DOI:
10.1039/C6RA25283D
(Paper)
RSC Adv., 2017,
7, 5039-5045
Band gap tuning from an indirect EuGa2S4 to a direct EuZnGeS4 semiconductor: syntheses, crystal and electronic structures, and optical properties†
Received
15th October 2016
, Accepted 24th December 2016
First published on 17th January 2017
Abstract
Four isostructural europium chalcogenides, EuZnGeS4 (1), EuGa2S4 (2), EuIn2S4 (3) and EuIn2Se4 (4), have been synthesized using a high-temperature solid-state method. Single-crystal X-ray diffraction analysis indicates that they crystallize in the orthorhombic space group Fddd with Z = 32. Their structures feature an MQ4 (M = Zn, Ge, Ga and In; Q = S and Se) tetrahedrally constructed 3D network, and Eu2+ ions occupy the bicapped trigonal prismatic cavities. Compounds 1–3 have optical band gaps of 2.26, 2.32 and 2.37 eV, respectively. Theory calculations indicate that 2, with an indirect band gap, can be tuned to 1, with a direct band gap, via simple chemical substitution.
Introduction
Rare-earth chalcogenides have been extensively explored in the past twenty years because of their rich structures and potential applications in the fields of magnetism,1 thermoelectricity,2 photoluminescence,3 second-order nonlinear optics,4 and so on. Among them, europium chalcogenides have been paid much attention as Eu ions can be divalent or trivalent. It is well known that the Eu2+ cation has a similar ionic radius to alkali-earth metal cations, so many Eu2+ compounds have alkali-earth metal analogues. Recently, many alkali-earth metal chalcogenides have been studied as second-order nonlinear optical materials as they have high nonlinear optical coefficients and laser induced damage thresholds, such as Ba8Sn4S15,5 Ba7Sn5S15,6 Ba6Sn6Se13,7 BaHgSe2,8 BaHgS2 (ref. 9) and BaCdSnSe4,10 which have encouraged many chemists and material scientists' to explore them in the field of laser frequency conversion.
When checking the database, it was discovered that there are a large group of ternary or quaternary compounds can be uniformly written as II-III2 (III-III′)-Ch4 or quaternary II-II′(I2)-IV-Ch4 (II = Mg, Ca, Sr, Ba, Eu and Yb; II′ = divalent transition metal; I = Li, Na, Cu and Ag; III = B, Al, Ga and In; III′ = Yb, Sb and Bi; IV = Ge and Sn; Ch = S, Se and Te).11 The representative members of this family are listed in Table 1. What's more, there are also many related II-III2-Ch4 compounds containing transition-metal reported, where II = divalent transition-metal Zn, Cd, Hg and Mn, III = Ga and In, and Ch = S and Se.12 If take EuGa2S4 as the parent compound for this series, the sites of Eu and S can be also occupied by divalent alkali-earth metal or Yb, and Se or Te, respectively. The Ga sites can be replaced by the same group element B, Al and In. Specially, two Ga sites can be also replaced by two different elements to form quaternary compound II-II′-IV-Ch4 or II-I2-IV-Ch4, in which one site is occupied by tetravalent cations like Ge and Sn, the other site occupied by divalent transition-metal cations or monovalent coinage or alkali metal cations.
Table 1 Known structure types of ternary II-III2 (III-III′)-Ch4 or quaternary II-II′(I2)-IV-Ch4 (II = Mg, Ca, Sr, Ba, Eu and Yb; II′ = divalent transition metal; I = Li, Na, Cu and Ag; III = B, Al, Ga and In; III′ = Yb, Sb and Bi; IV = Ge and Sn; Ch = S, Se and Te) compounds. All these data are from ICSD or Pearson's crystal dataa
| Compound |
Space group |
Crystal system |
Pearson code |
Phase prototype |
| As there are so many members can be classified to this family, only one compound was chosen as a representative for each type of structure. All these data are checked results from Inorganic Crystal Structure Data (ICSD) and Pearson's Crystal Data (PCD). |
| MgAl2S4 |
Pnma |
Orthorhombic |
oP28 |
Mg2SiO4 |
| MgAl2S4 |
R3m |
Trigonal |
hR7 |
ZnIn2S4 |
| MgAl2S4 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
Trigonal |
hR7 |
ZnIn2S4 |
| MgGa2S4 |
C2/c |
Monoclinic |
mS84 |
MgGa2S4 |
| MgIn2S4 |
Fdm |
Cubic |
cF56 |
MgAl2O4 |
| MgIn2Te4 |
I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m 2m |
Tetragonal |
tI14 |
Cu2HgI4 |
| SrB2S4 |
