
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eosin Y dye-based porous organic polymers for highly efficient heterogeneous photocatalytic dehydrogenative coupling reaction†
Chang-An Wang*,
Yan-Wei Li,
Xue-Li Cheng ,
Jian-Ping Zhang and
Yin-Feng Han*
,
Jian-Ping Zhang and
Yin-Feng Han*
College of Chemistry and Chemical Engineering, Taishan University, Tai'an, Shandong 271021, P. R. China. E-mail: wangcha_chem@163.com; hanyf1978@163.com
First published on 14th November 2016
Abstract
Construction of porous organic polymers (POPs) as metal-free heterogeneous organic photocatalysts for highly efficient catalytic organic transformations using visible light remains a key challenge. Herein, we report the “bottom-up” strategy to facilely synthesize two Eosin Y dye-based POP frameworks (EY-POPs) for highly efficient heterogeneous organic-photocatalysis. Owing to the high BET surface area and the built-in character of the covalently linked catalytic sites of EY-POPs, these photoactive polymers show excellent catalytic activity in photocatalyzing the aza-Henry reaction. The superior utility of the EY-POP-1 polymer in catalysis was demonstrated by the broad scope of the reactants and the high yield of the reaction products. Moreover, the EY-POP-1 polymer shows robust recycling capability with good retention of photoactivity over at least twelve cycles without any significant loss of the catalytic activity (94–98% yield).
Introduction
Organic transformations driven by visible light offer a sustainable alternative to many ever-pressing environmental issues. With the aim to exploit photons for synthetic purposes, visible-light photoredox catalysis has recently proven to be a powerful method to perform several organic reactions.1 Because the majority of organic substrates in these reactions do not readily absorb photons in the visible region, photocatalysts are often required in visible light driven organic reactions. To date, transition-metal-systems, such as ruthenium(II), mainly [Ru(bpy)3Cl2], or iridium(III) complexes have been applied to photoredox reactions2 such as radical addition and cycloaddition reactions,3 aza-Henry reactions,4 asymmetric alkylation of aldehydes,5 and other reactions. Recently, organic dyes have been also successfully applied as effective visible light metal-free photoredox organocatalysts.6 However, like other precious homogeneous catalysis, these homogeneous photocatalysts are generally considered of low efficiency due to the high catalyst loading and the difficulty in separation from the products, which restricts their practical application in large scale. Furthermore, their potential toxicity could lead to detrimental effects on public health and the environment. Accordingly, it is highly desirable to develop robust and reusable heterogeneous photocatalytic systems based on these homogeneous photocatalysts.7 The ability to recover and reuse such heterogeneous photocatalysts can both eliminate the contamination of organic products by trace amounts of heavy metals, and also reduce processing and waste disposal costs in large scale reactions.In recently, porous organic polymers (POPs), such as conjugated microporous polymers (CMPs),8 polymers of intrinsic microporosity (PIMs),9 hyper-cross-linked polymers (HCPs),10 and covalent organic frameworks (COFs),11 can be construct from well-designed organic monomers, have attracted significant scientific attention in the area of gas storage/separation,12 organic photoelectric,13 and heterogeneous catalysis.14 In particular, conjugated microporous polymers (CMPs) can serve as an ideal platform for incorporating photosensitive monomers into highly stable, recyclable, and reusable heterogeneous photocatalyst systems by taking advantage of their extended π-conjugation structure and the ability to tune their compositions and properties at the molecular level.15 For example, Lin et al. reported the highly stable and porous cross-linked polymers with phosphorescent [Ru(bpy)3]2+ and [Ir(ppy)2(bpy)]+ building blocks for efficient photocatalysis.7a Son et al. reported benzodifuran-based CMPs via post modification, and the resulting polymer showed high photocatalytic activity for the oxidative conversion of primary amines into imines.15d Wang and Zhang et al. synthesized conjugated polybenzodiazoles as organic heterogeneous photocatalyst for visible-light-promoted H2 evolution,15i and so on. Remarkable progress has been made in the development of photosensitive monomers based CMPs frameworks for efficient heterogeneous photocatalysis. However, most of these photosensitive monomers either require tedious experimental procedures to prepare or expensive, that limit low-cost preparation of the polymers on a large scale. In this context, the bottom-up construction of heterogeneous photocatalyst directly from the high activity and low cost monomers which without any further synthetic elaboration, may possibly overcome these drawbacks.
Organic dyes have been studied widely in the fields of dye-sensitized solar cells16 and homogeneous catalysis.6 Eosin Y (Scheme 1) and Rose Bengal, the most commonly used organic dyes, are demonstrated to be effective noble metal-free photo-organic-catalysts, and have been studied in the area of homogeneous photocatalysis due to their low cost and low toxicity.6a–d Eosin Y and Rose Bengal are the ideal candidates to construct photoactive porous organic polymers, the main reasons lie in that: (1) the structural rigidity of the monomers are readily offered by the benzene ring, which could contribute to the construction of rigid POPs frameworks; (2) the bromine or iodine atoms on the benzene ring (without any further synthetic elaboration) could be easily reacted with the structural building blocks; (3) high effective photocatalytic activity, low cost, and low toxicity. Herein, we report the bottom-up construction of robust photoactive porous organic polymers (EY-POPs) embedded with Eosin Y dye for highly efficient heterogeneous photocatalytic dehydrogenative coupling reaction (Scheme 1). Very recently, Jiang and Cooper et al. reported conjugated microporous polymers with Rose Bengal dye as heterogeneous organic-photocatalyst for highly efficient aza-Henry reaction.15c Unlike the significant work of Jiang and Cooper et al., we synthesized two Eosin Y-functionalized POPs frameworks with different π-conjugation structure, and preliminary investigated the influence of the extended π-conjugation structure on the catalytic activity.
