
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature controlled cationic photo-curing of a thick, dark composite†
Long Yanga,
Jinliang Yangb,
Jun Nieab and
Xiaoqun Zhu *ab
*ab
aState Key Laboratory of Chemical Resource Engineering & Beijing Laboratory of Biomedical Materials, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: zhuxq@mail.buct.edu.cn; Tel: +86 01064421310
bChangzhou Institute of Advanced Materials, Beijing University of Chemical Technology, Changzhou, Jiangsu 213164, P. R. China
First published on 16th January 2017
Abstract
In this research, the cationic photopolymerization of 1,4-butanediol diglycidyl ether was found to have an infinitely long induction period due to the sustained stability of the secondary oxonium ions species at low temperature. Based on this, the system could be separated into two steps: photolysis of the photoinitiator without polymerization under irradiation and polymerization in the dark. Under irradiation, only the secondary oxonium ion species was generated at low temperature, whereas the polymerization could proceed and auto-accelerate at room temperature without irradiation. The two steps could be monitored by a significant change of the temperature. The temperature controlled cationic mechanism resolved the issue of light penetration in colored thick composites. Through temperature control, the infinitely thick and dark material could be prepared by cationic photopolymerization.
Introduction
Attributed to different active centres, photopolymerization can be divided into two kinds of mechanisms: cationic polymerization and free radical polymerization. Much work on free radical photopolymerization has been reported because it has the advantages of fast polymerization and a large choice of initiator, monomer, and oligomer, and the technique of radical photopolymerization has been widely used in industry. Cationic photopolymerization, on the other hand, has a low rate of polymerization and requires a long induction period.1–6 However, cationic photopolymerization exhibits some advantages over free-radical photopolymerization,1,7,8 such as the absence of air inhibition, a “dark-curing” reaction, low levels of toxicity and irritation, a lower volume shrinkage during photopolymerization and so on. These peculiarities of cationic photopolymerization have attracted scientists' growing interest in researching it.Among these researches, many studies have focused on shortening the induction period and increasing the reaction rate of cationic photopolymerization,3,4,9–13 because the rate is one of the most important factors. The most common method of increasing the rate of cationic photopolymerization is by increasing the reaction temperature, because cationic photopolymerization is sensitive to temperature and the induction period could be shortened by a temperature increase.10,14 Alternatively, the monomer structure and formulation also have a strong effect on cationic polymerization. Crivello's studies showed that cycloaliphatic epoxy monomers exhibited essentially no induction period and a very high rate of ring-opening polymerization. In contrast, alkyl diglycidyl ethers displayed a long induction period.3,9,15 The studies showed that vinyl substituted epoxides markedly accelerated the cationic ring-opening photopolymerization of oxetane monomers with the addition of small amounts (3–5 wt%) of the epoxide to the oxetane monomer.11
On the other hand, the photogenerated active centres in cationic photopolymerization have long lifetimes and are essentially non-terminating, which means they have the potential to migrate out of the irradiation region into the shadow region of the sample, thereby polymerizing the unexposed monomer.1,8,14,16–19 However, the diffusion coefficient of these active centres is low,16,18,20,21 which means that curing thick samples, if it is possible at all, takes a long time or requires a high temperature. What is more, coloured additives like pigment or fibres, especially dark colours, further reduce the ability of light to penetrate the solution and the rate of production of active centres, and thus the maximum thickness of dark-coloured samples that can be cured is limited.16 Possibly, post-curing might be effective for dark-coloured or thick samples. Taking advantage of the diffusion of active centres, Sangermano and coworkers successfully prepared antistatic epoxy coatings with 1 wt% carbon nanotubes and a thickness of more than 1 cm through cationic photopolymerization.16