P21/c |
Monoclinic |
mP28 |
SrB2S4 |
| SrB2S4 |
R |
Trigonal |
hR63 |
SrB2S4 |
| SrAl2S4 |
Cccm |
Orthorhombic |
oS28 |
BaGe2Se4 |
| SrAl2S4 |
Fddd |
Orthorhombic |
oF224 |
EuGa2S4 |
| SrGa2Te4 |
I4/mcm |
Tetragonal |
tI14 |
CaIn2Te4 |
| SrCu2GeSe4 |
Ama2 |
Orthorhombic |
oS32 |
SrCu2GeSe4 |
| SrCu2SnS4 |
P31 |
Trigonal |
hP24 |
SrCu2SnS4 |
| SrCu2SnS4 |
P3221 |
Trigonal |
hP24 |
SrCu2SnS4 |
| BaB2S4 |
Cc |
Monoclinic |
mS28 |
BaB2S4 |
| BaAl2S4 |
Pa |
Cubic |
cP84 |
BaAl2S4 |
| BaAl2Se4 |
P4/nnc |
Tetragonal |
tP28 |
BaAl2Se4 |
| BaAl2Te4 |
P4/nbm |
Tetragonal |
tP14 |
BaAl2Te4 |
| BaSbBS4 |
Pnma |
Orthorhombic |
oP28 |
KBaPO4 |
| BaBiBS4 |
C2/m |
Monoclinic |
mS28 |
BaBiBS4 |
| BaAg2GeS4 |
I2m |
Tetragonal |
tI16 |
K3VO4 |
| BaAg2SnS4 |
I222 |
Orthorhombic |
oI16 |
BaAg2SnS4 |
| BaAu2SnS4 |
P21212 |
Orthorhombic |
oP32 |
BaAu2SnS4 |
| BaCdSnS4 |
Fdd2 |
Orthorhombic |
oF224 |
BaCdSnS4 |
| BaHgSnS4 |
Pnn2 |
Orthorhombic |
oP28 |
BaHgSnS4 |
| EuNa2SiSe4 |
R3c |
Trigonal |
hR48 |
EuNa2SiSe4 |
| EuNa2GeSe4 |
I3m |
Cubic |
cI16 |
Tl3VS4 |
| YbCaInS4 |
Pnma |
Orthorhombic |
oP28 |
Mg2SiO4 |
Stimulated by their versatile structure types and potential applications and our rich experience in rare-earth chalcogenides,13 we recently synthesized four such compounds, EuZnGeS4 (1), EuGa2S4 (2), EuIn2S4 (3) and EuIn2Se4 (4). Compounds 1, 3 and 4 can be viewed as the derivants of 2. Compound 1 is firstly obtained, 3 and 4 are new phases of EuIn2S4 and EuIn2Se4, respectively, different from the known Cccm ones.14 Compound 2 was only characterized using powder X-ray diffraction data.15 Here, we studied their syntheses, crystal and electronic structures, and optical properties.
Experimental section
Materials
Eu2O3 (99.9%, Aladdin), Zn (99.99%, Aladdin), Ga2O3 (99.999%, Aladdin), In2O3 (99.9%, Aladdin), Ge (99.999%, Aladdin), S (99.95%, Aladdin), Se (99.999%, Aladdin), B powder (99%, Aladdin) and KI (99.0%, China Sinopharm Chem.).
Syntheses and analyses
All starting materials were used as received without further purification. Single crystals of the title compounds were obtained by solid-state reactions with KI as flux. The starting materials are stoichiometric mixture of Eu2O3, Zn, Ge and S for 1, Eu2O3, Ga2O3 and S for 2, Eu2O3, In2O3 and S for 3, and Eu2O3, In2O3 and Se for 4. A certain amount of boron powder and KI were added to each sample as reducing reagent and flux, respectively. Each sample has a total mass of 500 mg and 400 mg KI additional. The mixtures of starting materials were ground into fine powder in an agate mortar and pressed into pellets, followed by being loaded into quartz tubes. The tubes were evacuated to be 1 × 10−4 torr and flame-sealed. The samples were placed into a muffle furnace, heated from room temperature to 573 K in 5 h and equilibrated for 10 h, followed by heating to 923 K in 5 h and equilibrated for another 10 h, then heated to 1223 K in 5 h and homogenized for 10 days, finally cooled down to 573 K in 5 days and powered off. All the crystals of 1–4 stable in moisture and air were obtained and hand-picked under a microscope since the yields were not high, then washed using ethanol and water under ultrasonic wave, whose purities were confirmed by powder X-ray diffraction (PXRD) study. The PXRD patterns were collected with a Bruker D8 Advance diffractometer at 40 kV and 100 mA for CuKα radiation (λ = 1.5406 Å) with a scan speed of 5° min−1 at room temperature. The simulated patterns were produced using Mercury v2.3 program provided by the Cambridge Crystallographic Data Center and single-crystal reflection data. A representative PXRD pattern of 3 corresponds well with the simulated one (Fig. 1), indicating it is phase-pure of the selected crystals. Semiquantitative microscope element analysis on the as-prepared single crystals was performed on a field-emission scanning electron microscope (FESEM, HITACHI S-4800II) equipped with an energy dispersive X-ray spectroscope (EDS, Bruker, Quantax), which confirmed the presence of each elements with the approximate compositions of 1–4. The exact compositions were established from X-ray structure determination.
