectively). Thus, the regioselective seven-membered iridacycle 6d is favoured over 6c. The slight energy difference between TS5c-6c and TS5d-6d (1.0 kcal mol−1) is attributed to the congestion of their substituent groups (see Fig. S1b in ESI†). In comparison to TS5c-6c, the entropy of TS5d-6d is higher by 3.9 cal mol−1 K−1. Taken together, the coordination and migratory insertion of the alkene b1 are endergonic by about 13.0 kcal mol−1 with an entropy change of −58.1 cal mol−1 K−1. In 6d, the Ir atom is coordinated by a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π bond (Ir–C1 = 2.25 Å and Ir–C2 = 2.30 Å) to satisfy the 18-electron rule. The CC π-bond coordination to Ir is confirmed by the WBIs of the Ir–C1 (0.40) and Ir–C2 (0.37) bonds. The Ir–C1 (C2) bonding orbital is mainly formed by the valence p orbital on C1 (or C2) and the valence d orbital on Ir. The WBIs of the Ir–C1 (0.50), Ir–C5 (0.68) and C6–C1 (0.66) bonds in TS5d-6d are close to those in 6d (0.40, 0.72, and 0.97, respectively), indicating that TS5d-6d is a late transition state. The calculated energy barriers for the migratory insertion of alkene into Ir–C in the iridacycle complex 4 are about 5.0 kcal mol−1 higher than those for the migratory insertion into Rh–C in the corresponding rhodacycle.36 However, between the corresponding iridacycle and rhodacycle complexes, the energy required for the coordination and migratory insertion of the alkene is only about 1.5 kcal mol−1 higher in the former. From Fig. 1a, it can be found that the dispersion effects increase the energy barrier and endergonicity of migratory insertion, especially for cases 5c and 5d. 6d undergoes a conformational change through TS6d-7d (Ir–C1 = 3.36 Å and Ir–C2 = 3.35 Å) to form another isomer 7d featuring an uncoordinated CC π bond, with the endergonicity of 8.6 kcal mol−1 (Fig. 1b) due to the lower stability of 7d compared to 6d. Compared to the migratory insertion of alkene, the conformational change requires less energy (12.3 versus 25.1 kcal mol−1) and is less affected by the dispersion effects (0.6 versus 1.7 kcal mol−1). The value of 12.3 kcal mol−1 is 0.7 kcal mol−1 higher than that for the conformational change in the corresponding cyclometalated, CF3-substituted Cp*Rh complex (11.6 kcal mol−1).36 Through rotation about the N–O bond, the O atom of the pivaloyl in 7d coordinates to the Ir centre to form 8 (Ir–O2 = 2.20 Å and N–O1 = 1.46 Å) that satisfies the 18-electron rule. Through this step, the system is exergonic by 8.0 kcal mol−1 and the entropy is lowered by 2.0 cal mol−1 K−1. The conformational change and coordination of the O atom prepare for the subsequent pivaloyl transfer to the Ir centre through the N–O bond cleavage. 8 is converted to 9 with 16 valence electrons (Ir–O2 = 2.09 Å and Ir–N = 1.83 Å) via TS8-9 (Ir–O2 = 2.14 Å and N–O1 = 1.84 Å) by breaking the N–O bond, with an energy barrier of 4.5 kcal mol−1. At the same time, the Ir(III) centre in iridacycle 8 is oxidised to Ir(V). The system is exergonic by 6.1 kcal mol−1, accompanied by an entropy increase of 4.2 cal mol−1 K−1. In this process, natural charge gradually accumulates on the Ir centre (0.387 for 8, 0.483 for TS8-9, and 0.613 for 9), indicating changing oxidation state of the Ir centre. In 8, there is a stronger interaction (75.8 kcal mol−1) between the lone pair on N and the sp3 hybrid orbital on Ir with a WBI value of 0.51, while in 9 the Ir–N is a polarised σ-bond (73.7% from the sp hybrid orbital on N) and the WBI is 1.18. The transfer of the pivaloyl in the iridacycle is more favourable here than in the corresponding rhodacycle complex (4.5 versus 9.4 kcal mol−1).36 The dispersion effects are marginal for these elementary reactions, e.g. isomerisation of the seven-membered iridacycle 7d and pivaloyl transfer to the Ir centre. Then, 9 is converted to 10 via TS9-10 (Ir–N = 1.87 Å, Ir–C5 = 2.47 Å, and N–C5 = 2.36 Å), accompanied by the formation of the C–N and breakage of the Ir–C bonds. This reductive elimination step has an energy barrier of 7.6 kcal mol−1. This energy barrier is increased by 2.2 kcal mol−1, owing to the dispersion effects. It furnishes the product-ligated species, and the exergonicity of 48.4 kcal mol−1 makes the reaction irreversible. The large exergonicity comes from releasing the tension in the seven-membered ring, and reducing the steric congestion in 9. However, the dispersion effects make the system less exergonic by 4.6 kcal mol−1. For this process, the system entropy change is −2.1 cal mol−1 K−1. In TS9-10, the natural charge on Ir is 0.525 below that in the case of 9. The WBIs of the Ir–C5, Ir–N and C5–N bonds are 0.42, 0.94, and 0.35, respectively, indicating that TS9-10 is an early transition state. In comparison, the reductive elimination in the corresponding rhodacycle complex is more favourable, as it has a lower energy barrier (3.0 kcal mol−1) and a larger exergonicity (59.4 kcal mol−1).36 When 10 with the ƞ1-pivaloyl is converted to 11 with the ƞ2-pivaloyl to satisfy the 18-electron rule, the system continues to release energy (6.3 kcal mol−1) and the entropy is slightly increased by 0.3 cal mol−1 K−1. Subsequently, 11 dissociates into complex 12 and 2,3-dihydropyridine 13, with the endergonicity of 11.7 kcal mol−1. The entropy of the system is increased by 49.3 cal mol−1 K−1. In order to close the catalytic cycle, the pivaloyl is replaced by an external acetate anion to regenerate the active species 1-CF3, and the system is endergonic by 6.5 kcal mol−1.