Normally, for cationic photopolymerization, after irradiation, the sample can have a long induction period and can remain quiescent for more than 1 h at 25–28 °C without an appreciable change in appearance or viscosity.22,23 Crivello's studies showed that the photopolymerization of many alkyl glycidyl ethers displayed a prominent induction period at room temperature as a result of the formation of long-lived, relatively stable secondary oxonium ion species that are formed as intermediates, through protonation of the monomer by photogenerated Brønsted acid. Furthermore, it has been proposed that the protonated glycidyl ethers are stabilized through the formation of bidentate hydrogen-bonded species, as depicted in Scheme 1. Because the basicity of the oxygen atoms of open chain ethers and epoxides is similar,24 the proton in such structures is intramolecular hydrogen bonded to both oxygen atoms of the glycidyl ether moiety. The more the oxygen atoms are coordinated to the proton, the more stable the secondary oxonium ions species.3 Further studies revealed that secondary oxonium ions species are much more stable under low temperature conditions.10
 | ||
| Scheme 1 Hydrogen bonding in secondary oxonium ions species. | ||
The disadvantage of a long induction period of cationic photopolymerization has been recognized and much attention has been paid to shortening it. However, few studies have focused on taking advantage of the long induction period of cationic photopolymerization to prepare thick and/or dark-coloured materials in a short time. In this research, we took advantage of the long induction period of cationic photopolymerization and even prolonged it; we then separated the irradiation process and the curing process of the cationic photopolymerization completely by controlling the temperature. At the low temperature, through stirring and irradiating the monomer solution with a cationic initiator, the cationic initiator could be photolyzed completely and stable secondary oxonium ion species were formed. If the temperature control was removed, the temperature of the sample increased to room temperature naturally, the secondary oxonium ions species turned into active centres, which quickly induced polymerization, and the released heat propelled the reaction further, and then the sample was cured. In theory, the thicker the material, the better the curing, because the heat could be retained well in the thick material and the material could be self-heated; in turn, the polymerization of the epoxide was auto-accelerated by the heat. The whole process had a “snowballing” effect. From this process, material with infinite thickness and/or dark colour could be cationically photopolymerized in theory. This kind of cationic photopolymerization is described in Scheme 2.
 | ||
| Scheme 2 Temperature control of cationic photopolymerization of epoxy. | ||
Experimental
Materials
1,4-Butanediol diglycidyl ether (BDE, 98%) was obtained from Kegonghua (Beijing) Chemical Technology Co. Before use, the monomer was dried by calcium hydride and then filtered to remove the excess inorganic drying agent and its hydrolysis products. The photoinitiator triphenylsulfonium hexafluoroantimonate (50 wt% solution in propylene carbonate, PAG201) was obtained from Changzhou Tronly New Electronic Materials Co. All concentrations of initiator used in this article are given as weight percentages based on the monomer used.Sample preparation and temperature measurement
The conversion of photopolymerization can be characterized well by photo-DSC. However, if the sample is large and is irradiated under stirring, photo-DSC cannot be used for its characterization. In this research, except for the characterization of the induction period by photo-DSC, the temperature–time curve that was collected by the type T thermocouple in real time was used to describe the extent of photopolymerization.The photo-DSC experiment was performed by using a photo-differential scanning calorimeter (DSCQ2000 series, TA instruments) equipped with a photo-calorimetric accessory. Approximately 7 mg of sample (BDE and PAG201) was placed as a thin layer in a standard aluminium pan. The pan was placed on the thermostatic head of the device and sample was irradiated by a UV lamp (EFOS Lite, 100 W miniature arc lamp with a wavelength of 200–800 nm and 5 mm crystal optical fibre, Canada). The DSC cell was purged with ultra-high-purity nitrogen gas during the polymerization, and the nitrogen flow was 50 mL min−1. According to our selected programme, the sample was cooled to the assigned temperature, kept isothermally for 5 min, then subjected to irradiation in isothermal conditions, and finally kept isothermally for 30 min after the irradiation was terminated.