 | ||
| Scheme 1 Design and synthesis of Eosin Y dye-embedded porous organic polymers (EY-POPs) via the bottom-up strategy. | ||
Results and discussion
Design, synthesis and characterization of Eosin Y dye-based porous organic polymers (EY-POPs)
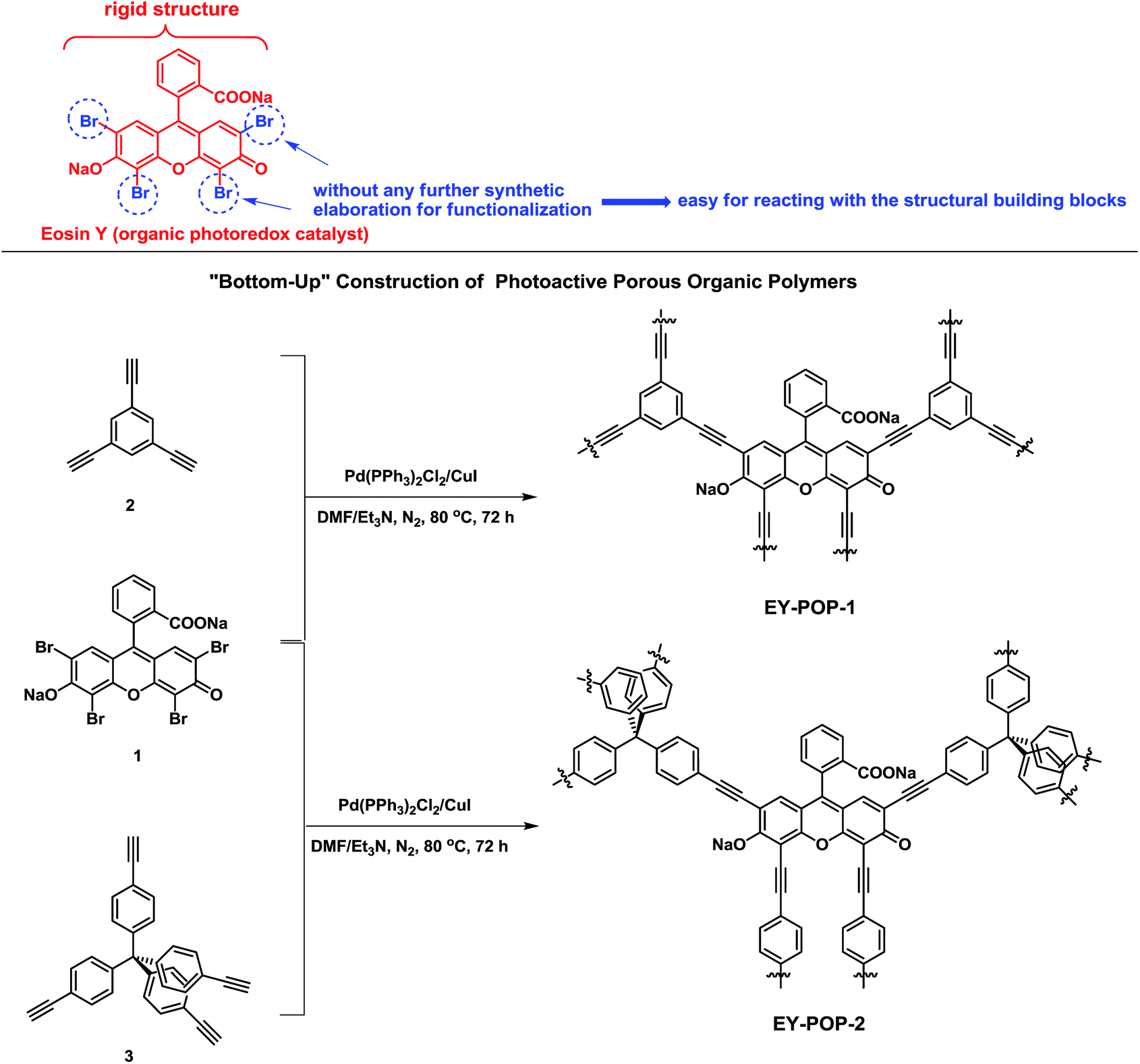
One of the key concerns for constructing functional POPs is the rational design of the functional building blocks (FBBs). The desired FBBs candidates should be rigid in structure, suitable in size, easy to prepare, ready to react with the structural building blocks. With these considerations in mind, we accordingly select the skeleton of Eosin Y as the FBB candidate to construct the photoactive porous organic polymers. As illustrated in Scheme 1, the main reasons lie in that: (1) the structural rigidity of organic photoredox catalyst is readily offered by the benzene ring, which could contribute to the construction of rigid POPs frameworks; (2) the bromine atoms on the benzene ring could be easily reacted with the structural building blocks; (3) Eosin Y is demonstrated to be effective noble metal-free photo-organic-catalyst, together with low cost and low toxicity. The polymer networks were easily prepared through palladium-catalyzed Sonogashira–Hagihara cross-coupling reaction of Eosin Y dye (EY) with different structural building blocks (see the Experimental section for experimental details). Reaction of 1,3,5-triethynylbenzene (2) with Eosin Y gave the polymer EY-POP-1 with extended π-conjugation structure; and the equivalent reaction of tetra(4-ethynylphenyl)methane (3) with Eosin Y gave the copolymer EY-POP-2 with no extended π-conjugation structure. All the constructed POPs networks are expected to be three-dimensional with varied porosity due to the three-pronged aryleneethynylene linkages and the rigid tetrahedral structure. After the Sonogashira–Hagihara coupling reactions (see Experimental section), the desired Eosin Y-embedded POPs networks were readily precipitated from the solution as red colored solids, both of these polymers are insoluble in all organic solvents tested due to their highly cross-linked structures. The thermogravimetric analysis (TGA) revealed that the EY-POPs networks were stable up to 350 °C under a nitrogen atmosphere (Fig. 1a). Power X-ray diffraction measurements indicate that the EY-POPs networks are amorphous in nature, as for other reported POPs networks in our laboratory.17 The UV-visible absorption spectra of EY-POPs networks recorded for solid state powders demonstrates that both polymers show broad absorption in the range from 250 to 600 nm (Fig. 1b), which is similar to that of the Eosin Y monomer, indicating that the EY-POPs networks can absorb light across a wide range of the visible spectrum.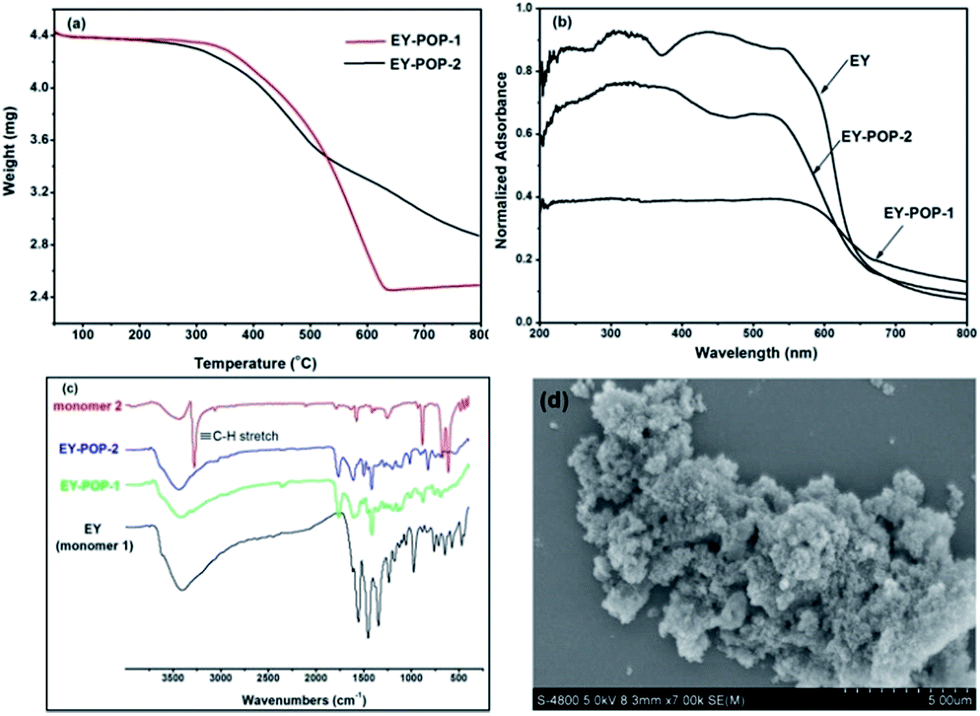 | ||
| Fig. 1 (a) Thermogravimetric analysis (TGA) curves for EY-POPs networks. (b) UV-visible absorption spectra for the monomer of EY and the EY-POPs networks as solid state powders. (c) FT-IR comparison of monomer 1 (in black), monomer 2 (in red), EY-POP-1 (in green), and EY-POP-2 (in blue). (d) SEM image for the EY-POP-1 network. | ||