C π bond (Ir–C1 = 2.25 Å and Ir–C2 = 2.30 Å) to satisfy the 18-electron rule. The CC π-bond coordination to Ir is confirmed by the WBIs of the Ir–C1 (0.40) and Ir–C2 (0.37) bonds. The Ir–C1 (C2) bonding orbital is mainly formed by the valence p orbital on C1 (or C2) and the valence d orbital on Ir. The WBIs of the Ir–C1 (0.50), Ir–C5 (0.68) and C6–C1 (0.66) bonds in TS5d-6d are close to those in 6d (0.40, 0.72, and 0.97, respectively), indicating that TS5d-6d is a late transition state. The calculated energy barriers for the migratory insertion of alkene into Ir–C in the iridacycle complex 4 are about 5.0 kcal mol−1 higher than those for the migratory insertion into Rh–C in the corresponding rhodacycle.36 However, between the corresponding iridacycle and rhodacycle complexes, the energy required for the coordination and migratory insertion of the alkene is only about 1.5 kcal mol−1 higher in the former. From Fig. 1a, it can be found that the dispersion effects increase the energy barrier and endergonicity of migratory insertion, especially for cases 5c and 5d. 6d undergoes a conformational change through TS6d-7d (Ir–C1 = 3.36 Å and Ir–C2 = 3.35 Å) to form another isomer 7d featuring an uncoordinated CC π bond, with the endergonicity of 8.6 kcal mol−1 (Fig. 1b) due to the lower stability of 7d compared to 6d. Compared to the migratory insertion of alkene, the conformational change requires less energy (12.3 versus 25.1 kcal mol−1) and is less affected by the dispersion effects (0.6 versus 1.7 kcal mol−1). The value of 12.3 kcal mol−1 is 0.7 kcal mol−1 higher than that for the conformational change in the corresponding cyclometalated, CF3-substituted Cp*Rh complex (11.6 kcal mol−1).36 Through rotation about the N–O bond, the O atom of the pivaloyl in 7d coordinates to the Ir centre to form 8 (Ir–O2 = 2.20 Å and N–O1 = 1.46 Å) that satisfies the 18-electron rule. Through this step, the system is exergonic by 8.0 kcal mol−1 and the entropy is lowered by 2.0 cal mol−1 K−1. The conformational change and coordination of the O atom prepare for the subsequent pivaloyl transfer to the Ir centre through the N–O bond cleavage. 8 is converted to 9 with 16 valence electrons (Ir–O2 = 2.09 Å and Ir–N = 1.83 Å) via TS8-9 (Ir–O2 = 2.14 Å and N–O1 = 1.84 Å) by breaking the N–O bond, with an energy barrier of 4.5 kcal mol−1. At the same time, the Ir(III) centre in iridacycle 8 is oxidised to Ir(V). The system is exergonic by 6.1 kcal mol−1, accompanied by an entropy increase of 4.2 cal mol−1 K−1. In this process, natural charge gradually accumulates on the Ir centre (0.387 for 8, 0.483 for TS8-9, and 0.613 for 9), indicating changing oxidation state of the Ir centre. In 8, there is a stronger interaction (75.8 kcal mol−1) between the lone pair on N and the sp3 hybrid orbital on Ir with a WBI value of 0.51, while in 9 the Ir–N is a polarised σ-bond (73.7% from the sp hybrid orbital on N) and the WBI is 1.18. The transfer of the pivaloyl in the iridacycle is more favourable here than in the corresponding rhodacycle complex (4.5 versus 9.4 kcal mol−1).36 The dispersion effects are marginal for these elementary reactions, e.g. isomerisation of the seven-membered iridacycle 7d and pivaloyl transfer to the Ir centre. Then, 9 is converted to 10 via TS9-10 (Ir–N = 1.87 Å, Ir–C5 = 2.47 Å, and N–C5 = 2.36 Å), accompanied by the formation of the C–N and breakage of the Ir–C bonds. This reductive elimination step has an energy barrier of 7.6 kcal mol−1. This energy barrier is increased by 2.2 kcal mol−1, owing to the dispersion effects. It furnishes the product-ligated species, and the exergonicity of 48.4 kcal mol−1 makes the reaction irreversible. The large exergonicity comes from releasing the tension in the seven-membered ring, and reducing the steric congestion in 9. However, the dispersion effects make the system less exergonic by 4.6 kcal mol−1. For this process, the system entropy change is −2.1 cal mol−1 K−1. In TS9-10, the natural charge on Ir is 0.525 below that in the case of 9. The WBIs of the Ir–C5, Ir–N and C5–N bonds are 0.42, 0.94, and 0.35, respectively, indicating that TS9-10 is an early transition state. In comparison, the reductive elimination in the corresponding rhodacycle complex is more favourable, as it has a lower energy barrier (3.0 kcal mol−1) and a larger exergonicity (59.4 kcal mol−1).36 When 10 with the ƞ1-pivaloyl is converted to 11 with the ƞ2-pivaloyl to satisfy the 18-electron rule, the system continues to release energy (6.3 kcal mol−1) and the entropy is slightly increased by 0.3 cal mol−1 K−1. Subsequently, 11 dissociates into complex 12 and 2,3-dihydropyridine 13, with the endergonicity of 11.7 kcal mol−1. The entropy of the system is increased by 49.3 cal mol−1 K−1. In order to close the catalytic cycle, the pivaloyl is replaced by an external acetate anion to regenerate the active species 1-CF3, and the system is endergonic by 6.5 kcal mol−1.
