
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating energy storage characteristics of ultrathin boron carbide monolayer under varied scandium doping†
S. R. Naqvia,
T. Hussain *c,
P. Panigrahid,
W. Luoa and
R. Ahujaab
*c,
P. Panigrahid,
W. Luoa and
R. Ahujaab
aCondensed Matter Theory Group, Department of Physics and Astronomy, Uppsala University, Box 516, SE-75120 Uppsala, Sweden
bApplied Materials Physics, Department of Materials and Engineering, Royal Institute of Technology (KTH), SE-100 44 Stockholm, Sweden
cCentre for Theoretical and Computational Molecular Science, Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, Qld 4072, Australia. E-mail: t.hussain@uq.edu.au
dCentre for Clean Energy and Nano Convergence (CENCON), Hindustan University, Padur, Kelambakkam, Chennai, India
First published on 26th January 2017
Abstract
We report, for the first time we believe, a detailed investigation on hydrogen storage efficiency of scandium (Sc) decorated boron carbide (BC3) sheets using spin-polarized density functional theory (DFT). We analyzed the energetics of Sc adsorption and explored the most favorable adsorption sites of Sc on BC3 sheets with 3.12%, 6.25%, and 12.5% coverage effects. Our investigations revealed that Sc strongly binds on pristine BC3 sheet, with a minimum binding energy of ∼5 eV, which is robust enough to hinder Sc–Sc metal clustering. Sc, the lightest transition metal, adsorbs a large number of H2 molecules per atom, resulting in a reasonable storage capacity. With 12.5% Sc-coverage, functionalized BC3 sheets could attain a H2 storage capacity of 5.5 wt% with binding energies suitable for a practical H2 storage medium.
Introduction
Hydrogen (H2) has emerged as an alternative energy carrier with a strong potential to serve a vibrant economy in the future particularly owing to its application in the transportation sector.1–3 However, H2 economy presents challenges when it comes to the fabrication of a solid-state material for H2 storage with high gravimetric/volumetric density that performs efficiently under ambient conditions.4,5 Previously, several new materials have been explored and studied for practical H2 storage but the criteria of the U.S. Department of Energy (DOE) for the year of 2017 have yet to be accomplished.6 Thus, it is important to design and explore novel solid-state materials with the desired characteristics to serve as economical, efficient and viable solutions for reversible H2 storage.Carbon-based nanomaterials, owing to their low cost, light weight, large surface area and novel H2 adsorption, have emerged as a subject of further research interest.7–15 However, in their pristine forms such nanomaterials show very feeble binding affinities as a result of weak van der Waals interactions between H2 and the host material.16,17 For practical purposes the binding energy of H2 with the respective storage material should be in the range of −0.2 eV to −0.6 eV.18 To achieve this criterion, metal decorated carbon-based nanomaterials have been investigated as potential candidates for H2 storage. The binding affinity could be further tuned by introducing isolated charged sites. However, the bonding mechanism and the binding enthalpy for the interaction of H2 with metal adatom depend on the type of metallic site, the extent of polarizability and on charge localization. It has been shown theoretically that charge transfer between the alkali metal (AM) centers and the host material leaves the AM adatom in the cationic state. The H2 adsorption enthalpy is enhanced due to transfer of charge between H2 and the metallic species.19 A rapid decrease in adsorption energy due to increasing temperature has also been reported, which limits the room temperature H2 storage of AM functionalized carbonaceous materials.20–23 To enhance the binding aptitude of H2, both alkaline earth metal (AEM) and transition metal (TM) coated materials have been extensively studied in the recent past.26–34 Smaller adsorption energies of H2 for AEM than TM decorated storage media have been reported.26 Functionalization with TMs leads to strong hybridization between the hydrogen σ- or σ*-orbitals and low-lying empty d-orbitals in the TM atoms.19 This kind of interaction is typically observed in the Kubas complexes,24,25 and is responsible for enhanced binding affinity towards H2 molecules. However, heavier TMs lead to lower gravimetric density for H2 storage. In addition, clustering is also a crucial problem for TM-doped carbon-based materials, leading to lower storage efficiency.27,29 Thus, H2 storage efficiency could be drastically improved by using light TM atoms and further preventing the metal clustering effect.
In this context, Nachimuthu et al. have reported a H2 storage capacity of 6.4 wt% in Ni–Ti–Mg functionalized boron-doped graphene. Their results indicate that the boron content in graphitic materials could successfully hinder the formation of metallic clusters.26,33 In contrast, Beheshti et al. suggested that substitutional doping of boron atoms in graphene also efficiently prevents Ca atoms from forming clusters, which is unavoidable in the case of pure graphitic materials.30
Recent investigations on boron carbide (BC3) nanomaterials in pure,35 titanium-doped,32 calcium-doped42 and lithium-doped34 systems have urged the scientific community to unravel the potential of this novel material for the purpose of practical H2 storage. In the present work, we are interested in exploring the H2 storage potential of scandium (Sc) functionalized boron carbide (BC3) nanosheets using spin-polarized density functional theory (DFT) calculations. Our investigations include structure analysis, bonding characteristics, charge transfer mechanisms and hydrogenation of functionalized BC3 sheets. The novelty of the present study lies in the fact that even in its pristine form BC3 sheet holds multiple Sc dopants with much higher binding energies than graphene, graphane, h-BN sheet, silicene and many other materials, which make this material superior to these 2D materials.