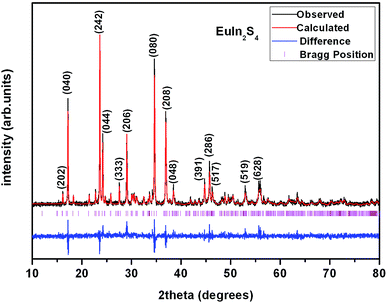 |
| | Fig. 1 Powder XRD pattern of 3. Calculated, experimental and residual diffraction patterns are marked with red, black and blue lines, respectively. Bragg reflections are indicated with vertical ticks. The relatively big residuals are caused by the orientation of the crystalline sample. | |
Structure determination. The intensity data set was collected on Bruker D8 QUEST X-ray diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least-squares techniques on F2 with anisotropic thermal parameters for all atoms. All the calculations were performed with Siemens Shelxtl-97 crystallographic software package.16 The final refinement included anisotropic displacement parameters for all atoms and a secondary extinction correction. The crystallographic data, atomic coordinates and equivalent isotropic displacement parameter, and bond lengths are listed in Tables 2, S1 and S2,† respectively. Their CIF documents have also been deposited with Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany [fax: +49-7247-808-666; e-mail: E-mail: crysdata@fiz.karlsruhe.de] with depository number CSD-432057 for 1, CSD-432055 for 2, CSD-432056 for 3 and CSD-432058 for 4.†
Table 2 Crystal data and structure refinement parameters of 1–4
| R1 = ||Fo| − |Fc||/|Fo|; wR2 = [w(Fo2 − Fc2)2]/[w(Fo2)2]1/2. |
| Chemical formula |
EuZnGeS4 (1) |
EuGa2S4 (2) |
EuIn2S4 (3) |
EuIn2Se4 (4) |
| Fw |
418.16 |
419.64 |
509.84 |
697.44 |
| T (K) |
296 |
| Crystal system |
Orthorhombic |
| Space group |
Fddd |
| Z |
32 |
| a (Å) |
12.234(4) |
12.209(11) |
13.016(3) |
13.490(1) |
| b (Å) |
20.398(6) |
20.445(18) |
20.772(5) |
21.748(1) |
| c (Å) |
20.682(6) |
20.680(19) |
21.076(5) |
21.859(1) |
| V (Å3) |
5161(3) |
5162(8) |
5699(2) |
6412.9(6) |
| Dcalcd (g cm−3) |
4.305 |
4.320 |
4.754 |
5.779 |
| μ (mm−1) |
19.076 |
19.048 |
16.166 |
31.487 |
| F(000) |
6048 |
6048 |
7200 |
9504 |
| θ range (°) |
2.18 to 25.50 |
2.18 to 25.49 |
2.08 to 25.48 |
2.01 to 27.50 |
| Measd. reflns |
5315 |
2843 |
9320 |
7650 |
| Indep. reflns/Rint |
1203/0.0283 |
1171/0.0303 |
1329/0.0581 |
1844/0.0403 |
| R1/wR2 (I > 2σ(I))a |
0.0483/0.1245 |
0.0342/0.0841 |
0.0327/0.0793 |
0.0287/0.0595 |
| R1/wR2 (all data)a |
0.0553/0.1279 |
0.0421/0.0894 |
0.0428/0.0878 |
0.0493/0.0666 |
| GOF on F2 |
1.129 |
1.145 |
1.090 |
1.093 |
| Δρmax/Δρmin, e Å−3 |
2.571/−1.815 |
3.368/−2.110 |
1.627/−2.332 |
2.042/−1.457 |
UV-Vis-NIR diffuse reflectance spectroscopies. The diffuse reflectance spectra were recorded at the room temperature on a computer-controlled Lambda 900 UV-Vis-NIR spectrometer equipped with an integrating sphere in the wavelength range of 200–1700 nm. A BaSO4 plate was used as a reference, on which the finely ground powdery sample was coated. The absorption spectra were calculated from reflection spectrum by the Kubelka–Munk function.17
Calculation details. The calculation models were built directly from the single-crystal diffraction data of 1 and 2. The electronic structure calculation including band structure and density of states based on density functional theory (DFT) was performed using Material Studio.18 The generalized gradient approximation (GGA) was chosen as the exchange–correlation functional and a plane wave basis with the projector-augmented wave (PAW) potentials was used. The plane-wave cutoff energies of 1 and 2 are 480.0 eV and the threshold of 10−5 eV were set for the self-consistent-field convergence of the total electronic energy. The electronic configurations for Eu, Zn, Ge, Ga and S were 4f and 6s, 3d and 4s, 4s and 4p, 4s and 4p, and 3s and 3p, respectively. The numerical integration of the Brillouin zone was performed using 2 × 2 × 2 Monkhorst–Pack k-point meshes and the Fermi level (Ef = 0 eV) was selected as the reference.