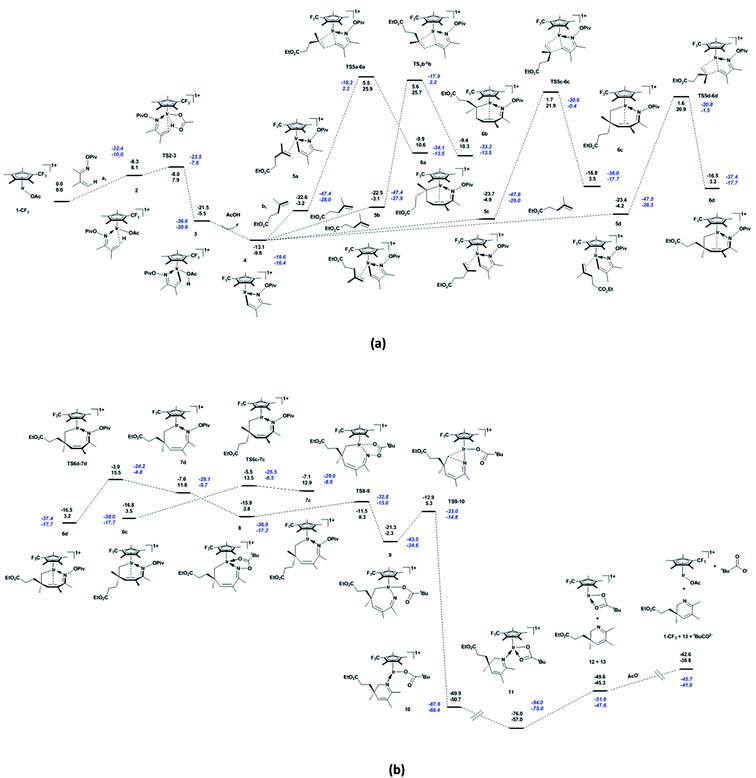 |
| | Fig. 1 (a) Energetic profile for the reaction catalysed by 1-CF3, including C–H activation and migratory insertion etc., and (b) conformational change, pivaloyl transfer, and reductive elimination. Born–Oppenheimer energies (top) and Gibbs free energies (bottom) are given in kcal mol−1, and values in blue italics include the dispersion correction. | |
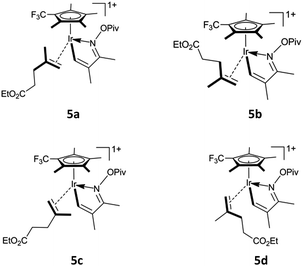 |
| | Scheme 2 Four approaching pathways for alkene b1. | |
By analysing the energetic profiles of the reaction, we find that the migratory insertion of alkenes into Ir–C is the rate-determining step, and the five-membered iridacycle 4 is a crucial intermediate for determining the apparent activation energy of the reaction. The differences between the calculated results for Cp*Ir and Cp*Rh complexes may be ascribed to the larger size of the Ir atom and the stronger relativistic effects in Ir compared to Rh.47 Based on the energetic span model and owing to the relatively smaller exergonicity, Cp*Ir is more efficient than Cp*Rh in the catalytic cycle of synthesising 2,3-dihydropyridines.48 Complex 6c undergoes similar processes, thus its energetic profile is not given here.
Effects of substituent groups on Cp*
Next, we replaced the active species 1-CF3 with 1-CH3, 1-NH2, and 1-N(CH3)2, which bear a Cp*, a NH2-substituted Cp*, and a N(CH3)2-substituted Cp*, respectively. The corresponding reaction steps, such as C–H activation and migratory insertion of the alkene b1 into Ir–C, were computationally evaluated. As shown in Fig. 2a, for 1-CH3, the acetate-assisted CMD process via TS14-15 (Ir–C1 = 2.17 Å, Ir–H1 = 2.21 Å, C1–H1 = 1.29 Å, and O4–H1 = 1.40 Å; see Fig. S2 in ESI†) requires more energy than for 1-CF3 (8.9 versus 1.8 kcal mol−1). These calculated results are in accordance with other reports for similar processes.5b,g,49 Compared to 1-CF3, the system with 1-CH3 is much less exergonic (11.6 versus 4.9 kcal mol−1). Similarly, for 1-N(CH3)2, the acetate-assisted CMD process via TS24-25 (Ir–C1 = 2.15 Å, Ir–H1 = 2.15 Å, C1–H1 = 1.34 Å, and O4–H1 = 1.36 Å) has an energy barrier of 8.1 kcal mol−1 (Fig. 2b), and releases a small amount of energy (1.7 kcal mol−1). Through comparing these substituted catalysts, the conclusion is that introducing electron-donating substituent group into Cp* increases the energy barrier of the acetate-assisted CMD process, and decreases the exergonicity of the system. The higher energy barriers for 1-CH3 than 1-N(CH3)2 are due to the higher stability of 14 (Ir–O3 = 2.22 Å and Ir–O4 = 2.25 Å) with the ƞ2-OAc. The energy barrier of these two steps, namely the coordination of unsaturated oxime a1 and subsequent CMD, is 15.8 kcal mol−1 higher for 1-N(CH3)2 than for 1-CH3 (13.2 kcal mol−1). In 14 and 24, the Ir centre is located far away from the C1–H1 bond (Ir–H1 = 3.27 Å for 14 and Ir–H1 = 3.22 Å for 24), indicating that they do not interact with each other. Introducing a stronger electron-donating substituent group into Cp* makes the Ir centre more electron-rich, and thereby has less stable intermediates, more difficult C–H activation, and stronger influence on the subsequent migratory insertion process. For 1-CH3, 1-NH2, and 1-N(CH3)2, the energy changes caused by the coordination of the alkene b1 to the five-membered iridacycles (16, 21, and 26) are 3.5, 8.5 (Fig. 2b), and 11.7 kcal mol−1; and the energy barriers of the subsequent migratory insertions are 26.8, 27.9, and 27.5 kcal mol−1, respectively. From 17 to 22 to 27, the congestion between the substituent groups increases (as evidenced by the decreasing Ir–C5 distance: 2.22 to 2.21 to 2.20 Å), which accounts for the trend of energy change during the coordination of the alkene. The small change in activation energies is ascribed to a concerted process of migratory insertion. The energy changes related to the coordination of the alkene are responsible for the different apparent activation energies of the reaction (31.3, 36.4, and 39.2 kcal mol−1, respectively). Obviously, the apparent activation energy of the reaction is increased by stronger electron-donating abilities of the substituent groups on Cp*. In addition, when using the bulky and more electron-donating tBu group in place of CH3, the required activation energy for the migratory insertion of the alkene b1 is lowered from 26.8 to 25.5 kcal mol−1. The reduced energy barrier in comparison to the case of 1-CH3 is ascribed to the larger size of tBu. Thus, bulky substituent groups on Cp* can reduce the activation energy barrier of migratory insertion of the alkenes for the title reaction.