Because the cationic photopolymerization of epoxy is an exothermic reaction, the time evolution of the temperature is directly proportional to the conversion of epoxy groups, assuming no heat loss.25,26 For our experiments, the temperature was measured by a type T thermocouple. The thermocouple, which was electrically insulated from the mixture with PFA Teflon, was connected to a data logger (TP9008U, Toprie, Shenzhen, CT), which was set at a frequency of 1 Hz. The details of the operation are described as follows.
The system of cationic photopolymerization was stirred during the irradiation process under low temperature conditions. Unless otherwise stated, in this paper, the mass of the sample was 4.0 g, the concentration of photoinitiator was 0.1 wt%, and the light intensity was 1 mW cm−2. The sample was put in a glass bottle sealed by a rubber plug with an inside diameter of 20 mm. The bottle was immerged into the double bottle filled with circulating coolant by the precise cryogenic thermostat on the magnetic stirrers. The thickness of the sample was about 10 mm. The sample was irradiated by a UV spot light source with an optical fibre that penetrated the rubber stopper for the assigned time (EFOS Lite, 100 W miniature arc lamp with a wavelength of 200–800 nm and 5 mm crystal optical fibre, Canada) and the temperature was measured using a thermocouple at a depth of 5 mm. The light intensity on the surface of samples was detected by UV radiometer (Beijing Normal University, China).
Under stirring, the sample of BDE with the photoinitiator was cooled to an assigned temperature, kept isothermally for at least 30 min, irradiated for some time in isothermal conditions, kept isothermally for 60 min after the irradiation (for working process), and then put into an incubator rising to an assigned temperature through natural heating to research the “dark reaction”. The set-up is shown in Fig. 1(a). The whole process of the temperature change could be divided into different stages and is shown as Fig. 1(b).
 | ||
| Fig. 1 (a) Schematic diagram of experimental set-up. (b) Scheme of experimental process: cooling, irradiation, isothermality and dark reaction. | ||
Results and discussion
Many researches have reported the effect of temperature on cationic photopolymerization. These works mainly considered static thin films.1,4,10,17 Due to the limited penetration of UV radiation into organic materials, it is difficult to prepare sample of super thickness or a large quantity of samples by cationic photopolymerization. However, provided that a large quantity of sample is irradiated without curing, it is possible to photolyze all the photoinitiator in it with stirring. Thus, we proposed to separate cationic photopolymerization into two independent and discontinuous parts, which are the photolysis of the cationic initiator without polymerization under irradiation and then the polymerization process in the dark. We tried to turn the hypothesis into reality by controlling the temperature.Effect of different cooling temperature on the induction period
To demonstrate how the temperature influences the length of the induction period, 0.1 wt% photoinitiator PAG201 in BDE (4.0 g) was irradiated under different cooling temperatures (from −20 °C to 15 °C) and the change of the temperature was monitored by a type T thermocouple in real time. The results are shown in Fig. 2.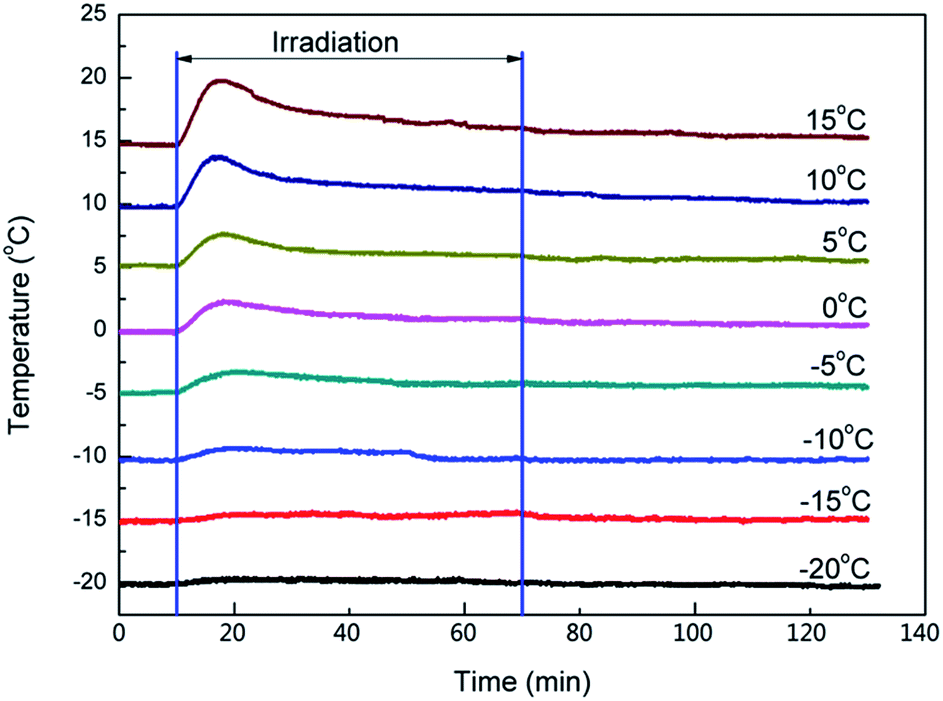 | ||