Results and discussion
Crystal structure
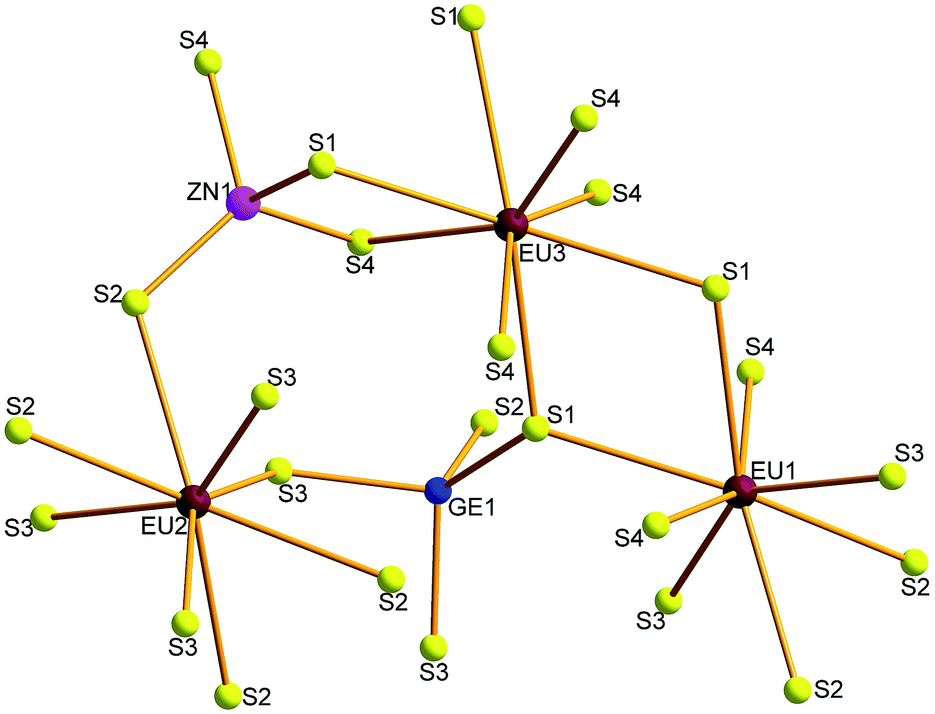
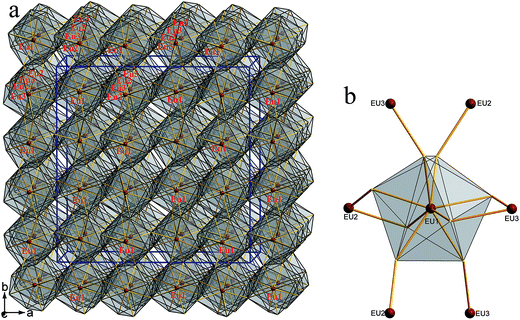
Compounds 1–4 crystallize in the orthorhombic space group Fddd. The crystal structure of 1 is illustrated as a representative as they are isostructural. There are three Eu, one Zn, one Ge and four S atoms in the crystallographically independent unit. Its coordination geometry is shown in Fig. 2. All the three Eu atoms coordinated with eight neighboring S atoms to form EuS8 bicapped trigonal prisms, Zn and Ge are fourfold-coordinated with four S atoms to form ZnS4 or GeS4 tetrahedron.
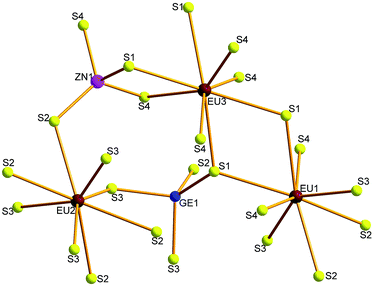 |
| | Fig. 2 Coordination geometry of 1. | |
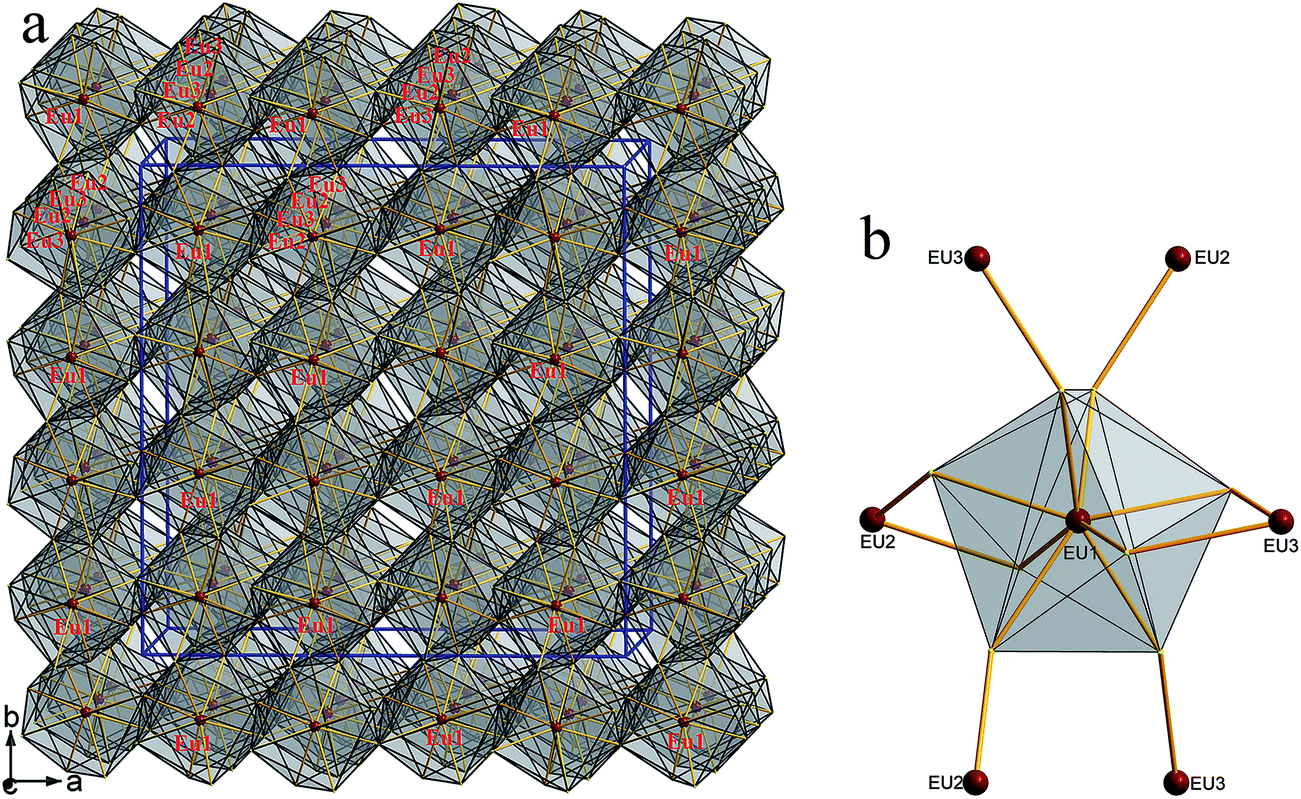
The structure can be viewed as layered if no consideration of the connection between Eu and S atoms (Fig. 3a). The layers parallel to the ac plane are constructed by the connection between ZnS4 and GeS4 tetrahedra (Fig. 3b). It can be observed that either ZnS4 or GeS4 tetrahedra form dimers via sharing edges, respectively, namely, [Zn2S6]8− or [Ge2S6]8− units. Both of which are isolated and alternately linked to form the 2D structure. Take the whole structure consideration, the structure of 1 can be described as Eu–S bonds constructed 3D structure, and Zn2+ and Ge4+ cations occupy the tetrahedral cavities (Fig. 4a). It is necessary to understand how the EuS8 bicapped trigonal prisms connect if we want to know how the 3D structure forms. The Eu atoms arrange regularly along the c direction. Eu(1) atoms arrange in a line along the c direction, while Eu(2) and Eu(3) atoms alternatively arrange in another line along the c direction. As shown in Fig. 4b, each EuS8 units has six neighboring EuS8 units, four of them connect with the central one via sharing edges, while the other two connected via sharing corners. The former and latter have the Eu–Eu distances of 5.208(2) and 5.947(1) Å, respectively.
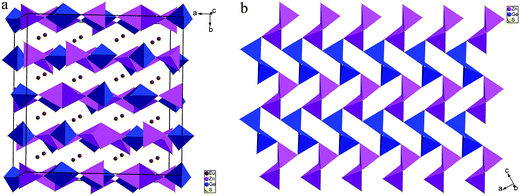 |
| | Fig. 3 (a) 2D structure of 1 constructed by the connection between GaS4 and ZnS4 tetrahedra. Eu–S bonds are omitted for clarity. (b) The [ZnGeS4]2− layer viewed along the b direction. GaS4 and ZnS4 tetrahedra are represented as pink and blue polyhedra, respectively. | |
 |
| | Fig. 4 (a) 3D framework constructed by the three types of EuS8 bicapped trigonal prisms viewed along the c direction; (b) the neighboring Eu(2) and Eu(3) atoms around one Eu(1)S8 unit. | |