 |
| | Fig. 2 Energetic profile for the reactions catalysed by (a) 1-CH3, (b) 1-NH2, and 1-N(CH3)2, including the C–H activation and migratory insertion steps. Born–Oppenheimer energies (top) and Gibbs free energies (bottom) are given in kcal mol−1, and values in blue italics include the dispersion correction. | |
Effects of different substituent groups on the substrates
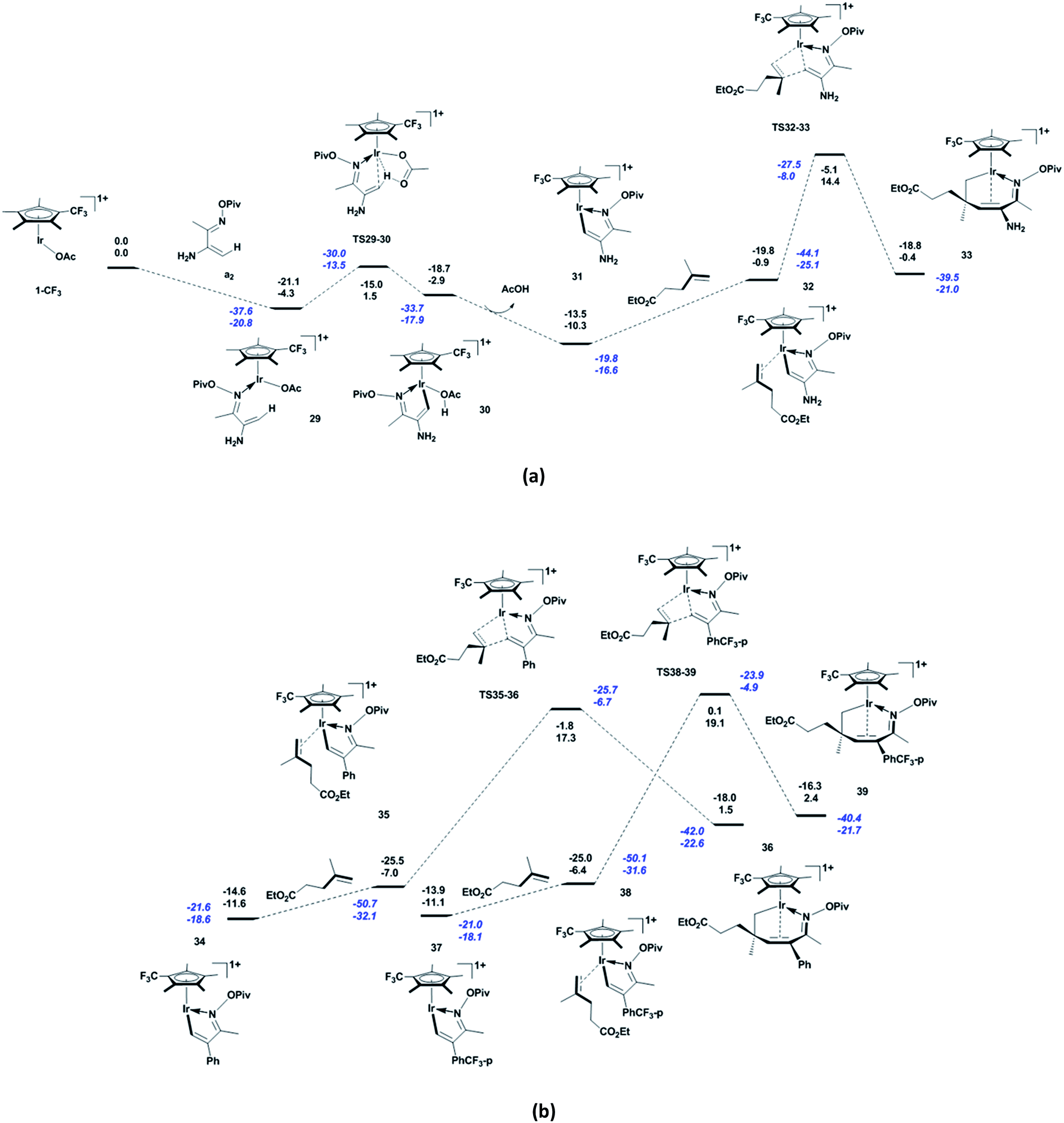
Compared to α,β-unsaturated oxime pivalate a1, the NH2-substituted a2 changes the shape of the energetic profile slightly. As shown in Fig. 3a, in contrast to a1, a2 coordinates to the active species 1-CF3 with the exergonicity of 4.3 kcal mol−1. In 29, the Ir and C1 atoms are close to each other (Ir–C1 = 2.21 Å, see Fig. S3 in ESI†), indicating their strong interaction. The subsequent acetate-assisted C–H activation via TS29-30 (Ir–C1 = 2.11 Å, Ir–H1 = 2.53 Å, C1–H1 = 1.35 Å, and O4–H1 = 1.26 Å) requires 5.8 kcal mol−1 energy to furnish the complex 30, and the system is 1.4 kcal mol−1 less endergonic than the case with a1. The distance between the Ir and H1 atoms indicates that Ir does not mediate the C–H activation. The release of the formed acetic acid molecule gives five-membered iridacycle 31 with the exergonicity of 7.4 kcal mol−1. Then, the alkene b1 coordinates to complex 31 to form 32 (Ir–C5 = 2.21 Å, C1–C6 = 2.91 Å, and C5–C6 = 1.41 Å), making the system endergonic by 9.4 kcal mol−1. To our delight, the migratory insertion of b1 into Ir–C via TS32-33 (Ir–C5 = 2.17 Å, C1–C6 = 2.15 Å, and C5–C6 = 1.45 Å) requires much less energy (i.e., 15.3 kcal mol−1) compared to a1. This is ascribed to the strong electron-donating ability of NH2 through conjugation with the system, which stabilises the transition state. In 33, the Ir–C1 and Ir–C2 distances (2.23 and 2.57 Å, respectively) show that the Ir atom interacts strongly with C1 but not with C2. Owing to the replacement of CH3 with NH2, the apparent activation energy of the reaction is reduced to 24.7 kcal mol−1. From the kinetic viewpoint, 1-CF3 is a more robust catalyst for substrate a2 than CF3-substituted Cp*Rh, because reaction with the latter requires a slightly higher apparent activation energy (25.5 kcal mol−1).36 Herein, we did not consider other possible reactions of NH2, because the aim here is understanding the influences of electronic effects of substituent groups on the title reaction. We believe that introducing a strong electron-donating substituent group into the 2-position of α,β-unsaturated oxime pivalate can allow the reaction to proceed at room temperature. In the case of phenyl-substituted unsaturated oxime pivalate, the alkene b1 coordinates to the five-membered iridacycle 34 to give 35 (Ir–C5 = 2.22 Å, C1–C6 = 2.91 Å, and C5–C6 = 1.40 Å), with the endergonicity of 4.6 kcal mol−1 (Fig. 3b). The subsequent migratory insertion of b1 into Ir–C via TS35-36 (Ir–C5 = 2.11 Å, C1–C6 = 1.86 Å, and C5–C6 = 1.49 Å) requires an energy of 24.3 kcal mol−1. In the case of unsaturated oxime pivalate with p-CF3-phenyl substitution, the formation of 38 (Ir–C5 = 2.22 Å, C1–C6 = 2.91 Å, and C5–C6 = 1.40 Å) through coordination of b1 to the five-membered iridacycle 37 has almost the same endergonicity (4.7 kcal mol−1) as that for phenyl-substituted unsaturated oxime pivalate. The migratory insertion of b1 into Ir–C via TS38-39 has