| Fig. 2 Effect of cooling temperature on irradiation and isothermality process, irradiation time 60 min, light intensity of 1 mW cm−2. | ||
When the cooling temperature was lower than −15 °C (from −20 to −15 °C), the temperature of the samples showed almost no change throughout the irradiation and remained the same as the cooling temperature, which meant there was no photopolymerization. As reported, the Bronsted acid that was produced by the photolysis of PAG201 could be formed as secondary oxonium ion species, which are relatively stable, through protonation of the glycidyl ethers. Our research further demonstrates that the secondary oxonium ion species remained stable enough or had no ability to trigger the polymerization at low temperature. However, when the temperature was higher than −15 °C (from −10 to 15 °C), there was an obvious peak on the temperature–time curves during irradiation and the peak became stronger as the temperature increased. It can be speculated that the stability of the secondary oxonium ion species declined and some of the species decomposed into active centres, which initiated the polymerization of the epoxide, because the polymerization of epoxide is an exothermic reaction, which raises the temperature of the sample.
Photo-DSC is one of the most accurate methods of characterization of the conversion by detecting the change of the heat flow. In order to verify that this method was suitable to characterize the induction period of cationic photopolymerization, the method was performed to check the cationic photopolymerization of BDE with 1 wt% PAG201 at different temperatures. When photopolymerization of BDE was characterized by photo-DSC, we found that the sample with 0.1 wt% photoinitiator could not be polymerized. We suppose that the quantity of photoinitiator PAG201 may be too small to induce photopolymerization. The result of photo-DSC proved there was no photopolymerization of BDE at the temperature of −20 °C from the change of the reaction energy (Fig. 3). This is in agreement with the tendency of time evolution of the temperature. This result proved that the secondary oxonium ion species could remain stable enough and the induction period could be infinitely long at low temperature.
 | ||
| Fig. 3 Heat flux vs. time in the irradiation process from −30 °C to 70 °C, irradiated 30 min, light intensity of 1 mW cm−2, 1 wt% PAG201. | ||
The samples transformed from liquid to gel during irradiation when the cooling temperature was more than 5 °C (from 5 to 15 °C), which indicated that the secondary oxonium ion species were more unstable and the polymerization was accelerated. Fig. 4 shows the appearance of the sample before the dark reaction at different temperatures. The sample was always liquid at low temperature, and the viscosity increased until it turned into solid with the increase of the temperature. Based on the above results, it could be concluded that the induction period of the cationic photopolymerization could be tuned by temperature and the period could be prolonged infinitely if the temperature was reasonable.