The Zn–S and Ge–S bond lengths in 1 are in the range of 2.247(4)–2.303(5) and 2.246(4)–2.315(4) Å, respectively, which are in good agreement with 2.243(2)–2.305(2) Å of Ga–S bonds in 2. These values are consistent with 2.242(1)–2.342(1) Å of Ga–S bonds in (K3I)[SmB12(GaS4)3].19 The Eu–S bonds in 1 have the distances of 3.070(4)–3.112(5) Å, similar with those discovered in RbEuGeS4.20
As compounds 2–4 have more similar compositions. Here only the comparison is performed between 1 and 2. There are two Ga sites in 2, which can be substituted by one divalent and one tetravalent cations, respectively. So far, there are no quaternary II-III2 (III-III′)-Ch4 or II-II′(I2)-IV-Ch4 compounds reported with Fddd structure. Compound 1 represents the first one with this structure. It is interesting to study which one of the two Ga sites will be replaced by divalent cation. First, it has to be mentioned that both of the two Ga atoms occupy 32h sites. Second, both Zn and Ge positions are close to that of Ga in periodic table of the elements, so it is almost impossible to distinguish them by atomic weights when solving the structure. Third, bond-valence calculation was performed on 2 to help determine which site will be occupied by Zn2+ ion, and the other one by Ge4+ ion. If Ga(1) and Ga(2) atoms are replaced by Zn and Ge atoms, respectively, their bond valences calculated to be 2.368 and 3.267, respectively. If Zn and Ge exchange their sites, their bond valences are 2.329 and 3.331, respectively. Even the calculation precision is considered, it is still difficult to determine that Ga(1) site is occupied by Zn or Ge atom. The most reasonable solution is that Zn and Ge atoms statistically co-occupy both of two Ga sites according to the bond-valence calculation results. For simplicity and convenience, the structure of 1 is described above as Zn and Ge atom completely occupy one Ga site, respectively. Definitely, the molar ratio of Zn and Ge should be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to maintain electric neutrality.
:1 to maintain electric neutrality.
Optical properties
The UV-Vis-NIR diffuse reflectance spectra of 1–3 are shown in Fig. 5. Their optical band gaps are determined to be 2.26, 2.32 and 2.37 eV, respectively, which are consistent with their orange or yellow colors. Comparing these values, it may be proposed that there will be no big band gap change when the Ga atoms in EuGa2S4 are replaced by II + IV two types of atoms or by heavier III atoms like In atoms. This supposition may help design II-III2 (III-III′)-Ch4 or II-II′(I2)-IV-Ch4 compounds with tunable band gaps.