Methodology
The computational analysis of structure, energetics, and charge transfer mechanisms was carried out within the framework of spin-polarized DFT using the Vienna Ab-initio Simulation Package (VASP).36,37 For electrons, exchange and correlation interactions were treated with generalized gradient approximation (GGA) as suggested by Perdew–Bruke–Ernzerhof,38 whereas the description of ion–electron interactions was provided by the projector augmented wave method (PAW), which treats the following electronic states as valence: Sc 3p4s3d, B s2p1 and C s2p2.39 For a geometrical description, GGA serves quite well except that the actual energies are underestimated. In order to give an accurate description of the energetics of our systems, we need to account for van der Waals interactions. Therefore, we included the semi-empirical corrections of Grimme40 as available in VASP. Owing to the sensitiveness of H2 binding energies with functionalized BC3@Sc systems, we have also compared the performance of DFT-D2 with that of non-local van der Waals functional. For all the computations, the energy cut-off was set to 500 eV. Our initial designed system was a 2 × 2 × 1 supercell, which consisted of 8 B and 24 C atoms. A vacuum thickness of 20 Å was inserted normal to the surface in order to decouple the periodically recurring images. The structures were allowed to relax until the total energy and force converged to 10−5 eV per atom and 0.05 eV Å−1 respectively. The Brillouin zone was sampled using a Γ-point scheme with 5 × 5 × 1 mesh for structural relaxation. The density of states calculations with a denser 11 × 11 × 1 mesh were performed using the tetrahedron method. The charge transfer mechanism between the metal adatoms and BC3 sheet was investigated using Bader charge analysis.41Results and discussion
To functionalize BC3 we have carefully selected the lightest transition metal Sc, which could result in a high gravimetric density and adequate binding enthalpy for H2. Boron content in BC3 sheets is expected to hinder clustering of Sc atoms over the sheets. The optimized structure of the (2 × 2 × 1) supercell of BC3 is presented in Fig. 1. The supercell of pristine BC3 monolayer consists of 8 B and 24 C atoms. The primitive cell of BC3 sheet (marked with dotted lines) consists of 2 B and 6 C atoms. | ||
| Fig. 1 (a) Top and side view of BC3 sheet; the primitive cell is marked with dotted lines. High symmetry sites available for adatom occupation are indicated as T1, T2, H1, H2, B1 and B2. (b) Binding energy for occupied binding sites. | ||
Structural analysis
Structural analysis of optimized BC3 reveals the C–C and B–C bond lengths to be 1.42 Å and 1.56 Å respectively, which agree well with previously reported results.42,43To functionalize pure BC3 sheet with Sc atoms, we have investigated the adsorption behavior of a single Sc atom on all available high symmetry sites on the single side of the sheet. The six available adsorption sites for the Sc atom are designated (i) top of B atom (T1), (ii) top of C atom (T2), (iii) hollow site in BC ring (H1), (iv) hollow site in C ring (H2), (v) bridge between B and C atoms (B1) and (vi) bridge between two C atoms (B2), as shown in Fig. 1. For each possible site, the Sc atom is initially kept at a vertical distance of 2 Å. However, the Sc atom initially on T2 and B2 sites migrates towards the more favorable H2 site. The Sc atom binds to H2 sites with a binding energy of 7.11 eV at a vertical distance (dSc–sheet) of 1.45 Å. Initial and final geometries of all the possible configurations of Sc on BC3 are given in S1 (ESI†). The adsorption energies for all Sc occupied sites are presented in Fig. 1(b). The adsorption energy of the Sc atom is defined as:
| Eb = [E(BC3) + E(Sc)] − E(BC3@Sc) | (1) |
The strong binding of single Sc on BC3 encouraged us to introduce more Sc dopant on the sheet. Thus, in a similar fashion, we found the most favorable site for the second Sc atom (6.25% Sc-coverage) on the BC3 sheet, as shown in Fig. 2. The most preferred binding site for the second Sc atom turns out to be the hollow site of the C ring (H2) exactly opposite to the first Sc atom. The binding energy per Sc is found to be 6.02 eV, which is slightly lower than for 3.12% Sc-coverage. However, it is still much higher than Sc–Sc cohesive energy, indicating that BC3 could be further exposed to Sc adatoms. The distance dSc–sheet has now increased by 0.19 Å due to the weakening of the bond as compared to the single Sc case (see Table 1).