 | ||
| Fig. 4 The photographs of the samples irradiated at different temperature before dark reaction. | ||
Effect of different cooling temperature on the polymerization
In order to prove that secondary oxonium ion species would initiate cationic photopolymerization once the temperature was suitable and also to prove that the cationic photopolymerization could be separated into two independent processes by controlling the temperature, a series of the experiments were carried out in which the sample was kept cool at different temperatures for 60 min after irradiation and then allowed to warm up naturally to room temperature (about 27 °C). The results are shown in Fig. 5. | ||
| Fig. 5 The effect of temperature of isothermally to dark reaction, irradiation time 60 min, light intensity of 1 mW cm−2, isothermality time 60 min. | ||
Firstly, the temperature of samples with the initial temperature below 0 °C rose rapidly to a maximum temperature (Tmax) of more than 100 °C. When the temperature reached room temperature, the active centres were discharged together in short time. More importantly, the active centres were uniformly distributed in the sample. Therefore, the active centres initiated the polymerization evenly and simultaneously. Furthermore, the polymerization of the epoxide groups is an exothermic reaction. This instantaneous collective heat caused the temperature to rise rapidly, and conversely, the increase of the temperature accelerated the polymerization of the epoxide groups. This phenomenon has a “snowballing” effect and the reaction is self-accelerated. Along with the consumption of the epoxide groups, the heat release was smaller than its dissipation, and the temperature of the sample declined gradually. As the temperature rose, the liquid sample turned into solid. This result proved our hypothesis that the secondary oxonium ion species was only dormant at the low temperature and the function of initiation could be triggered once the temperature was reasonable. Thus, the cationic photopolymerization could be separated into two independent processes by controlling the temperature.
Secondly, there was another peculiar phenomenon whereby the lower the cooling temperature, the shorter the time taken to reach Tmax. It took 16, 18.5, and 20 min when the cooling temperature was −20, −15, and −10 °C respectively. This also indicated that the lower the cooling temperature, the more stable the secondary oxonium ion species and the fewer species were decomposed. Once the temperature increased, more active centres were released almost at the same time, triggering the polymerization of the epoxide group together. Thus, the instantaneous collective heat was more significant and the temperature rose quickly. Although it took a short time for the higher cooling temperature to rise to room temperature, some of the active centres and epoxide groups were consumed before the temperature rise and the instantaneous collective heat declined, which caused the Tmax to be reached later.
Thirdly, Tmax declined with the increase of the cooling temperature. Similarly, it was also determined by the effect of the instantaneous collective exothermic. The more clearly instantaneous the collective heat was, the higher Tmax was. If the cooling temperature was high, some secondary oxonium ion species were decomposed and the epoxide groups were consumed too. Thus, in the process of curing, the amount of secondary oxonium ion species and epoxide groups was lower compared with the low cooling temperature, which resulted in a low Tmax with the high cooling temperature.
Thus, the cooling temperature determined the amount of secondary oxonium ion species, and the oxonium ion species determined the amount of active centres released simultaneously, which determined the rate of ring-opening polymerization and the level of instantaneous collective heat. Inversely, the heat could accelerate the rate of ring-opening polymerization of epoxide groups. This was like a “snowballing” effect. Because the irradiation and polymerization were separated, cationic polymerization of a thick sample was possible.
Effect of different photoinitiator contents on photopolymerization
Since the amount of the secondary oxonium ion species affected Tmax significantly, it can be speculated that the concentration of cationic photoinitiator should also affect the Tmax because the secondary oxonium ion species are produced by photolysis of the cationic photoinitiator. Therefore, it is necessary to investigate how different concentrations of photoinitiator affect the Tmax. The results of the temperature versus time profiles of cationic photopolymerization with different photoinitiator concentrations ranging from 0.05 to 5 wt% are shown in Fig. 6. | ||
| Fig. 6 The effects of photoinitiator concentration on temperature during irradiation, isothermal and dark reaction irradiated 30 min with 1 mW cm−2 at 20 °C. | ||
With an increase of the photoinitiator, Tmax first increased and then declined. When the temperature was −20 °C, under irradiation, there was almost no temperature change when the concentration of photoinitiator was less than 0.5 wt%. The secondary oxonium ion species and epoxide groups were not consumed until the temperature was raised. Thus, with the increase of the concentration of photoinitiator, the instantaneous collective heat was more and more obvious and Tmax increased. However, when the concentration of the photoinitiator increased further (>0.5 wt%), it was found that the temperature increased during the irradiation, which demonstrated that photopolymerization proceeded due to the high concentration of the secondary oxonium ion species, and part of them decomposed into active centres, triggering the photopolymerization. Although the concentration of the secondary oxonium ion species may still be higher than the low concentration of the photoinitiator, the quantity of the epoxide groups in the sample declined because some of them were consumed during the irradiation. Besides, the high concentration of active centres caused the gel point to be reached early. It confined the activity of the epoxide groups or active centres, which resulted in a decrease of the conversion too. Because the total heat release decreased with a high concentration of photoinitiator, finally, Tmax was reached early but then declined.