The absence of the carbon–hydrogen stretching peak of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H group around 3280 cm−1 in the IR spectra of the EY-POPs suggested that most of the alkyne group in the starting materials have been consumed to form –CC– groups in the POPs networks. The high degree of spectral similarity of EY-POP-1 and EY-POP-2 also demonstrates the structural consistency of these materials (Fig. 1c). The SEM image of EY-POPs displayed rather rough surfaces and the particles were on the order of micrometer in size (Fig. 1d).
C–H group around 3280 cm−1 in the IR spectra of the EY-POPs suggested that most of the alkyne group in the starting materials have been consumed to form –CC– groups in the POPs networks. The high degree of spectral similarity of EY-POP-1 and EY-POP-2 also demonstrates the structural consistency of these materials (Fig. 1c). The SEM image of EY-POPs displayed rather rough surfaces and the particles were on the order of micrometer in size (Fig. 1d).
The permanent porosity of EY-POPs networks were investigated by nitrogen adsorption/desorption measurements at 77 K, and the obtained results are listed in Table S1.† As shown in Fig. 2a and b, both of the EY-POPs networks showed similar adsorption and desorption isotherms, and gave rise to type I nitrogen gas sorption isotherms according to the IUPAC classification.18 The adsorption shows a steep nitrogen gas uptake at low relative pressure, indicating that the materials are microporous. The application of the Brunauer–Emmett–Teller (BET) modal resulted in the surface areas of 587 and 718 m2 g−1 for EY-POP-1 (Fig. S1, ESI†) and EY-POP-2 (Fig. S2, ESI†), respectively. As calculated by non-local density functional theory (NLDFT), the pore size distribution (PSD) of EY-POP-1 and EY-POP-2 mainly distributed around 0.7 and 1.3 nm, respectively (Fig. 2c and d). This demonstrates a significant proportion of micropores in the EY-POPs networks.
 | ||
| Fig. 2 Nitrogen adsorption/desorption isotherms of EY-POP-1 (a) and EY-POP-2 (b). Pore size distribution (PSD) for EY-POP-1 (c) and EY-POP-2 (d), calculated with non-local density functional theory (NLDFT). | ||
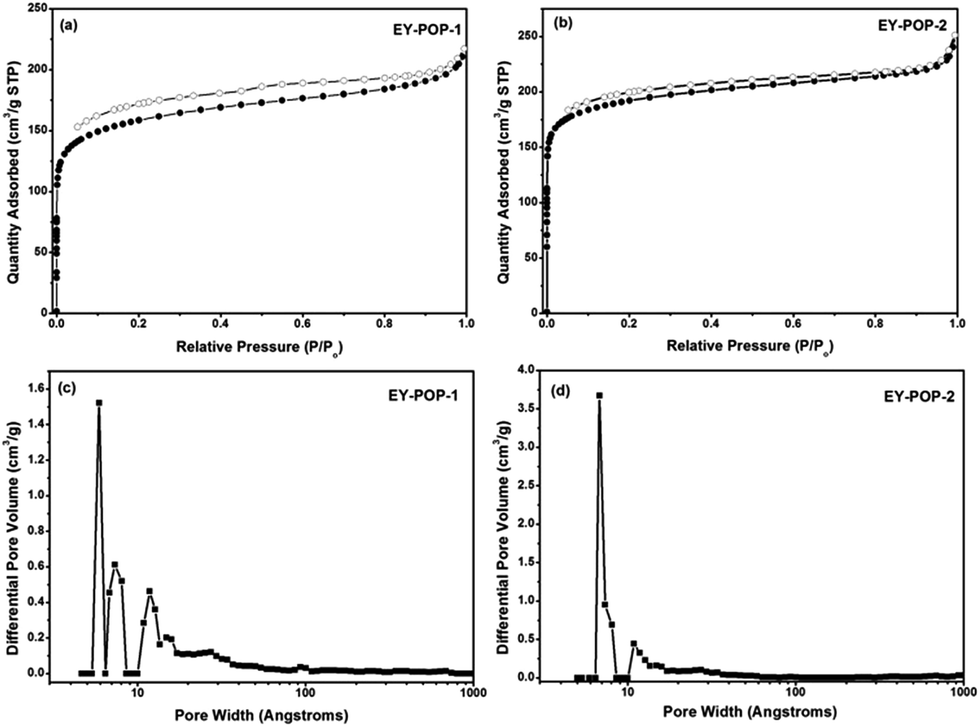
The chemical composition of EY-POPs networks were characterized by solid-state 13C cross-polarization magic-angle spinning (CP/MAS) NMR spectroscopy. Fig. 3 shows the 13C CP/MAS NMR spectra recorded for EY-POP-1 and EY-POP-2. The signals at δ = 168, 156, 147, 129 and 102 ppm confirmed that the Eosin Y monomer skeleton has well been embedded into the polymer networks, while the peak at approximately 82 and 93 ppm represent the alkynyl groups in the EY-POPs networks. The peak at about 129 and 125 ppm confirm that the 1,3,5-triethynylbenzene skeleton has been successful embedded into the EY-POP-1 network, and the peak at about 65, 129 and 147 ppm also confirm that the tetra(4-ethynylphenyl)methane skeleton has been successful embedded into the EY-POP-2 network. All of the results verified the bottom-up construction of the photoactive EY-POPs networks with the Eosin Y skeleton embedded into the frameworks.