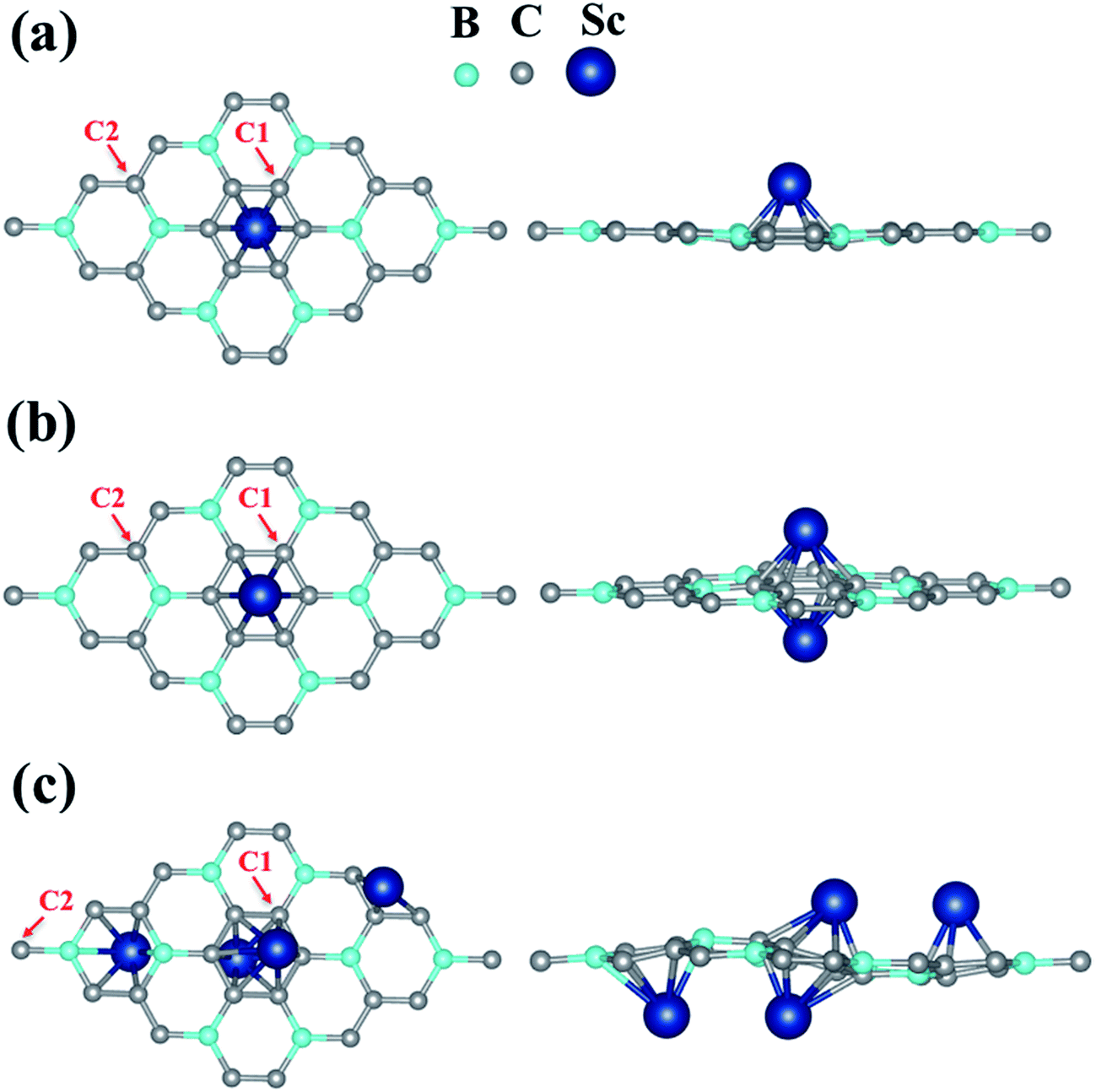 | ||
| Fig. 2 Top (left) and side (right) views of BC3 sheet functionalized with (a) 1 Sc [3.12% Sc-coverage], (b) 2 Sc [6.25% Sc-coverage] and (c) 4 Sc [12.5% Sc-coverage]. C1 and C2 indicate the carbon atoms that were chosen for the plotting of density of states. | ||
| System | Binding energy per Sc Eb (eV) | Average Sc to BC3 distance dSc–sheet (Å) | Minimum dopant–dopant distance dSc–Sc (Å) | Charge per Sc (e−) |
|---|---|---|---|---|
| BC3–1Sc | 7.11 | 1.45 | — | +1.444 |
| BC3–2Sc | 6.02 | 1.64 | 3.28 | +1.390 |
| BC3–4Sc | 5.45 | 1.95 | 3.65 | +1.251 |
In the case of 4 Sc on BC3 sheet (12.5% Sc-coverage), the most favorable binding configuration is shown in Fig. 2(c). The optimized geometry of this configuration is found to be a little degraded owing to the relatively high Sc doping. Here the binding energy per Sc is found to be 5.45 eV, still almost 40% higher than Sc–Sc cohesive energy. The energetics analysis reveals that even at a significantly high Sc doping concentration, the metal dopants bind strongly enough to nullify cluster formation and make a uniform distribution over the BC3 monolayer.