Effect of different light intensity and irradiation time on polymerization
For the photopolymerization, the light is one of the key factors because it controls the photolysis of the photoinitiator. It is necessary to investigate the effects of the irradiation intensity and time on the dark reaction. The temperature of the dark reaction with different irradiation times and intensities is shown in Fig. 7. When the light intensity was 1 mW cm−2 and the irradiation time was increased from 5 to 15 and 30 min, respectively, Tmax increased from 39 to 118 and 183 °C, and the time taken to reach Tmax decreased from 31 to 12 and 9 min (Fig. 7a). This is because the photoinitiator could be photolyzed completely when it was irradiated for a long time. If the irradiation time was insufficient, some photoinitiator was not photolyzed, reducing the quantity of the secondary oxonium ion species.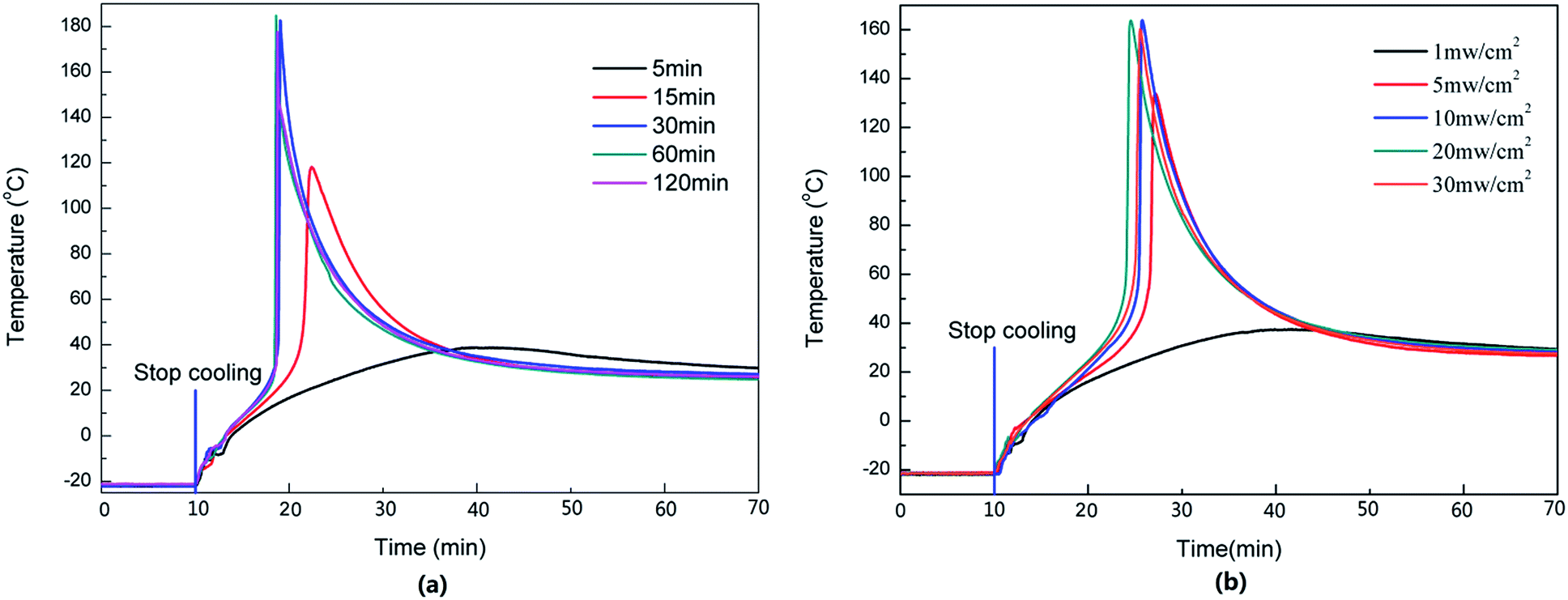 | ||
| Fig. 7 Effects of the light exposure on dark reaction. (a) Light intensity at 1 mW cm−2 at 20 °C; (b) irradiated 5 min at −20 °C; 0.1 wt% initiator. | ||
Similarly, the light intensity could also affect the photolysis of the photoinitiator. As shown in Fig. 7b, when the light intensity ranged from 1 to 30 mW cm−2, irradiation was carried out for 5 min, and Tmax was increased from 38 to 134 and 164 °C, respectively, the time required to reach Tmax was shortened from 30 to 18 and 16 min. A further increase of light intensity did not change Tmax or the time taken to reach Tmax much, which meant that a light intensity of 10 mW cm−2 and an irradiation time of 5 min were sufficient to photolyze all the photoinitiator in this sample.
Effect of temperature of curing process on polymerization
Cationic photopolymerization is sensitive to the temperature, and the reaction rate increases as the temperature rises.14 The photoinitiator triphenylsulfonium hexafluoroantimonate was stable up to 300 °C without decomposition. When dissolved in multifunctional epoxy resins, it may be kept at 120 °C for several hours without initiating polymerization.27,28 The change of the dark reaction when the temperature ranged from 12 °C to 64 °C is shown in Fig. 8. When the temperature of the dark reaction was 12 °C, Tmax only increased to 41 °C. However, when the temperature was higher than 25 °C, the Tmax could reach over 150 °C and the time taken to reach the Tmax was clearly shortened by the increase of the temperature. As we know, the polymerization of epoxide is an exothermic reaction, and the high temperature could raise the polymerization rate. At the same time, the thermal dissipation also decreases. Thus, the high temperature brings a high Tmax and causes Tmax to be reached early. | ||