an energy barrier of 25.5 kcal mol−1 (Ir–C5 = 2.10 Å, C1–C6 = 1.84 Å, and C5–C6 = 1.50 Å). Thus, introducing a more electron-withdrawing substituent group into the 2-position of unsaturated oxime pivalate increases the energy barrier of migratory insertion of alkenes, and further increases the apparent activation energy of the reaction. In the case of CF3-substituted Cp*Rh, similar effects of phenyl and p-CF3-phenyl substituent groups at the 2-position of α,β-unsaturated oxime pivalate have been confirmed by the Hammett study (ρ = −1.2).34
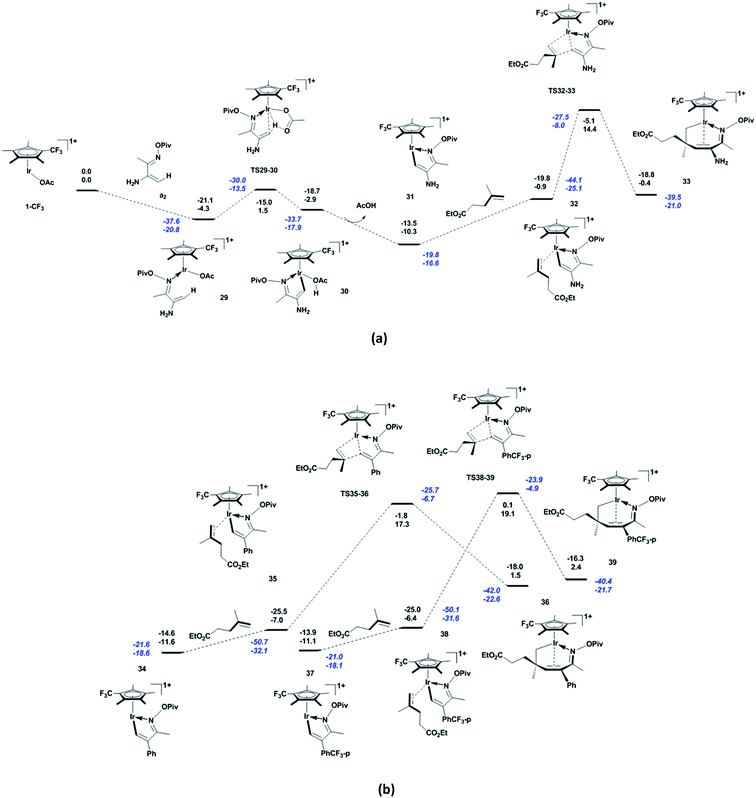 |
| | Fig. 3 Energetic profile for the reactions of (a) NH2-substituted and (b) phenyl-substituted and p-CF3-phenyl-substituted α,β-unsaturated oxime pivalates with alkene b1, including the C–H activation and migratory insertion of alkene. Born–Oppenheimer energies (top) and Gibbs free energies (bottom) are given in kcal mol−1, and values in blue italics include the dispersion correction. | |
When the alkene b2 or Cl,Cl,Cl-trisubstituted alkene b3 are used as substrates, the endergonicity of the system increases (4.2 and 5.2 kcal mol−1, respectively; Fig. 4a) through coordination of the alkenes to the five-membered iridacycle 4, and the energy barrier of migratory insertion of alkene into Ir–C also increases to 25.3 and 25.5 kcal mol−1, respectively. That is to say, these polarised alkenes increase the apparent activation energy of the reaction (29.5 and 30.7 kcal mol−1, respectively) through their electron-withdrawing substituent groups. As shown in Fig. 4b, the migratory insertion of electron-donating NH2-substituted b4 into Ir–C (25.0 kcal mol−1) requires less energy than that for b2.
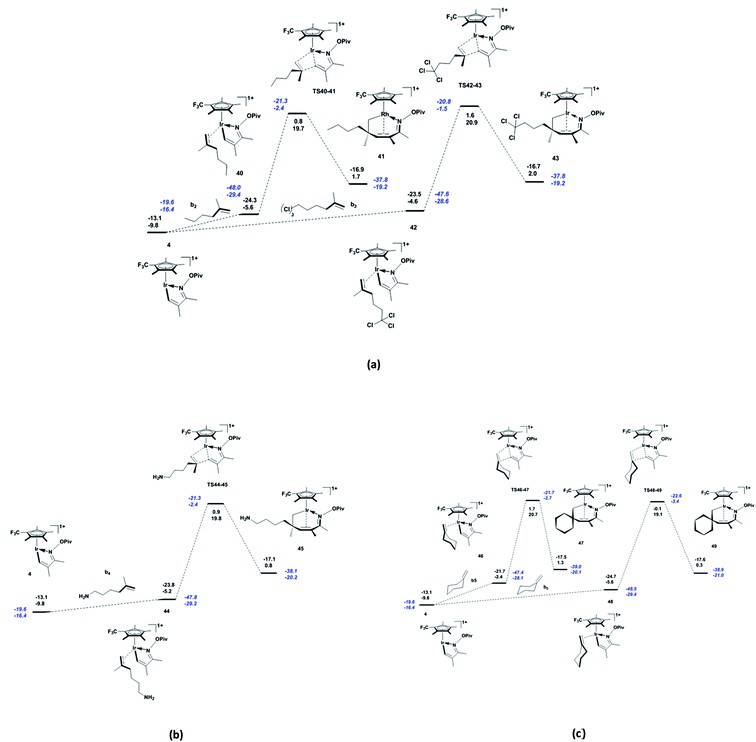 |
| | Fig. 4 Energetic profile for the reactions of the α,β-unsaturated oxime pivalate a1 with alkenes (a) b2, b3, (b) b4, and (c) b5, including the coordination and migratory insertion of alkene. Born–Oppenheimer energies (top) and Gibbs free energies (bottom) are given in kcal mol−1, and values in blue italics include the dispersion correction. | |
Diastereoselectivity of the reaction
The diastereoselectivity of the title reaction was explored by using cyclohexylethylene b5 as the substrate. As shown in Fig. 4c, b5 can approach the five-membered iridacycle 4 with different faces to generate the two species 46 (Ir–C5 = 2.21 Å and C1–C6 = 3.03 Å; see Fig. S3 in ESI†) and 48 (Ir–C5 = 2.21 Å and C1–C6 = 2.91 Å), with the former is higher in energy by 3.2 kcal mol−1. The subsequent migratory insertion of cyclohexylethylene through TS46-47 (C1–C6 = 1.88 Å) and TS48-49 (C1–C6 = 1.83 Å) generates the diastereoisomeric seven-membered rhodacycles 47 and 49, respectively, eventually leading to diastereoisomeric 2,3-dihydropyridine derivatives. Consequently, it can be inferred that TS46-47 and TS48-49 are responsible for the diastereoisomeric products. As can be seen from Fig. S3 in ESI,† the energy difference between TS46-47 and TS48-49 (1.6 kcal mol−1 higher in the former) stems from the clash between the steric structures of cyclohexyl and α,β-unsaturated oxime pivalate. Therefore, we predict that the diastereoselectivity of the reaction can be controlled by tuning the clash between the alkene and α,β-unsaturated oxime pivalate, through introducing appropriate substituent groups into the substrates.