The bonding mechanism between Sc and BC3 monolayer could be explained by studying the charge transfer mechanism. We employed Bader charge analysis for this purpose, which depicts a transfer of 1.251e− of charge from Sc atom to BC3 sheet due to the lower electronegativity of the former than the latter. The bulk portion of this transferred charge goes to the C atoms of BC3 that are in close vicinity to Sc dopant. This would transform Sc into a partially positive charged state, which proves to be extremely helpful in anchoring the incident H2 molecules. The accumulation and depletion of charges were described by plotting the isosurface charge densities for 3.12%, 6.25% and 12.5% Sc-coverage, as shown in Fig. 3(a–c). The charge density can be calculated by the following relation:
| Δρ = ρ(BC3@nSc) − ρ(BC3) − ρ(nSc) | (2) |
| {n = 1, 2, 4} |
 | ||
| Fig. 3 Top (left) and side (right) views of isosurface charge densities of BC3 with (a) 1 Sc atom [3.12% Sc-coverage], (b) 2 Sc [6.25% Sc-coverage] and (c) 4 Sc [12.5% Sc-coverage]. Cyan and yellow indicate the accumulation and depletion of charge, respectively. | ||
The minimum distance between Sc atoms (dSc–Sc) in the case of 2 Sc (6.25% coverage) and 4 Sc (12.5% coverage) doping is found to be 3.28 Å and 3.65 Å respectively. The binding energy per Sc atom decreases, whereas the optimum distance between the Sc and BC3 sheet increases with the increase in Sc dopant. Though the bond elongation for increased Sc-coverage (dSc–sheet) justifies comparatively weaker binding between the sheet and Sc atoms, the binding is still strong enough to prevent the formation of Sc–Sc clusters. Results from structural and charge analysis are presented in Table 1.
Electronic structure analysis
In order to investigate the electronic structure of the designed Sc decorated BC3 system, we have plotted and explained the partial electronic density of states (PDOS) of pure and functionalized BC3 sheets (Fig. 4). PDOS of pure BC3 (Fig. 4(a)) reveals a small bandgap of ∼0.65 eV, which is in reasonable agreement with the previously reported value.43 The PDOS of Sc functionalized BC3 sheet (3.12% coverage) is shown in Fig. 4(b). C1 indicates the C atom that is closer to the Sc atom, and C2 resides at a greater distance from Sc, as indicated in Fig. 2. The Sc(d) state is found to be strongly hybridized with B(p) and C1(p) states of the BC3 sheet. With 6.25% coverage, the extent of Sc(d) to B(p) and C1(p) hybridization decreases, which is reflected in a decrease in binding energy, as presented in Table 1. With higher coverage, the Sc to Sc attraction weakens the binding energy of BC3 sheet to Sc.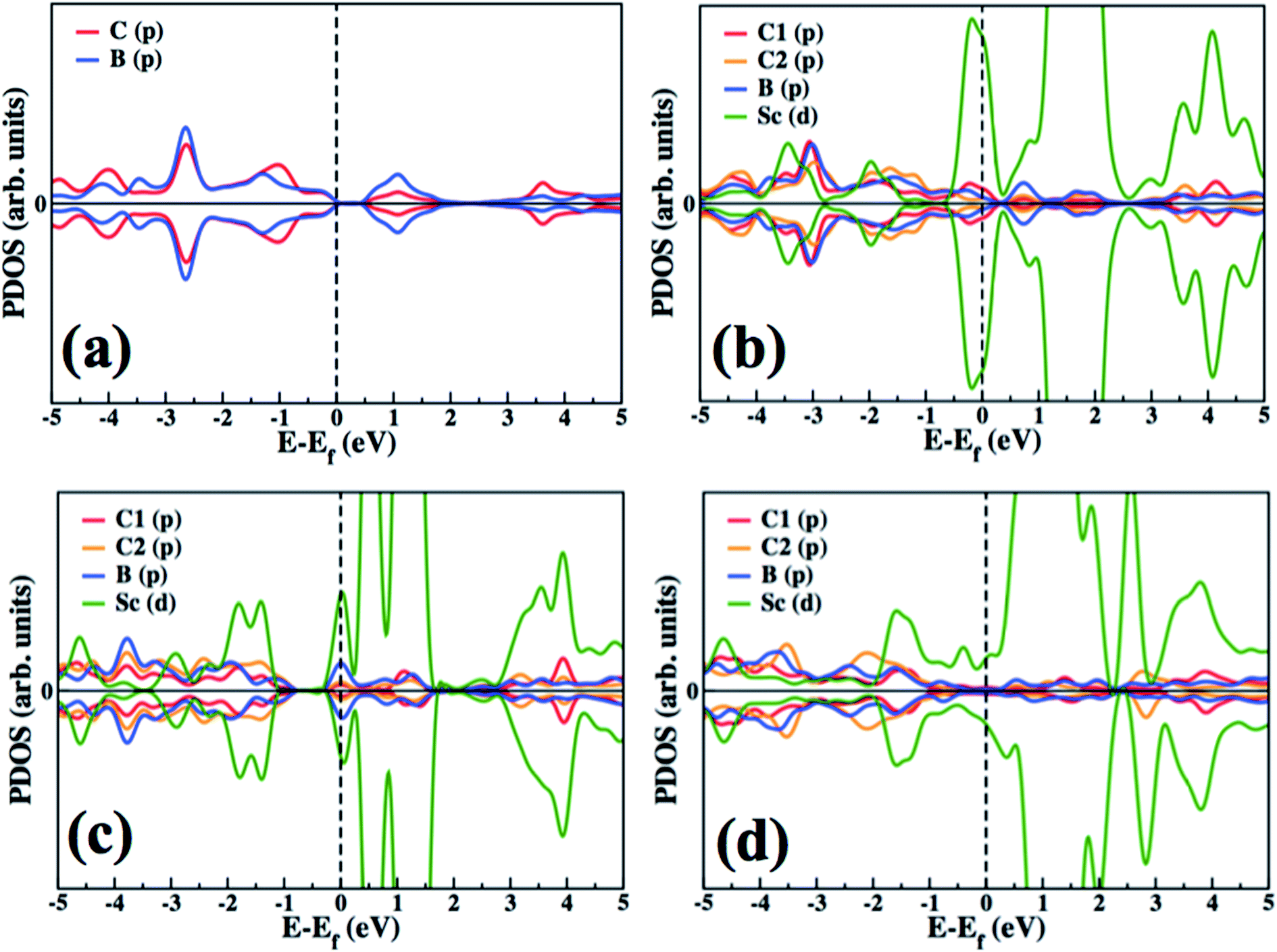 | ||