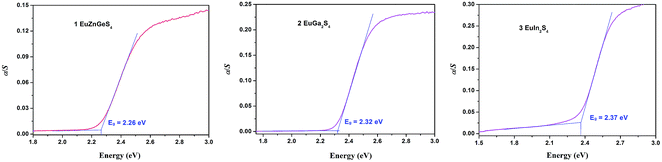 |
| | Fig. 5 Diffuse reflection spectra of 1–3. | |
Theory investigation
To investigate the electronic structures of 1 and 2, their band structures together with densities of states (DOS) computations based on the DFT theory were performed using Material Studio software.18 The calculated band structures along high symmetry points of the first Brillouin zone are shown in Fig. 6, from which it can be seen that the band gaps of 1 and 2 are calculated to be 1.32 and 2.69 eV (Table 3), respectively, the former one is underestimated but reasonable in view of the calculation precision.21 Both the lowest conduction band (CB) and highest valence band (VB) of 1 are located at Γ point, indicating that 1 has a direct band gap. Different from 1, the CB and VB of 2 are located at Γ and X points, respectively, showing that 2 has an indirect band gap.
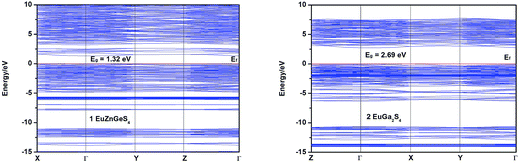 |
| | Fig. 6 Calculated band structures of 1 and 2. The Fermi level is chosen as the energy reference at 0 eV. | |
Table 3 Experimental and calculated optical band gaps of 1–3
| |
|
EuZnGeS4 (1) |
EuGa2S4 (2) |
EuIn2S4 (3) |
| Band gap (eV) |
Experimental |
2.26 |
2.32 |
2.37 |
| Calculated |
1.32 |
2.69 |
— |
The total and partial densities of states (DOS and PDOS) of 1 and 2 are plotted in Fig. 7. For 1, the highest VB is mainly constituted of S-3p, and the lowest CB is mainly composed of Ge-4s and S-3p orbitals. The VBs between 0 and –5.0 eV are originated mainly from S-3p and Eu-4f orbitals. The CBs ranging from 2.5 to 7.5 eV are primarily consisted of Ge-4p, and minor Zn-4s and S-3p orbitals. Briefly, the band gap of 1 is determined by the S-3p and Ge-4s orbitals, the respective valence orbitals of S2− and Ge4+ ions. For 2, the highest VB is mainly constituted of S-3p, and the lowest CB is mainly composed of Ga-4s, S-3p and Ga-4p orbitals, so its band gap is determined by valence orbitals of S2− and Ga3+ ions. The VBs from 0 to −4.0 eV are primarily consisted of S-3p, Eu-4f and minor Ga-4p orbitals. Comparing the PDOS of 1 and 2, it can be concluded that the optical absorptions of 1 and 2 are mainly ascribed to the charge transitions from S-3p to valence orbitals of Ge4+ or Ga3+ cations.
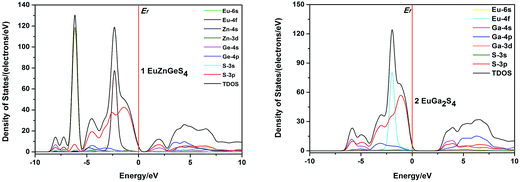 |
| | Fig. 7 Calculated density of states (DOS) of 1 and 2. The Fermi level is chosen as the energy reference at 0 eV. | |
It is well known that direct or indirect semiconductors have different applications in the optoelectronics research field. From EuGa2S4 to EuZnGeS4, two Ga3+ sites are replaced by one Zn2+ and one Ge4+ cations, which is a simple substitution but the parent structure EuGa2S4 can be maintained and the measured band gap have a little change. Together with the above discussion, it should be interesting to understand why semiconductor type can be changed while crystal structure and measured band gap keep stable. Besides, it seems the optical absorptions of 1 and 2 have little relationship with the valence orbitals of Eu2+, which may give us some rules to explore II-III2 (III-III′)-Ch4 or II-II′(I2)-IV-Ch4 compounds with desirable band gaps for their further photoelectric applications. One example might be used to verify this speculation. Other II-III2-Ch4 compounds containing divalent transition-metal, CdGa2Se4 and HgGa2Se4, have direct band gaps of 2.40 and 1.93 eV, respectively, close to our sulfides.22 Besides, it is interesting to find that the calculated band gaps for compounds EuZnGeS4, CdGa2Se4 and HgGa2Se4 with direct band gaps are much lower than their experimental values.
Conclusions
Four isostructural europium chalcogenides were synthesized using solid-state reactions. Their measured band gaps indicate that they are semiconductors. Interestingly, the band gap type can be tuned from indirect to direct when using one divalent Zn2+ and one tetravalent Ge4+ cations to substitute the two trivalent Ga3+ cations sites in EuGaS2. This study may give some experience how to tune structures and band gaps of II-III2 (III-III′)-Ch4 or quaternary II-II′(I2)-IV-Ch4 compounds (II = Mg, Ca, Sr, Ba, Eu and Yb; II′ = divalent transition metal; I = Li, Na, Cu and Ag; III = B, Al, Ga and In; III′ = Yb, Sb and Bi; IV = Ge and Sn; Ch = S, Se and Te).