 | ||
| Fig. 3 Solid-state 13C CP/MAS NMR spectra for EY-POP-1 and EY-POP-2. The assignments for the 13C MAS NMR signals are indicated (top). | ||
Photocatalytic activity of EY-POPs in the heterogeneous photocatalytic aza-Henry reaction
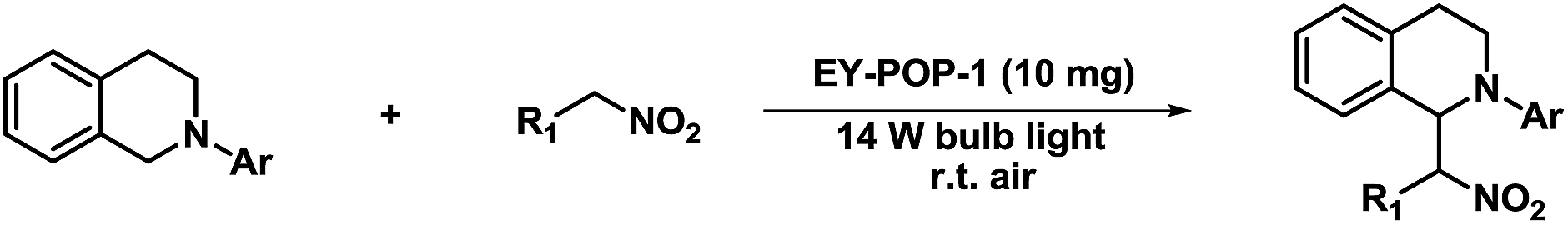
The dehydrogenative coupling reactions, performed under oxidative conditions, are an important C–C bond formation methodology.19 Significant progress has been made recently in achieving visible-light-promoted photocatalytic dehydrogenative coupling reactions with transition metal complexes4 or organic dyes6b,6d as the homogeneous photoredox catalysts. However, the practical application of homogeneous photoredox catalysis is generally hindered due to the requirement for high catalyst loadings and the difficulty in separating the catalyst from the product. To address these limitations, heterogenization of the homogeneous catalyst is a promising option. We investigated the aza-Henry reaction of 2-phenyl-1,2,3,4-tetrahydroisoquinoline with nitromethane as a modal reaction to estimate the photocatalytic activities of the EY-POPs networks (Table 1). In an initial experiment, the oxidative coupling of 2-phenyl-1,2,3,4-tetrahydroisoquinoline (0.20 mmol) with nitromethane (1.0 mL) in the presence of EY-POPs (5 mg), visible light (14 W fluorescent lamp) and air was investigated. Both of the EY-POPs polymers (5.0 mg) showed approximately similar photocatalytic activities for this aza-Henry reaction, the reaction proceeded smoothly and gave the desired product in moderate yield after 36 h (Table 1, entries 1 and 2). Strikingly, we found that the reaction proceeded much faster by increasing the catalyst loading of EY-POPs polymers (10 mg), giving the product in high yield after 24 h (Table 1, entries 3 and 4). Amongst the EY-POPs polymers tested, the EY-POP-1 polymer with the extended π-conjugation structure showed the best reactivity, catalysing almost a complete conversion after 24 h with 10 mg catalyst (entry 3). A number of control experiments were carried out to demonstrate the heterogeneous and photocatalytic nature of the reaction. Nature light gave low yield (35%) and there was no reaction in the dark (Table 1, entries 5 and 6), indicating that light is necessary to promote the electron