| Fig. 4 Partial electronic density of state plots for (a) pure BC3, (b) BC3 with 1 Sc atom [3.12% Sc-coverage], (c) BC3 with 2 Sc [6.25% Sc-coverage] and (d) BC3 with 4 Sc [12.5% Sc-coverage]. The dashed line indicates the Fermi level. | ||
Hydrogenation
After establishing the stability, the Sc functionalized BC3 sheets were exposed to H2 molecules. We consider placing H2 molecules in different possible configurations whilst performing a set of structural relaxations. The average adsorption energies of H2 were calculated using the following formula:| Eads = [E(BC3@mSc + nH2) − E(BC3@mSc) − nE(H2)]/n | (3) |
The H2 gravimetric density (storage capacity) can be calculated by the following relation:
| H2 storage capacity = NH × WH/[(NB × WB) + (NC × WC) + (NSc × WSc) + (NH × WH)] |
In the above relation N represents the number of each type of atoms and W represents the molecular weight of each element present in the system.
The number of H2 molecules around Sc was gradually increased from 1 to the maximum adsorption limit. For the lowest Sc-coverage (3.12%), a maximum of five H2 molecules per Sc atom could be adsorbed to the functionalized sheet, which resulted in a relatively small H2 storage capacity of 2.364 wt%. Top and side views of the lowest energy hydrogenated systems are represented in Fig. 5(a) whereas all the other possible configurations are given in S2 (ESI†). The average distance between adsorbed H2 and Sc atoms (dH2–Sc) and the average binding energy for maximum H2 uptake are 2.2 Å and −0.34 eV respectively.
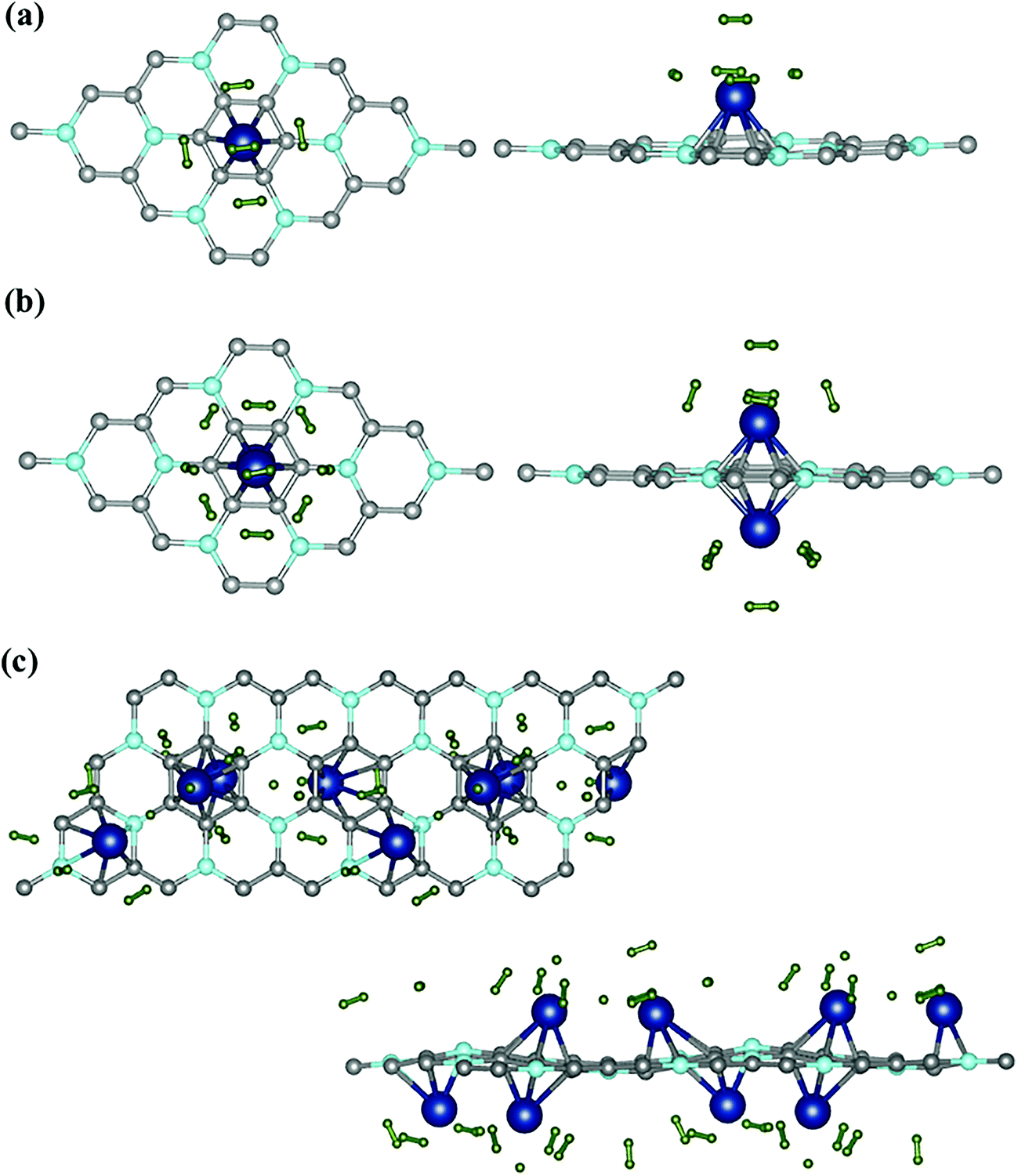 | ||
| Fig. 5 Hydrogenated BC3 sheets. Top view and side view for (a) 1 Sc [3.2% Sc] and (b) 2 Sc [6.4% Sc]. (c) Extended top and side view for 4 Sc [12.5% Sc]. | ||
It is important to mention here that the H2 adsorption energies are quite sensitive to van der Waals functionals, so a careful choice of a functional needs to be made in order to achieve unvarying values of adsorption energies. The DFT-D2 method of Grimme used in the present study has been extensively used and it has yielded reliable results consistent with the literature.45–48 For a comparison we have calculated the adsorption energies of H2 and 2 H2 adsorbed on BC3@Sc by using the non-local van der Waals functional optPBe-vdW49 and found that the adsorption energies of H2 and 2 H2 around BC3@Sc are −0.310 eV and −0.293 eV respectively. These values are roughly 23% lower than those calculated by DFT-D2.
The bond length of H2 has increased slightly to 0.76 Å. In the case of two Sc atoms, each Sc could bind 5 H2 (shown in Fig. 5(b)), which yielded a H2 storage capacity of 4.184 wt%, significantly higher than that of the BC3@Sc system. The isosurface charge density of BC3@Sc–H2 and BC3@Sc–5H2 is shown in S3 (ESI†). The adsorption energies of H2 with the functionalized BC3 material again show a decreasing trend with an increasing