Conclusions
2,3-Dihydropyridines are important starting materials for nitrogen heterocycles that are prevalent in pharmaceuticals. Since the Cp*Ir complexes are extensively used as catalysts, exploring their application in the catalysed synthesis of 2,3-dihydropyridines has significant importance. Using DFT methods, we calculated the potential energy surfaces of the Cp*Ir-catalysed reactions of α,β-unsaturated oxime pivalates with alkenes to furnish 2,3-dihydropyridines. The overall reaction includes several elementary reactions, such as a CMD process, migratory insertion of the alkene into Ir–C, pivaloyl transfer to the Ir centre, and reductive elimination. The results showed that the CMD process is reversible, and the migratory insertion of the alkene into Ir–C is rate-determining. The reductive elimination furnishes the product-ligated complex, and the higher exergonicity of this step makes the reaction irreversible. With stronger electron-donating substituent groups on Cp*, the apparent activation energy of the reaction is increased and the exergonicity of C–H activation process decreased. However, the energy barrier of migratory insertion of the alkene is less affected. By introducing strong electron-donating groups such as the amido group into the 2-position of α,β-unsaturated oxime pivalates, the energy barrier of migratory insertion of alkene and the apparent energy barrier of the reaction are greatly reduced (to 15.3 and 24.7 kcal mol−1, respectively), so that the reaction can possibly proceed at room temperature. Compared to phenyl, the more electron-deficient p-CF3-phenyl at the 2-position of α,β-unsaturated oxime pivalate increases the apparent activation energy of the reaction. When more polarised alkenes with electron-withdrawing substituent groups are used as substrates, the apparent energy barrier of the reaction also increases. Finally, diastereoselectivity of the reaction was examined using cyclohexylethylene as the substrate, and a method to enhance the diastereocontrol of the reaction is suggested.
Acknowledgements
We are grateful for the financial support from The Natural Sciences Foundation of Anhui Province (1608085MB42), Scientific Research Foundation of Anhui University of Science and Technology (ZY538), Open Projects of State Key Laboratory of Supramolecular Structure and Materials (sklssm201631), and Beijing National Laboratory for Molecular Sciences (1G129).
Notes and references
-
(a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed;
(b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed;
(c) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed;
(d) J. Liu, X. Wu, J. A. Iggo and J. Xiao, Coord. Chem. Rev., 2008, 252, 782–809 CrossRef CAS;
(e) Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC;
(f) R. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef PubMed;
(g) M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed.
-
(a) C.-X. Zhuo, X. Zhang and S.-L. You, ACS Catal., 2016, 6, 5307–5310 CrossRef;
(b) A. Ros, R. López-Rodríguez, B. Estepa, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 4573–4576 CrossRef CAS PubMed;
(c) J. A. Porras, I. N. Mills, W. J. Transue and S. Bernhard, J. Am. Chem. Soc., 2016, 138, 9460–9472 CrossRef PubMed;
(d) E. Benedetto, M. Tredwell, C. Hollingworth, T. Khotavivattana, J. M. Brown and V. Gouverneur, Chem. Sci., 2013, 4, 89–96 RSC;
(e) S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith III, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed;
(f) S. Kawamorita, R. Murakami, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2013, 135, 2947–2950 CrossRef CAS PubMed;
(g) D. Y. Wang, Y. Choliy, M. C. Haibach, J. F. Hartwig, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2016, 138, 149–163 CrossRef CAS PubMed;
(h) D. L. Huang, R. Beltrán-Suito, J. M. Thomsen, S. M. Hashmi, K. L. Materna, S. W. Sheehan, B. Q. Mercado, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 2016, 55, 2427–2435 CrossRef CAS PubMed;
(i) F. He, P. Braunstein, M. Wesolek and A. A. Danopoulos, Chem. Commun., 2015, 51, 2814–2817 RSC;
(j) H. Taguchi, D. Sasaki, K. Takeuchi, S. Tsujimoto, T. Matsuo, H. Tanaka, K. Yoshizawa and F. Ozawa, Organometallics, 2016, 35, 1526–1533 CrossRef CAS.
-
(a) J.-W. Park, S. J. Park and C.-H. Jun, Org. Lett., 2012, 14, 1468–1471 CrossRef CAS PubMed;
(b) S. Nie, X. Sun, W. Wei, X. Zhang, M. Yan and J. Xiao, Org. Lett., 2013, 15, 2394–2397 CrossRef CAS PubMed;
(c) S. Pan, K. Endo and T. Shibata, Org. Lett., 2012, 14, 780–783 CrossRef CAS PubMed;
(d) C. Xu, H.-M. Li, Z.-Q. Xiao, Z.-Q. Wang, S.-F. Tang, B.-M. Ji, X.-Q. Hao and M.-P. Song, Dalton Trans., 2014, 43, 10235–10247 RSC;
(e) S. Hohloch, S. Kaiser, F. L. Duecker, A. Bolje, R. Maity, J. Košmrlj and B. Sarkar, Dalton Trans., 2015, 44, 686–693 RSC;
(f) S. Pan, N. Ryu and T. Shibata, Org. Lett., 2013, 15, 1902–1905 CrossRef CAS PubMed;
(g) H. Li and C. Mazet, Org. Lett., 2013, 15, 6170–6173 CrossRef CAS PubMed;
(h) A. Joliton and E. M. Carreira, Org. Lett., 2013, 15, 5147–5149 CrossRef CAS PubMed;
(i) M. Oishi, M. Oshima and H. Suzuki, Inorg. Chem., 2014, 53, 6634–6654 CrossRef CAS PubMed.