transfer. On the other hand, the background reaction in the absence of EY-POPs polymers but in the presence of light showed only around <5% yield after 36 h (Table 1, entry 7), verifying that the EY-POPs polymers played a catalytic role in the reaction. The effect of oxygen on the reaction was also examined. The reaction proceeded much slower under an N2 atmosphere excluding oxygen, giving the product in moderate yield (60%) after 36 h (Table 1, entry 8). For the purpose of comparison, we also investigated the catalytic performance of the homogeneous Eosin Y catalyst under the same reaction condition, it can be seen that the efficiency of EY-POP-1 polymer compares well to that of the homogeneous Eosin Y catalyst (Table 1, entry 9).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (× mg) | Light source | Time (h) | Yieldb |
| a Reaction condition: 2-phenyl-1,2,3,4-tetrahydroisoquinoline (41.8 mg, 0.2 mmol), nitromethane (1.0 mL), and EY-POPs, r.t., 14 W household bulb, in air.b Isolated yield after silica gel column chromatography.c The reaction was carried out under N2. | ||||
| 1 | EY-POP-1 (5) | 14 W bulb | 36 | 65 |
| 2 | EY-POP-2 (5) | 14 W bulb | 36 | 54 |
| 3 | EY-POP-1 (10) | 14 W bulb | 24 | 98 |
| 4 | EY-POP-2 (10) | 14 W bulb | 24 | 87 |
| 5 | EY-POP-1 (10) | Natural light | 36 | 35 |
| 6 | EY-POP-1 (10) | Dark | 36 | Trace |
| 7 | — | 14 W bulb | 36 | <5 |
| 8c | EY-POP-1 (10) | 14 W bulb | 36 | 60 |
| 9 | Eosin Y (5 mol%) | 14 W bulb | 24 | 98 |
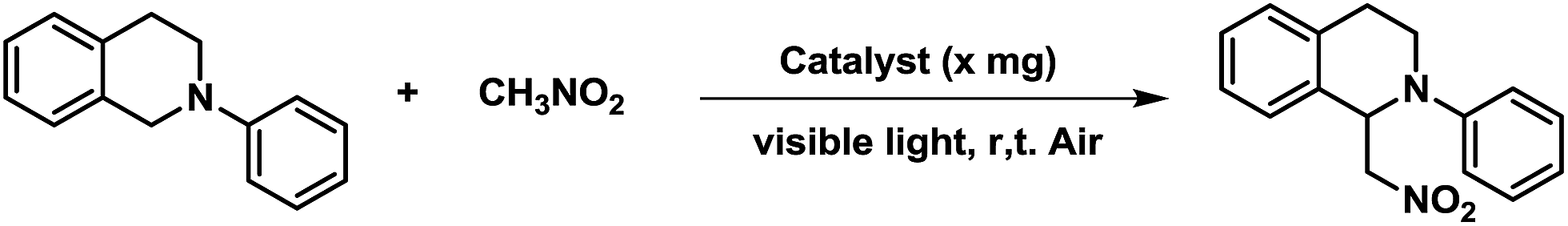
The substrate scope of the aza-Henry reaction catalyzed by EY-POP-1 was examined with a variety of tetrahydroisoquinolines and nitromethane/nitroethane (Table 2). As shown in Table 2, good to excellent isolated yields (90–99%) were obtained for all of the aza-Henry reaction of a variety of N-aryltetrahydroisoquinolines with nitromethane (Table 2, entries 1–8) and nitroethane (Table 2, entries 9–12) catalyzed by the EY-POP-1 polymer, which are comparable to those using the Ir- or Ru-loaded PCPs,7a the RB-CMP network15c and the homogeneous organic dye counterpart.6b,6d These results identified that the EY-POP-1 nanoporous polymer works as a high efficient heterogeneous organic-photocatalyst for the aza-Henry reaction.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Yieldb (%) |