number of H2. However, the binding energy of H2 for maximum hydrogen coverage is −0.3 eV, which still lies in the energy window above weak physisorption (Fig. 6). The average distance between each H2 and Sc atom is 2.39 Å and the average bond length of H2 is 0.77 Å. We employed a similar procedure to hydrogenate the BC3 sheet with 12.5% Sc-coverage. In total, 16 H2 molecules could be incorporated in the functionalized system, giving a gravimetric density of 5.5 wt% (Fig. 5(c)).
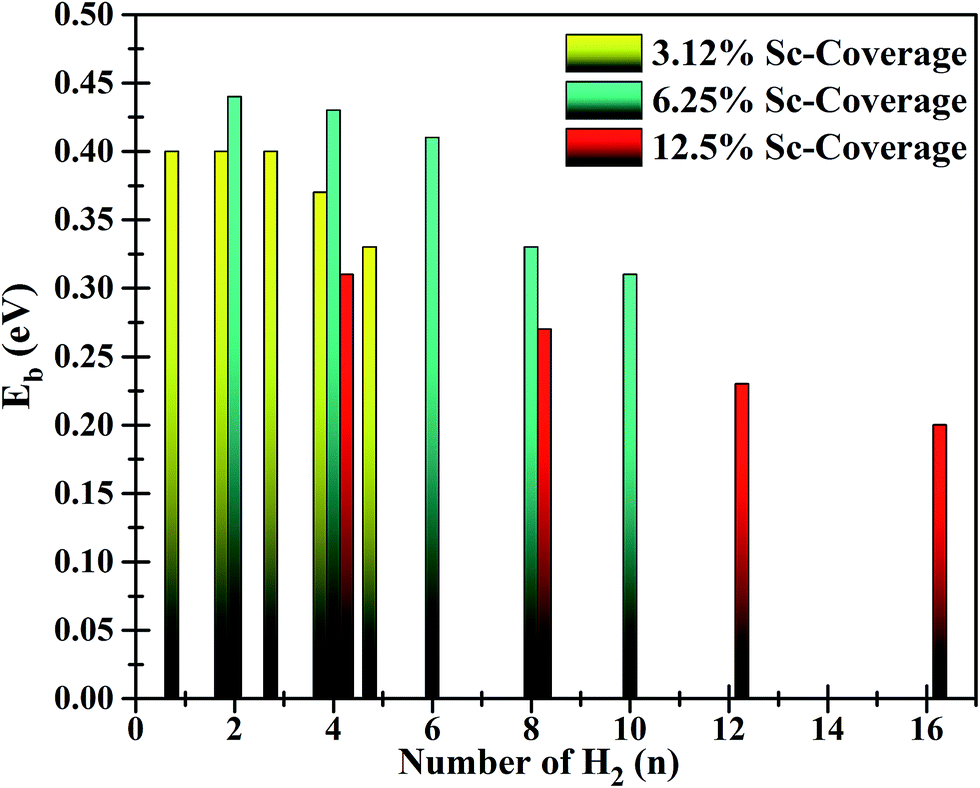 | ||
| Fig. 6 Adsorption energies of H2 on functionalized BC3 sheets for three different values of Sc-coverage. | ||
Interestingly, the first H2, introduced in between 2 Sc atoms (on each side of the sheet), experiences dissociative adsorption. The two H atoms locate to an optimal distance apart (dH–H) of 2.2 Å and each H attains an electronic charge of −0.495e−. The bond dissociation leads to adsorption of one H per Sc atom (H/Sc) at an average distance (dH–Sc) of 1.93 Å and average binding energy of −0.9 eV. Further addition of H2 in the vicinity of Sc atoms leads to associative adsorption of H2. In the first step, the H2 introduced in between 2 Sc atoms gets dissociated due to the presence of enough charge to contribute to the σ*-antibonding orbitals. To support this argument, Bader charge analysis also reveals partially polarized H2 molecules in the vicinity of Sc atoms. However, for successive adsorption of H2, there is not enough charge available to destabilize the H2 molecule. It is evident from Fig. 6 that for each Sc-coverage value, the average adsorption energy of H2 monotonically decreases with increasing number of H2 molecules.
By gradually increasing the number of H2 from 1 H2/Sc to the maximum limit of adsorption, the average binding energy for associative adsorption varies from −0.31 to −0.20 eV for 12.5% Sc-coverage (Fig. 6). The average bond length of adsorbed H2 molecules increased to 0.78 Å which is slightly larger than for free H2 (0.75 Å). Bond elongation is typically observed in Kubas interaction where an amount of charge is donated from H2 to the empty d-orbital of TM followed by back-donation of charge from TM to an anti-bonding orbital of H2. Such donation and back-donation of charge governed by Kubas interaction has been extensively studied in the literature.8,50–52 The greater charge transfer results in a longer bond length of H2. Successive addition of H2 molecules to the functionalized BC3 sheet (with 12.5% Sc-coverage) reveals that a total of 15 H2 (including 2 dissociated molecules) could be adsorbed in the vicinity of Sc atoms, where one H2 directly binds to the sheet at a distance (dH2–sheet) of 2.7 Å above the B2 site. Top and side views of the functionalized system with maximum H2 uptake and average adsorption energies with increasing number of H2 are represented in Fig. 5(c).