-
(a) S. Takebayashi and T. Shibata, Organometallics, 2012, 31, 4114–4117 CrossRef CAS;
(b) J. Campos, R. Peloso, M. Brookhart and E. Carmona, Organometallics, 2013, 32, 3423–3426 CrossRef CAS;
(c) K. Q. Vuong, C. M. Wong, M. Bhadbhade and B. A. Messerle, Dalton Trans., 2014, 43, 7540–7553 RSC;
(d) L. E. E. Broeckx, W. Delaunay, C. Latouche, M. Lutz, A. Boucekkine, M. Hissler and C. Mϋller, Inorg. Chem., 2013, 52, 10738–10740 CrossRef CAS PubMed;
(e) M. Magre, O. Pàmies and M. Diéguez, ACS Catal., 2016, 6, 5186–5190 CrossRef.
-
(a) Y. Boutadla, D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2009, 5887–5893 RSC;
(b) W. J. Tenn III, K. J. H. Young, G. Bhalla, J. Oxgaard, W. A. Goddard III and R. A. Periana, J. Am. Chem. Soc., 2005, 127, 14172–14173 CrossRef PubMed;
(c) D. L. Davies, S. M. A. Donald, O. AI-Duaij, S. A. Macgregor and M. Pölleth, J. Am. Chem. Soc., 2006, 128, 4210–4211 CrossRef CAS PubMed;
(d) S. Qu, Y. Dang, C. Song, M. Wen, K.-W. Huang and Z.-X. Wang, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS PubMed;
(e) C. Cheng, B. G. Kim, D. Guironnet, M. Brookhart, C. Guan, D. Y. Wang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2014, 136, 6672–6683 CrossRef CAS PubMed;
(f) D. H. Ess, R. J. Nielsen, W. A. Goodard III and R. A. Periana, J. Am. Chem. Soc., 2009, 131, 11686–11688 CrossRef CAS PubMed;
(g) W. J. Tenn III, K. J. H. Young, J. Oxgaard, R. J. Nielsen, W. A. Goddard III and R. A. Periana, Organometallics, 2006, 25, 5173–5175 CrossRef;
(h) D. L. Davies, S. M. A. Donald, O. AI-Duaij, J. Fawcett, C. Little and S. A. Macgregor, Organometallics, 2006, 25, 5976–5978 CrossRef CAS;
(i) S. M. W. Rahaman, S. Dinda, A. Sinha and J. K. Bera, Organometallics, 2013, 32, 192–201 CrossRef CAS;
(j) D. R. Pahls, K. E. Allen, K. I. Goldberg and T. R. Cundari, Organometallics, 2014, 33, 6413–6419 CrossRef CAS;
(k) S. K. Meier, K. J. H. Young, D. H. Ess, W. J. Tenn III, J. Oxgaard, W. A. Goddard III and R. A. Periana, Organometallics, 2009, 28, 5293–5304 CrossRef CAS;
(l) R. Tanaka, M. Yamashita, L.
W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS;
(m) M. V. Jiménez, J. Fernández-Tornos, J. J. Pérez-Torrente, F. J. Modrego, P. García-Orduña and L. A. Oro, Organometallics, 2015, 34, 926–940 CrossRef;
(n) B. J. A. van Weerdenburg, A. H. J. Engwerda, N. Eshuis, A. Longo, D. Banerjee, M. Tessari, C. Fonseca Guerra, F. P. J. T. Rutjes, F. M. Bickelhaupt and M. C. Feiters, Chem.–Eur. J., 2015, 21, 10482–10489 CrossRef CAS PubMed.
- F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef CAS PubMed.
- H. Taguchi, D. Sasaki, K. Takeuchi, S. Tsujimoto, T. Matsuo, H. Tanaka, K. Yoshizawa and F. Ozawa, Organometallics, 2016, 35, 1526–1533 CrossRef CAS.
- Z. Tang, C. Tejel, M. M. D. S. Buchaca, M. Lutz, J. I. V. van der and B. de Bruin, Eur. J. Inorg. Chem., 2016, 963–974 CrossRef CAS.
- G. Mancano, M. J. Page, M. Bhadbhade and B. A. Messerle, Inorg. Chem., 2014, 53, 10159–10170 CrossRef CAS PubMed.
-
(a) J. Zhou, J. Shi, Z. Qi, X. Li, H. E. Xu and W. Yi, ACS Catal., 2015, 5, 6999–7003 CrossRef CAS;
(b) H. Kim, G. Park, J. Park and S. Chang, ACS Catal., 2016, 6, 5922–5929 CrossRef CAS;
(c) K. Nakao, G. Choi, Y. Konishi, H. Tsurugi and K. Mashima, Eur. J. Inorg. Chem., 2012, 1469–1476 CrossRef CAS;
(d) G. J. Sherborne, M. R. Chapman, A. J. Blacker, R. A. Bourne, T. W. Chamberlain, B. D. Crossley, S. J. Lucas, P. C. McGowan, M. A. Newton, T. E. O. Screen, P. Thompson, C. E. Willans and B. N. Nguyen, J. Am. Chem. Soc., 2015, 137, 4151–4157 CrossRef CAS PubMed.
-
(a) F. Li, J. Ma and N. Wang, J. Org. Chem., 2014, 79, 10447–10455 CrossRef CAS PubMed;
(b) A. M. Phelps, V. S. Chan, J. G. Napolitano, S. W. Krabbe, J. M. Schomaker and S. Shekhar, J. Org. Chem., 2016, 81, 4158–4169 CrossRef CAS PubMed;
(c) A. Gunay, M. A. Mantell, K. D. Field, W. Wu, M. Chin and M. H. Emmert, Catal. Sci. Technol., 2015, 5, 1198–1205 RSC.
-
(a) R. S. Phatake, P. Patel and C. V. Ramana, Org. Lett., 2016, 18, 292–295 CrossRef CAS PubMed;
(b) A. Bolje, S. Hohloch, M. van der Meer, J. Košmrlj and B. Sarkar, Chem.–Eur. J., 2015, 21, 6756–6764 CrossRef CAS PubMed;
(c) A. Ruff, C. Kirby, B. C. Chan and A. R. O'Connor, Organometallics, 2016, 35, 327–335 CrossRef CAS.