Acknowledgements
We gratefully acknowledge financial support by the NSF of China (21673203), the Higher Education Science Foundation of Jiangsu Province (15KJB150031), State Key Laboratory of Structural Chemistry Fund (20150009), Natural Science Foundation of Yangzhou (YZ2016122) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
-
(a) K. Mitchell and J. A. Ibers, Chem. Rev., 2002, 102, 1929–1952 CrossRef CAS PubMed;
(b) S.-P. Guo, G.-E. Wang, M.-J. Zhang, M.-F. Wu, G.-N. Liu, X.-M. Jiang, G.-C. Guo and J.-S. Huang, Dalton Trans., 2013, 42, 2679–2682 RSC;
(c) Y. Chi, S.-P. Guo, H.-J. Kong and H.-G. Xue, New J. Chem., 2016, 40, 6720–6727 RSC.
- C.-Y. Meng, H. Chen, P. Wang and L. Chen, Chem. Mater., 2011, 23, 4910–4919 CrossRef CAS.
- R. J. Yu, H. K. Yang, B. K. Moon, B. C. Choi and J. H. Jeong, Phys. Status Solidi A, 2012, 12, 2620–2625 CrossRef.
-
(a) H. J. Zhao, Y. F. Zhang and L. Chen, J. Am. Chem. Soc., 2012, 134, 1993–1995 CrossRef CAS PubMed;
(b) Y. F. Shi, Y. K. Chen, M. C. Chen, L. M. Wu, H. Lin, L. J. Zhou and L. Chen, Chem. Mater., 2015, 27, 1876–1884 CrossRef CAS;
(c) M. J. Zhang, B. X. Li, B. W. Liu, Y. H. Fan, X. G. Li, H. Y. Zeng and G. C. Guo, Dalton Trans., 2013, 42, 14223–14229 RSC.
- Z. Z. Luo, C. S. Lin, W. L. Zhang, H. Zhang, Z. Z. He and W. D. Cheng, Chem. Mater., 2013, 26, 1093–1099 CrossRef.
- Z. Z. Luo, C. S. Lin, W. D. Cheng, H. Zhang, W. L. Zhang and Z. Z. He, Inorg. Chem., 2012, 52, 273–279 CrossRef PubMed.
- K. Feng, X. Jiang, L. Kang, W. Yin, W. Hao, Z. S. Lin, J. Y. Yao, Y. C. Wu and C. T. Chen, Dalton Trans., 2013, 42, 13635–13641 RSC.
- C. Li, W. L. Yin, P. F. Gong, X. S. Li, M. L. Zhou, A. Mar, Z. S. Lin, J. Y. Yao, Y. C. Wu and C. T. Chen, J. Am. Chem. Soc., 2016, 138, 6135–6138 CrossRef CAS PubMed.
- K. Wu, X. Su, S. L. Pan and Z. H. Yang, Inorg. Chem., 2015, 54, 2772–2779 CrossRef CAS PubMed.
- K. Wu, X. Su, Z. H. Yang and S. L. Pan, Dalton Trans., 2015, 44, 19856–19864 RSC.
-
(a) N. Zhen, K. Wu, Y. Wang, Q. Li, W. H. Gao, D. W. Hou, Z. H. Yang, H. D. Jiang, Y. J. Dong and S. L. Pan, Dalton Trans., 2016, 45, 10681–10688 RSC;
(b) P. C. Donohue and J. E. Hanlon, J. Electrochem. Soc., 1974, 137–142 CrossRef CAS;
(c) M. Tampier and D. Johrendt, Z. Anorg. Allg. Chem., 2001, 627, 312–320 CrossRef CAS;
(d) T. Sasaki, H. Takizawa, T. Takeda and T. Endo, Mater. Res. Bull., 2003, 38, 33–39 CrossRef CAS;
(e) T. Takizawa, C. Komatsu-Hidaka, K. Asaka, T. Isomoto and H. Matsushita, Jpn. J. Appl. Phys., 2000,(Suppl. 39-1), 35–40 CrossRef CAS;
(f) A. Assoud, N. Soheilnia and H. Kleinke, Chem. Mater., 2005, 17, 2255–2261 CrossRef CAS.