The functionalized systems (BC3@Sc, BC3@2Sc, BC3@4Sc) reported in the present work could achieve a maximum H2 storage capacity of 5.5 wt%, which fulfills the DOE target to be met by the year 2017. However, the H2 capacity is smaller than given in a paper by Durgun et al.,7 which reported on the H2 storage properties of selected transition metals (Ti, Sc, V) decorated on carbon chain, graphene and carbon nanotubes, which attained high H2 storage capacities. However, to ensure the uniform distribution of metal dopants over the host materials, the binding energies of the former must be higher than their respective cohesive energies. In the paper mentioned above, the binding energies of TM dopants (Ti, Sc, V) fall short of their respective cohesive energies, which could allow the formation of TM clusters which significantly reduces the H2 storage capacity.
The novelty of our work lies in the fact that, despite lower H2 storage capacity as compared with the Durgun et al. paper, Sc, 2 Sc and 4 Sc bind strongly with BC3, nullifying the possibility of cluster formation and ensuring the formation of stable doped systems.
Conclusions
We have performed spin-polarized DFT calculations to study the H2 adsorption aptitude of Sc decorated BC3 sheet. Our calculations reveal that Sc binds strongly to the BC3 sheet with a binding energy that is much higher than its bulk cohesive energy. Hydrogen storage investigations performed with 3 different Sc-coverages (3.12%, 6.25%, and 12.5%) show that the binding energy of Sc on pristine BC3 sheet decreases from 7.11 eV to 5.45 eV as the Sc content increases from 3.12% to the maximum 12.5%. However, even in the case of high Sc doping, the calculated binding energies are still much higher than their corresponding cohesive energies. The density of states analysis shows that Sc adsorption tunes the electronic behavior of BC3 sheet and metallicity is enhanced with an increased Sc content. Adsorption energies of H2 on BC3–Scn systems lie within an energy window that is reasonable for practical H2 storage. Based on these results we expect that Sc decorated BC3 sheets can be used as a practical H2 storage medium.Acknowledgements
We acknowledge the Swedish Research Council (VR) and StandUp, Carl Trygress Foundation for financial support. SNIC and UPPMAX are acknowledged for providing computing time. We also acknowledge the assistance of resources provided at the NCI National Facility systems at the Australian National University through the National Computational Merit Allocation Scheme supported by the Australian Government and the University of Queensland Research Computing Centre.References
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef CAS PubMed.