-
(a) N. Onishi, M. Z. Ertem, S. Xu, A. Tsurusaki, Y. Manaka, J. T. Mucherman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2016, 6, 988–992 RSC;
(b) O. Prakash, K. N. Sharma, H. Joshi, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 983–993 CrossRef CAS;
(c) O. Prakash, H. Joshi, K. N. Sharma, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 3804–3812 CrossRef CAS.
-
(a) M. E. Kerr, I. Ahmed, A. Gunay, N. J. Venditto, F. Zhu, E. A. Ison and M. H. Emmert, Dalton Trans., 2016, 45, 9942–9947 RSC;
(b) G.-D. Tang, C.-L. Pan and F. Xie, Org. Biomol. Chem., 2016, 14, 2898–2904 RSC;
(c) B. Liu, X. Wang, Z. Ge and R. Li, Org. Biomol. Chem., 2016, 14, 2944–2949 RSC.
- M. W. Drover, H. C. Johnson, L. L. Schafer, J. A. Love and A. S. Weller, Organometallics, 2015, 34, 3849–3856 CrossRef CAS.
- D. L. Davies, O. AI-Duaij, J. Fawcett, M. Giardiello, S. T. Hilton and D. R. Russell, Dalton Trans., 2003, 4132–4138 RSC.
- Y.-F. Han, H. Li, P. Hu and G.-X. Jin, Organometallics, 2011, 30, 905–911 CrossRef CAS.
- Y. Boutadla, D. L. Davies, R. C. Jones and K. Singh, Chem.–Eur. J., 2011, 17, 3438–3448 CrossRef CAS PubMed.
- Y.-F. Han, Y.-J. Lin and G.-X. Jin, Dalton Trans., 2011, 40, 10370–10375 RSC.
- S. Kammer, I. Starke, A. Pietrucha, A. Kelling, W. Mickler, U. Schilde, C. Dosche, E. Kleinpeter and H.-J. Holdt, Dalton Trans., 2012, 40, 10219–10227 RSC.
-
(a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed;
(b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed;
(c) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
-
(a) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223–243 CrossRef CAS;
(b) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141–1156 RSC;
(c) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
-
(a) T. Yamakawa and N. Yoshikai, Org. Lett., 2013, 15, 196–199 CrossRef CAS PubMed;
(b) B. J. Fallon, J.-B. Garsi, E. Derat, M. Amatore, G. Aubert and M. Petit, ACS Catal., 2015, 5, 7493–7497 CrossRef CAS.
- X.-X. Zheng, C. Du, X.-M. Zhao, X. Zhu, J.-F. Suo, X.-Q. Hao, J.-L. Niu and M.-P. Song, J. Org. Chem., 2016, 81, 4002–4011 CrossRef CAS PubMed.
-
(a) F. J. Fañanás, T. Arto, A. Mendoza and F. Rodríguez, Org. Lett., 2011, 16, 4184–4187 CrossRef PubMed;
(b) T. Harschneck and S. F. Kirsch, J. Org. Chem., 2011, 76, 2145–2156 CrossRef CAS PubMed.
- H. Huang, J. Cai and G.-J. Deng, Org. Biomol. Chem., 2016, 14, 1519–1530 CAS.
- A. Saito, M. Hironaga, S. Oda and Y. Hanzawa, Tetrahedron Lett., 2007, 48, 6852–6855 CrossRef CAS.
- K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2008, 10, 325–328 CrossRef CAS PubMed.
- R. M. Martin, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2012, 77, 2501–2507 CrossRef CAS PubMed.
- T. Ehara, O. Irie, T. Kosaka, T. Kanazawa, W. Breitenstein, P. Grosche, N. Ostermann, M. Suzuki, S. Kawakami, K. Konishi, Y. Hitomi, A. Toyao, H. Gunji, F. Cumin, N. Schiering, T. Wagner, D. F. Rigel, R. L. Webb, J. Maibaum and F. Yokokawa, ACS Med. Chem. Lett., 2014, 5, 787–792 CrossRef CAS PubMed.
- M. G. P. Buffat, Tetrahedron, 2004, 60, 1701–1729 CrossRef CAS.
- N. Houllier, M. C. Lasne, R. Bureau, P. Lestage and J. Rouden, Tetrahedron, 2010, 66, 9231–9241 CrossRef CAS.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- F. Romanov-Michailidis, K. F. Sedillo, J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2015, 137, 8892–8895 CrossRef CAS PubMed.
- T. Zhao, W. Guo and Y. Xia, Chem.–Eur. J., 2015, 21, 9209–9218 CrossRef PubMed.
- X.-B. Zhang, Z.-Y. Hu, G. E Zhou and S. Wang, Eur. J. Org. Chem. DOI:10.1002/ejoc.201601247.
- L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414–12419 CrossRef CAS PubMed.
-
(a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed;
(b) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. Couty and M. B. Hall, J. Comput. Chem., 1998, 22, 1359–1370 Search PubMed.
-
(a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS;
(b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1972, 72, 650–654 CrossRef;
(c) V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
-
(a) R. Yamaguchi, C. Ikeda, Y. Takahashi and K. Fujita, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS PubMed;
(b) S. Li and M. B. Hall, Organometallics, 1999, 18, 5682–5687 CrossRef CAS.
-
(a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS;
(b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- V. Singh, Y. Nakao, S. Sakaki and M. M. Deshmukh, J. Org. Chem., 2017, 82, 289–301 CrossRef CAS PubMed.
-
(a) T. M. Figg, S. Park, J. Park, S. Chang and D. G. Musaev, Organometallics, 2014, 33, 4076–4085 CrossRef CAS;
(b) P. E. M. Siegbahn, J. Am. Chem. Soc., 1996, 118, 1487–1496 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
-
(a) K. J. H. Young, J. Oxgaard, D. H. Ess, S. K. Meier, T. Stewart, W. A. Goddard III and R. A. Periana, Chem. Commun., 2009, 3270–3272 RSC;
(b) D. L. Davies, S. A. Macgregor and A. I. Poblador-Bahamonde, Dalton Trans., 2010, 39, 10520–10527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated main bond parameters for Cp*Ir complex M, Fig. S1–S3, and optimised Cartesian coordinates and electronic energies (E) for relevant species. See DOI: 10.1039/c6ra25501a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Dan-Dan Zhanga and
Sheng-Gui He*c
*a,
Dan-Dan Zhanga and
Sheng-Gui He*c