| a Reaction conditions: 0.2 mmol substrate; 1.0 mL of nitroalkyl; 10 mg EY-POP-1; room temperature; 14 W household bulb; in air.b Isolated yield after silica gel column chromatography. | ||||
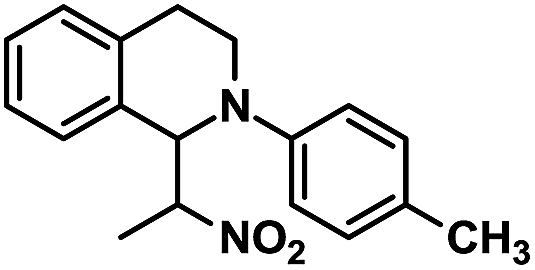
| 1 | 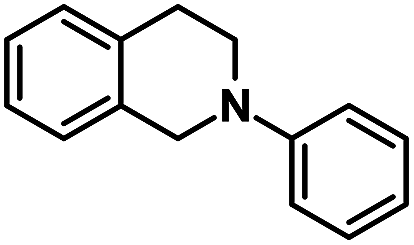 |
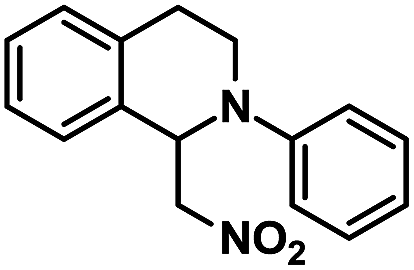 |
24 | 98 |
| 2 |  |
 |
36 | 94 |
| 3 |  |
 |
72 | 91 |
| 4 |  |
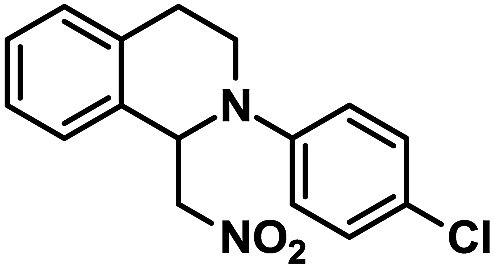 |
12 | 98 |
| 5 | 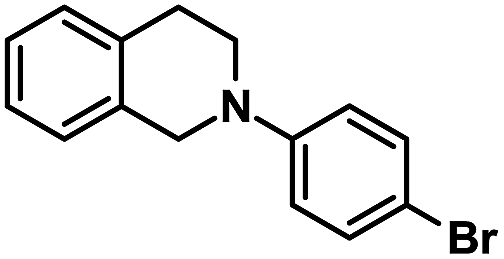 |
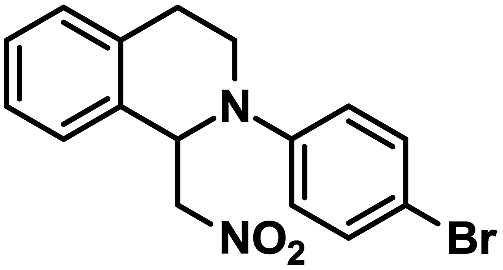 |
20 | 99 |
| 6 | 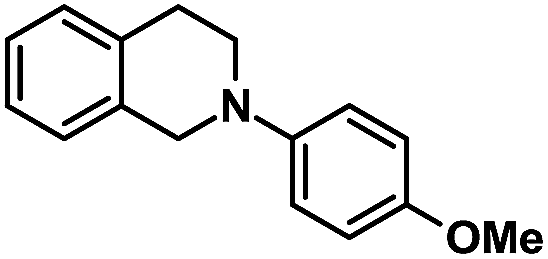 |
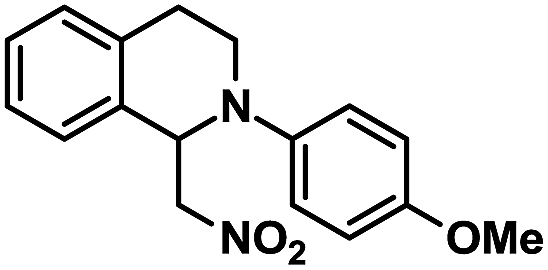 |
72 | 99 |
| 7 | 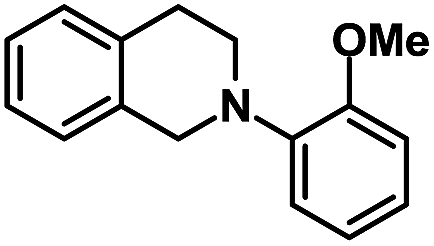 |
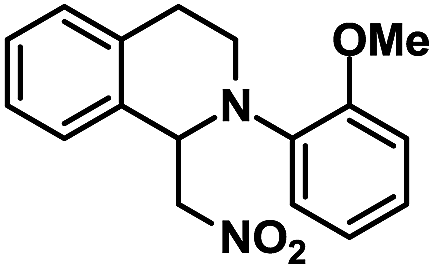 |
72 | 97 |
| 8 | 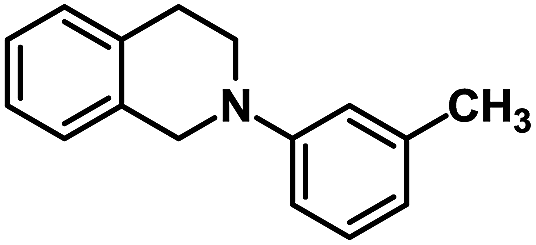 |
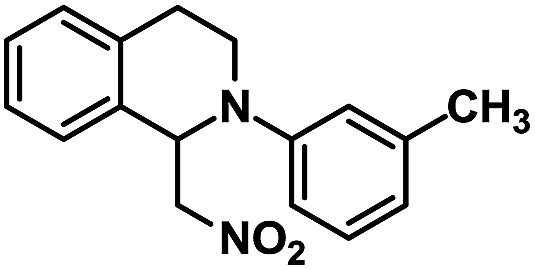 |
24 | 94 |
| 9 | 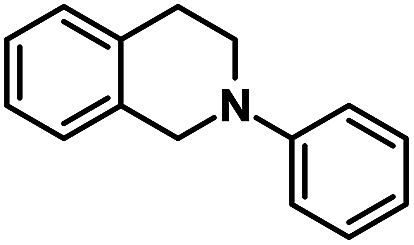 |
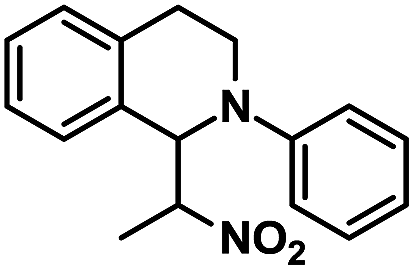 |
24 | 94 |
| 10 | 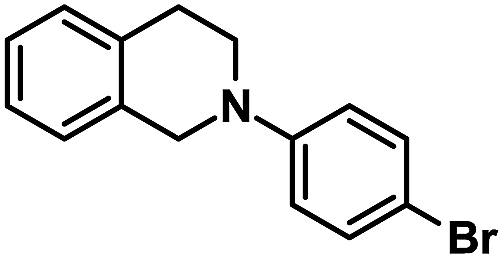 |
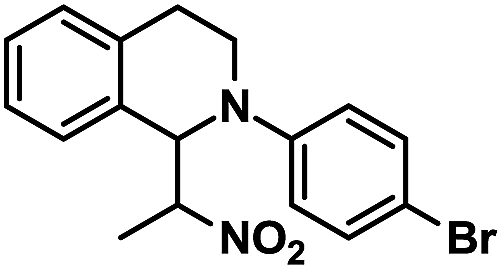 |
17 | 90 |
| 11 | 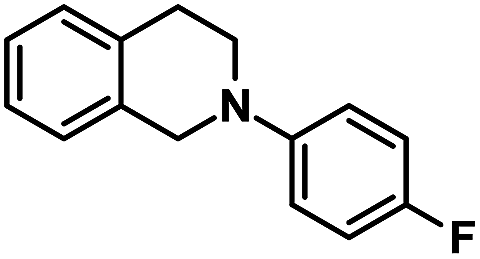 |
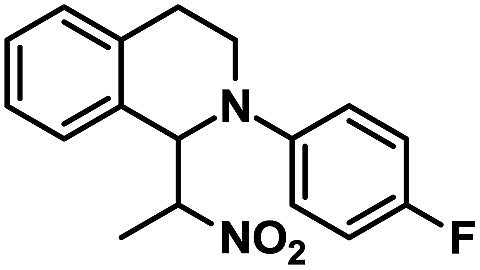 |
50 | 91 |
| 12 | 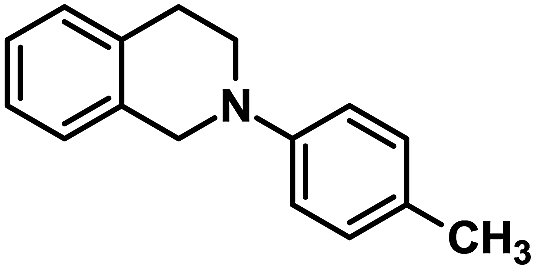 |
 |
24 | 92 |
We further investigated the recyclability of EY-POP-1 catalyst in the aza-Henry reaction of 2-(4-chlorophenyl)-1,2,3,4-tetrahydroisoquinoline with nitromethane (Table S2, ESI†). After the reaction, EtOAc was added to dilute the reaction mixture, and the EY-POP-1 catalyst was easily separated from the products by centrifugation and decantation of the reaction solution. The recycled catalyst was further washed with EtOAc to remove residual product, and simple dried in vacuum overnight before reuse. As in Fig. 4, the EY-POP-1 polymer could be reused at least twelve times without any significant loss of the photocatalytic activity (94–98% yield), which is attributed to the built-in character of the covalently linked catalytic sites of EY-POP-1 polymer. However, in the recycling experiments, the reaction time was prolonged after five cycles, which could be due to the impurities blocking the polymeric nanopores. We found that the BET surface area of EY-POP-1 from 587 m2 g−1 decrease to 347 m2 g−1 after the sixth recycle use (Fig. S3†).