| Fig. 8 The effects of the dark reaction temperature on the polymerization irradiated 30 min with 1 mW cm−2 at 20 °C, 0.1 wt% initiator. | ||
All the above results demonstrated that the cationic photopolymerization could be controlled by the temperature; however, they could not prove that the thickness and the colour of the sample were not directly confined by this kind of cationic photopolymerization. In order to confirm that temperature control of cationic photopolymerization could cure ultra-thick and deep colour samples, samples with a thickness of more than 3.5 cm and with 2 wt% of carbon black were cured and the videotaped curing process is shown in ESI Movies 1 and 2† respectively. The polymerization speed of the thick sample was very quick and the sample was cured within a few minutes. The interceptions from the video of the polymerization of the sample with 2% carbon black are shown in Fig. 9.
 | ||
| Fig. 9 The interception of segments from the video of the process of polymerization (the volume of solution is more than 45 mL) (a) pouring solution into beaker, (b) waiting for polymerization, (c) polymerized. | ||
Conclusions
Herein, the two steps of cationic photopolymerization of 1,4-butanediol diglycidyl ether (BDE) were investigated in the presence of the photoinitiator PAG201. The irradiation process and curing process were separated by control of the temperature. The experimental results showed that the secondary oxonium ion species remain stable enough and the system retains a low viscosity throughout the irradiation process under low temperature with stirring. The curing process was performed without irradiation and was controlled by temperature. This method completely overcomes the problems of photopolymerization such as light attenuation, additive interference, and difficulty of curing in shadow regions. Besides, the photo-cured production was almost uniform because the active centres were spread throughout the whole system by stirring. This temperature-controlled cationic photopolymerization is expected to be used in the fast curing of thick and dark-coloured materials.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 51373015, 51573011), Changzhou Sci & Tech Program (BY2014051) and Natural Foundation of Jiangsu Province (No. BK20150272). The authors also appreciate the support of the Beijing Laboratory of Biomedical Materials.Notes and references
- M. Atif, R. Bongiovanni and J. Yang, Polym. Rev., 2015, 55, 90–106 CrossRef CAS.