-
(a) O. Gomis, R. Vilaplana, F. J. Manjón, E. Pérez-González, J. López-Solano, P. Rodríguez-Hernández, A. Muñoz, D. Errandonea, J. Ruiz-Fuertes, A. Segura, D. Santamaría-Pérez, I. M. Tiginyanu and V. V. Ursaki, J. Appl. Phys., 2012, 111, 013518 CrossRef;
(b) O. Gomis, D. Santamaría-Pérez, R. Vilaplana, R. Luna, J. A. Sans, F. J. Manjón, D. Errandonea, E. Pérez-González, P. Rodríguez-Hernández, A. Muñoz and V. V. Ursaki, J. Alloys Compd., 2014, 583, 70–78 CrossRef CAS;
(c) O. Gomis, R. Vilaplana, F. J. Manjón, J. Ruiz-Fuertes, E. Pérez-González, J. López-Solano, E. Bandiello, D. Errandonea, A. Segura, P. Rodríguez-Hernández, A. Muñoz, V. V. Ursaki and I. M. Tiginyanu, Phys. Status Solidi B, 2015, 252, 2043–2051 CrossRef CAS;
(d) J. Ruiz-Fuertes, D. Errandonea, F. J. Manjón, D. Martínez-García, A. Segura, V. V. Ursaki and I. M. Tiginyanu, J. Appl. Phys., 2008, 103, 063710 CrossRef;
(e) O. Gomis, R. Vilaplana, F. J. Manjón, D. Santamaría-Pérez, D. Errandonea, E. Pérez-González, J. López-Solano, P. Rodríguez-Hernández, A. Muñoz, I. M. Tiginyanu and V. V. Ursaki, J. Appl. Phys., 2013, 113, 073510 CrossRef;
(f) D. Errandonea, R. S. Kumar, F. J. Manjón, V. V. Ursaki and I. M. Tiginyanu, J. Appl. Phys., 2008, 104, 063524 CrossRef;
(g) D. Errandonea, R. S. Kumar, O. Gomis, F. J. Manjón, V. V. Ursaki and I. M. Tiginyanu, J. Appl. Phys., 2013, 114, 233507 CrossRef;
(h) D. Santamaría-Pérez, M. Amboage, F. J. Manjón, D. Errandonea, A. Muñoz, P. Rodríguez-Hernández, A. Mújica, S. Radescu, V. V. Ursaki and I. M. Tiginyanu, J. Phys. Chem. C, 2012, 116, 14078–14087 CrossRef;
(i) O. Gomis, R. Vilaplana, F. J. Manjón, D. Santamaría-Pérez, D. Errandonea, E. Pérez-González, J. López-Solano, P. Rodríguez-Hernández, A. Muñoz, I. M. Tiginyanu and V. V. Ursaki, Mater. Res. Bull., 2013, 48, 2128–2133 CrossRef CAS.
-
(a) S.-P. Guo, Y. Chi and H.-G. Xue, Inorg. Chem., 2015, 54, 11052–11054 CrossRef CAS PubMed;
(b) Y. Chi and S.-P. Guo, J. Mol. Struct., 2017, 1127, 53–58 CrossRef CAS;
(c) Y. Chi, H.-J. Kong and S.-P. Guo, Inorg. Chim. Acta, 2016, 448, 56–60 CrossRef CAS;
(d) S.-P. Guo, Y. Chi, B.-W. Liu and G.-C. Guo, Dalton Trans., 2016, 45, 10459–10465 RSC;
(e) S.-P. Guo and G.-C. Guo, J. Mater. Chem. A, 2014, 2, 20621–20628 RSC;
(f) S.-P. Guo, Y. Chi, J.-P. Zou and H.-G. Xue, New J. Chem., 2016, 40, 10219–10226 RSC.
- O. M. Aliev, Inorg. Mater., 1980, 16, 1027–1031 Search PubMed.
-
(a) R. Roques, R. Rimet, J. P. Declercq and G. Germain, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 555–557 CrossRef;
(b) T. E. Peters and J. A. Baglio, J. Electrochem. Soc., 1972, 119, 230–236 CrossRef CAS.
- Siemens, SHELXTL™ Version 5 Reference Manual, Siemens Energy & Automation Inc., Madison, Wisconsin, USA, 1994 Search PubMed.
-
(a) W. W. Wendlandt and H. G. Hecht, Reflectance Spectroscopy, Interscience Publishers, New York, 1966 Search PubMed;
(b) G. Kortüm. Reflectance Spectroscopy, Springer, 1969 Search PubMed.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- S. P. Guo, G. C. Guo, M. S. Wang, J. P. Zou, H. Y. Zeng, L. Z. Cai and J. S. Huang, Chem. Commun., 2009, 4366–4368 RSC.
- A. Choudhury, L. A. Polyakova, I. Hartenbach, T. Schleid and P. K. Dorhout, Z. Anorg. Allg. Chem., 2006, 632, 2395–2401 CrossRef.
-
(a) D. Errandonea, D. Martínez-García, R. Lacomba-Perales, J. Ruiz-Fuertes and A. Segura, Appl. Phys. Lett., 2006, 89, 091913 CrossRef;
(b) R. Lacomba-Perales, D. Errandonea, A. Segura, P. Rodríguez-Hernández, S. Radescu, J. López-Solano, A. Mujica and A. Muñoz, J. Appl. Phys., 2011, 110, 043703 CrossRef;
(c) V. Panchal, D. Errandonea, A. Segura, P. Rodríguez-Hernández, A. Muñoz, S. Lopez-Moreno and M. Bettinelli, J. Appl. Phys., 2011, 110, 043723 CrossRef.
- F. J. Manjón, O. Gomis, P. Rodríguez-Hernández, E. Pérez-González, A. Muñoz, D. Errandonea, J. Ruiz-Fuertes, A. Segura, M. Fuentes-Cabrera, I. M. Tiginyanu and V. V. Ursaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 195201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic files in CIF format and additional tables. See DOI: 10.1039/c6ra25283d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Huai-Guo Xue
* and
Huai-Guo Xue