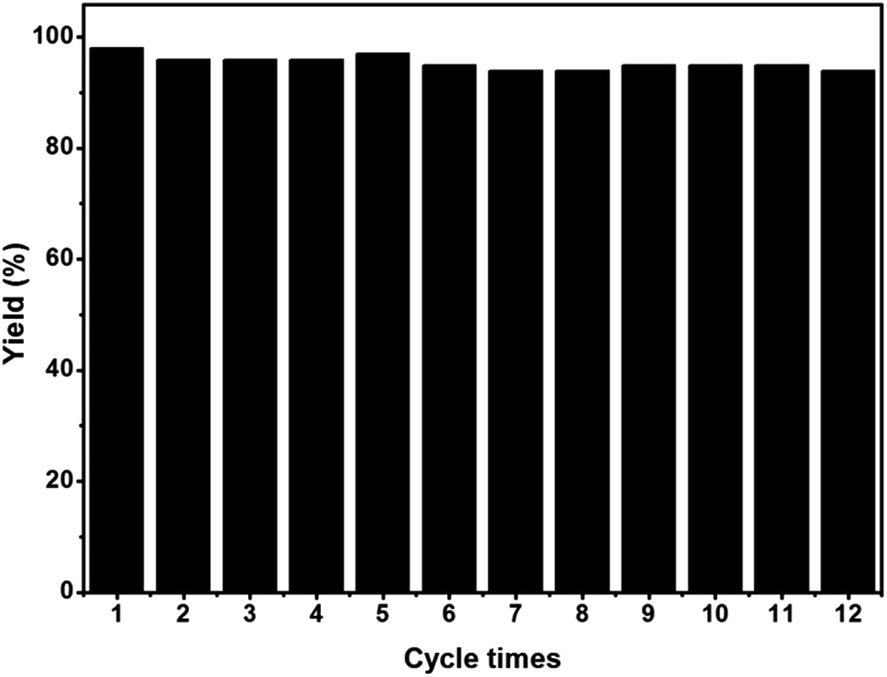 | ||
| Fig. 4 Recycling experiment of EY-POP-1 for the aza-Henry reaction of 2-(4-chlorophenyl)-1,2,3,4-tetrahydroisoquinoline and nitromethane. | ||
Conclusions
In conclusion, the Eosin Y, an organic dye, has been successfully embedded into a nanoporous network EY-POPs through a bottom-up strategy. The resultant framework materials are stable in various solvents and are thermally stable. Owing to the high BET surface area and the built-in character of the covalently linked catalytic sites of EY-POPs, these photoactive polymers were shown to be highly active, recyclable and reusable heterogeneous photocatalysts in aza-Henry reaction. Catalyzed by the EY-POP-1 catalyst, the aza-Henry reaction of tetrahydroisoquinolines to nitromethane/nitroethane gives the desired products in good to excellent yield (90–99%). The heterogeneous photocatalyst could be reused at least twelve times without any significant loss of the photocatalytic activity (94–98% yield). This work highlights the potential of using POPs polymers as a stable and molecularly platform for developing heterogeneous photocatalysts for other important organic transformations. Currently, research into the bottom-up construction of chiral porous organic photocatalysts for asymmetric catalysis is underway in our laboratory.Experimental section
Materials
All reagents were purchased from commercial sources. Et3N and DMF were dried through the standard procedures. Tetra(4-ethynylphenyl)methane and 1,3,5-triethynylbenzene were prepared according to the literature procedure.20 Pd(PPh3)2Cl2, PdCl2 and Eosin Y were purchased from J&K. A range of substrates were synthesized according to the literature procedure.21 All catalytic reactions were performed in a 15 mL glass tube.Synthesis of EY-POP-1
Eosin Y (692 mg, 1.0 mmol), 1,3,5-triethynylbenzene (200 mg, 1.33 mmol), bis-(triphenylphosphine)palladium(II) dichloride (30 mg), and copper(I) iodide (20 mg) were added into a dried round-bottom flask under nitrogen atmosphere. Anhydrous DMF (10 mL) and Et3N (10 mL) were added to the mixture via a syringe. The resulting mixture was heated to 80 °C and stirred for 72 h under nitrogen atmosphere. After cooling to room temperature, the precipitate network polymer was filtered and washed four times (once each) with chloroform, water, methanol and acetone to remove any unreacted monomer or catalyst residues. Further purification of the polymer was carried out by Soxhlet extraction with methanol for 48 h. The product was dried at 70 °C under vacuum for 6 h to give a red powder. Yield: 465 mg (82%). Anal. calcd for C36H10Na2O5: C, 76.06; H, 1.76. Found: C, 62.38, H, 1.25.Synthesis of EY-POP-2
Eosin Y (296 mg, 0.43 mmol), tetra(4-ethynylphenyl)methane (178 mg, 0.43 mmol), bis-(triphenylphosphine)palladium(II) dichloride (15 mg), and copper(I) iodide (10 mg) were added into a dried round-bottom flask under nitrogen atmosphere. Anhydrous DMF (5.0 mL) and Et3N (5.0 mL) were added to the mixture via a syringe. The resulting mixture was heated to 80 °C and stirred for 72 h under nitrogen atmosphere. After cooling to room temperature, the precipitate network polymer was filtered and washed four times (once each) with chloroform, water, methanol and acetone to remove any unreacted monomer or catalyst residues. Further purification of the polymer was carried out by Soxhlet extraction with methanol for 48 h. The product was dried at 70 °C under vacuum for 6 h to give a red powder. Yield: 299 mg (89%). Anal. calcd for C53H22Na2O5: C, 81.12; H, 2.81. Found: C, 69.94, H, 2.20.General procedure for the aza-Henry reaction catalyzed by EY-POP-1
In a typical run of catalytic activity test of EY-POP-1, N-aryltetrahydroisoquinolines (0.2 mmol), nitroalkane (1.0 mL), and EY-POP-1 (10 mg) were added into a 15 mL glass tube. The tube was sealed with a rubber stopper and the reaction mixture was connected to the air through a needle in the stopper. The tube was subsequently stirred at room temperature under irradiation with visible light using a cool daylight energy-saving bulb (14 W) until all starting material had been consumed. After the reaction was completed (monitored by TLC), the mixture was centrifugated and the solid was washed with EtOAc (3 × 5 mL). The combined organic phase was then concentrated and purified by flash column chromatography with petroleum ether/EtOAc = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as the eluent to give the pure product.
:1 as the eluent to give the pure product.