- R. Bongiovanni and M. Sangermano, Encyclopedia of Polymer Science and Technology, 2014 Search PubMed.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3036–3052 CrossRef CAS.
- R. Harikrishna, J. Therm. Anal. Calorim., 2015, 122, 1445–1454 CrossRef CAS.
- U. Bulut and J. V. Crivello, Macromolecules, 2005, 38, 3584–3595 CrossRef CAS.
- M. Atif, J. Yang, H. Yang, N. Jun and R. Bongiovanni, J. Compos. Mater., 2015, 0021998315616174 Search PubMed.
- X. Allonas, C. Croutxé-Barghorn, K. W. Bögl, N. Helle and G. A. Schreiber, Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- M. Sangermano, N. Razza and J. V. Crivello, Macromol. Mater. Eng., 2014, 299, 775–793 CrossRef CAS.
- J. D. Cho and J. W. Hong, J. Appl. Polym. Sci., 2005, 97, 1345–1351 CrossRef CAS.
- J. V. Crivello, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 591–598 CrossRef CAS.
- J. V. Crivello, Polymer, 2015, 64, 227–233 CrossRef CAS.
- I. E. dell'Erba, G. F. Arenas and W. F. Schroeder, Polymer, 2016, 83, 172–181 CrossRef.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(4), 594–601 CrossRef CAS.
- V. Sipani, A. Kirsch and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4409–4416 CrossRef CAS.
- M. Sangermano, G. Malucelli, R. Bongiovanni, A. Priola, U. Annby and N. Rehnberg, Eur. Polym. J., 2002, 38, 655–659 CrossRef CAS.
- M. Sangermano, S. Pegel, P. Pötschke and B. Voit, Macromol. Rapid Commun., 2008, 29, 396–400 CrossRef CAS.
- B. A. Ficek, The potential of cationic photopolymerization's long lived active centers, PhD thesis, University of Iowa, 2008.
- C. C. Hoppe, B. A. Ficek, H. S. Eom and A. B. Scranton, Polymer, 2010, 51, 6151–6160 CrossRef CAS.
- V. Sipani and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2064–2072 CrossRef CAS.
- G. Terrones and A. J. Pearlstein, Macromolecules, 2004, 37, 1565–1575 CrossRef CAS.
- B. A. Ficek, A. M. Thiesen and A. B. Scranton, Eur. Polym. J., 2008, 44, 98–105 CrossRef CAS.
- J. Crivello, B. Falk and M. Zonca, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1630–1646 CrossRef CAS.
- B. Falk, M. R. Zonca and J. V. Crivello, Macromol. Symp., 2005, 226(1), 97–108 Search PubMed.
- K. Matyjaszewski, M. Zieliński, P. Kubisa, S. Słomkowski, J. Chojnowski and S. Penczek, Macromol. Chem., 1980, 181, 1469–1482 CrossRef CAS.
- B. J. Falk, S. M. Vallinas and J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 579–596 CrossRef CAS.
- M. Retailleau, A. Ibrahim and X. Allonas, Polym. Chem., 2014, 5, 6503–6509 RSC.
- J. Crivello and J. Lam, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 2677–2695 CrossRef CAS.
- J. Crivello and J. Lam, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 2697–2714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25346f |
| This journal is © The Royal Society of Chemistry 2017 |