Typical recycling procedure for the aza-Henry reaction catalyzed by EY-POP-1
The recycling experiment was performed by recovering the EY-POP-1 catalyst using the centrifugation method. The recovered EY-POP-1 catalyst was washed with EtOAc to remove the residual product and simply dried before reuse. We chose the aza-Henry reaction of 2-(4-chlorophenyl)-1,2,3,4-tetrahydroisoquinoline to nitromethane to investigate the recyclability of EY-POP-1 catalyst, and the results are summarized in Table S2.†Physical characterization
Thin-layer chromatography (TLC) plates were visualized by exposure to ultraviolet light. Flash column chromatography (FCC) was carried out with silica gel (200–300 mesh). 1H and 13C liquid NMR spectra were recorded on a Bruker Avance III 400 MHz NMR spectrometer. The chemical shifts δ are given in ppm (parts per million) relative to tetramethylsilane (TMS) and the coupling constants J are given in Hz. All the spectra were recorded in CDCl3 as solvent at room temperature. TMS served as the internal standard (δ = 0.00 ppm) for 1H NMR, while CDCl3 as the internal standard (δ = 77.0 ppm) for 13C NMR. FT-IR spectra were recorded on a Nicolet NEXUS 670 instrument. N2 adsorption and desorption isotherms were measured at 77 K using a Micromeritics ASAP 2020M system. The pore-size-distribution curves were obtained from the adsorption branches using non-local density functional theory (NLDPT) method. Solid-state NMR experiments were performed on a Bruker Avance II WB 400 MHz NMR spectrometer. The 13C CP/MAS NMR spectra were recorded with the contact time of 3 ms (ramp 100) and the recycle delay of 2 s on a 2.5 mm double resonance probe. The thermal properties of the polymer networks were evaluated using a thermogravimetric analysis (TGA)-differential thermal analysis instrument (METTLER, TGA/DSC 1/1100) over the temperature range 25 to 800 °C under nitrogen atmosphere with a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) data were collected with a Rigaku D/MAX-2400 X-ray diffractometer operated at 40 kV and 100 mA with Cu Kα radiation at a scan rate of 15° min−1. Field emission scanning electron microscopy (SEM) observations were performed on Hitachi S-4800 microscope operated at an accelerating voltage of 5.0 kV. The UV-visible absorption spectra of the Eosin Y monomer and the EY-POPs networks were recorded on UV-Vis spectrometer (U-4100) in solid state powder at room temperature. Elemental analysis was performed on an Elementar Analysensysteme GmbH VarioEL V3.00 elemental analyzer.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21502136, 21571137), Higher Educational Science and Technology Program of Shandong Province (No. J15LC18), and the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Provine (No. BS2014CL035).Notes and references
- (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed; (b) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274–7276 CrossRef CAS PubMed; (b) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed; (c) T. Maji, A. Karmakar and O. Reiser, J. Org. Chem., 2011, 76, 736–739 CrossRef CAS PubMed; (d) J. Du, L. R. Espelt, I. A. Guzei and T. P. Yoon, Chem. Sci., 2011, 2, 2115–2119 RSC.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- (a) H. Liu, W. Feng, C. W. Kee, Y. Zhao, D. Leow, Y. Pan and C.-H. Tan, Green Chem., 2010, 12, 953–956 RSC; (b) D. P. Hari and B. König, Org. Lett., 2011, 13, 3852–3855 CrossRef CAS PubMed; (c) M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed; (d) Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682–2685 RSC; (e) Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341–3344 RSC; (f) K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC; (g) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169–171 CrossRef CAS; (h) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- (a) Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed; (b) P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed; (c) M. Liras, M. Iglesias and F. Sánchez, Macromolecules, 2016, 49, 1666–1673 CrossRef CAS.
- (a) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed; (b) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed; (c) A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- (a) N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC; (b) M. Hashem, C. Grazia Bezzu, B. M. Kariuki and N. B. McKeown, Polym. Chem., 2011, 2, 2190–2192 RSC; (c) J. Vile, M. Carta, C. G. Bezzu and N. B. McKeown, Polym. Chem., 2011, 2, 2257–2260 RSC; (d) P. Zhang, X. Jiang, S. Wan and S. Dai, J. Mater. Chem. A, 2015, 3, 6739–6741 RSC; (e) E. Madrid, P. Cottis, Y. Rong, A. T. Rogers, J. M. Stone, R. Malpass-Evans, M. Carta, N. B. McKeown and F. Marken, J. Mater. Chem. A, 2015, 3, 15849–15853 RSC; (f) S. Yi, X. Ma, I. Pinnau and W. J. Koros, J. Mater. Chem. A, 2015, 3, 22794–22806 RSC.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2006, 66, 768–779 CrossRef CAS.
- (a) X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC; (b) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC; (c) P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS PubMed.
- (a) T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed; (b) S. Yuan, S. Kirklin, B. Dorney, D.-J. Liu and L. Yu, Macromolecules, 2009, 42, 1554–1559 CrossRef CAS; (c) E. Klontzas, E. Tylianakis and G. E. Froudakis, Nano Lett., 2010, 10, 452–454 CrossRef CAS PubMed; (d) A. Li, R.-F. Lu, Y. Wang, X. Wang, K.-L. Han and W.-Q. Deng, Angew. Chem., Int. Ed., 2010, 49, 3330–3333 CrossRef CAS PubMed; (e) Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- (a) J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed; (b) L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed; (c) Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- (a) P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS; (b) Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC; (c) M. Rose, ChemCatChem, 2014, 6, 1166–1182 CAS; (d) C.-A. Wang and W. Wang, Acta Chim. Sin., 2015, 73, 498–529 CrossRef CAS.
- (a) M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Mullen, Chem. Commun., 2010, 46, 8932–8934 RSC; (b) K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed; (c) J.-X. Jiang, Y. Li, X. Wu, J. Xiao, D. J. Adams and A. I. Cooper, Macromolecules, 2013, 46, 8779–8783 CrossRef CAS; (d) N. Kang, J. H. Park, K. C. Ko, J. Chun, E. Kim, H.-W. Shin, S. M. Lee, H. J. Kim, T. K. Ahn, J. Y. Lee and S. U. Son, Angew. Chem., Int. Ed., 2013, 52, 6228–6232 CrossRef CAS PubMed; (e) Z. J. Wang, K. Garth, S. Ghasimi, K. Landfester and K. A. I. Zhang, ChemSusChem, 2015, 8, 3459–3464 CrossRef CAS PubMed; (f) R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed; (g) W. Liang, T. L. Church, S. Zheng, C. Zhou, B. S. Haynes and D. M. D'Alessandro, Chem.–Eur. J., 2015, 21, 18576–18579 CrossRef CAS PubMed; (h) L. Pan, M.-Y. Xu, L.-J. Feng, Q. Chen, Y.-J. He and B.-H. Han, Polym. Chem., 2016, 7, 2299–2307 RSC; (i) C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed; (j) L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef CAS PubMed; (k) R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- (a) C. A. Wang, Z. K. Zhang, T. Yue, Y. L. Sun, L. Wang, W. D. Wang, Y. Zhang, C. Liu and W. Wang, Chem.–Eur. J., 2012, 18, 6718–6723 CrossRef CAS PubMed; (b) C.-A. Wang, Y.-F. Han, Y.-W. Li, K. Nie, X.-L. Cheng and J.-P. Zhang, RSC Adv., 2016, 6, 34866–34871 RSC; (c) C.-A. Wang, Y.-W. Li, X.-M. Hou, Y.-F. Han, K. Nie and J.-P. Zhang, ChemistrySelect, 2016, 1, 1371–1376 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Prcrotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- (a) T. Kuroda, Y. Sakurai, Y. Suzuki, A. O. Nakamura, M. Kuwahara, H. Ozaki and H. Sawai, Chem.–Asina J., 2006, 1, 575–580 CrossRef CAS PubMed; (b) S. Yuan, S. Kirklin, B. Dorney, J.-J. Liu and L. Yu, Macromolecules, 2009, 42, 1554–1559 CrossRef CAS.
- F. Y. Kwong, A. Klapars and S. L. Buchwald, Org. Lett., 2002, 4, 581–584 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25123d |
| This journal is © The Royal Society of Chemistry 2017 |