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39 CrossRef CAS.
- S. W. Jorgensen, Curr. Opin. Solid State Mater. Sci., 2011, 15, 39 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024 CrossRef CAS.
- A. W. Van den Berg and C. O. Arean, Chem. Commun., 2008, 6, 668 RSC.
- Targets for Onboard Hydrogen Storage Systems for Light-duty Vehicles, http://www1.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage_explanation.pdf, accessed October 2016.
- E. Durgun, S. Ciraci and T. Yildirim, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085405 CrossRef.
- T. Yildirim and S. Ciraci, Phys. Rev. Lett., 2005, 94, 175501 CrossRef CAS PubMed.
- W. Liu, Y. H. Zhao, Y. Li, Q. Jiang and E. J. Lavernia, J. Phys. Chem. C, 2009, 113, 2028 CAS.
- Q. Sun, P. Jena, Q. Wang and M. Marquez, J. Am. Chem. Soc., 2006, 128, 9741 CrossRef CAS PubMed.
- S. A. Shevlin and Z. X. Guo, Chem. Soc. Rev., 2009, 38, 211 RSC.
- M. Yoon, S. Y. Yang, C. Hicke, E. Wang, D. Geohegan and Z. Y. Zhang, Phys. Rev. Lett., 2008, 100, 206806 CrossRef PubMed.
- K. R. S. Chandrakumar and S. K. Ghosh, Nano Lett., 2008, 8, 13 CrossRef CAS PubMed.
- Q. Wang, Q. Sun, P. Jena and Y. Kawazoe, J. Chem. Theory Comput., 2009, 5, 374 CrossRef CAS PubMed.
- K. Takahashi, Y. Wang, S. Chiba, Y. Nakagawa, S. Isobe and S. Ohnuki, Sci. Rep., 2014, 4, 4598 Search PubMed.
- M. Ritschel, M. Uhlemann, O. Gutfleisch, A. Leonhardt, A. Graff, C. Taschner and J. Fink, Appl. Phys. Lett., 2002, 80, 2985 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 43, 2209 CrossRef CAS.
- Y.-H. Kim, Y. Zhao, A. Williamson, M. J. Heben and S. B. Zhang, Phys. Rev. Lett., 2006, 96, 016102 CrossRef PubMed.
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357 RSC.
- W. Q. Deng, X. Xu and W. A. Goddard, Phys. Rev. Lett., 2004, 92, 166103 CrossRef PubMed.
- G. E. Froudakis, Nano Lett., 2001, 1, 531 CrossRef CAS.
- Y. L. Zhao, R. Q. Zhang and R. S. Wang, Chem. Phys. Lett., 2004, 398, 62 CrossRef CAS.
- Z. H. Zhu, G. Q. Lu and S. C. Smith, Carbon, 2004, 42, 2509 CrossRef CAS.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152 CrossRef CAS PubMed.
- G. J. Kubas, J. Organomet. Chem., 2001, 635, 37 CrossRef CAS.
- S. Nachimuthu, P.-J. Lai, E. G. Leggesse and J.-C. Jiang, Sci. Rep., 2015, 5, 16797 CrossRef CAS PubMed.
- Q. Sun, Q. Wang, P. Jena and Y. Kawazoe, J. Am. Chem. Soc., 2005, 127, 14582 CrossRef CAS PubMed.
- Y. Suttisawat, P. Rangsunvigit, B. Kitiyanan, M. Williams, P. Ndungu, M. V. Lotototsky and S. Kulprathipanja, Int. J. Hydrogen Energy, 2009, 34, 6669 CrossRef CAS.
- S. Li and P. Jena, Phys. Rev. Lett., 2006, 97, 209601 CrossRef CAS PubMed.
- E. Beheshti, A. Nojeh and P. Servati, Carbon, 2011, 49, 1561 CrossRef CAS.
- C. M. Ramos-Castillo, J. U. Reveles, M. E. Cifuentes-Quintal, R. R. Zope and R. de Coss, J. Phys. Chem. C, 2016, 120, 5001 CAS.
- L. Ma, J. Wang, Y. Liang, G. Wang, Chinese Scientific Papers Online, 2015, www.paper.edu.cn/index.php/default/releasepaper/downPaper/201503-287, accessed October 2016.
- S. Nachimuthu, P. J. Lai and J. C. Jiang, Carbon, 2014, 73, 132 CrossRef CAS.
- Z. Yang and J. Ni, Appl. Phys. Lett., 2012, 100, 183109 CrossRef.
- F.-C. Chuang, Z.-Q. Huang, W.-H. Lin, M. A. Albao and W.-S. Su, Nanotechnology, 2011, 22, 135703 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: a Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- Z. Yang and J. Ni, Appl. Phys. Lett., 2010, 97, 253117 CrossRef.
- D. Tománek, R. M. Wentzcovitch, S. G. Louie and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 3134 CrossRef.
- C. Kittel, Introduction to Solid State Physics, Wiley, New York, 7th edn, 1960 Search PubMed.
- T. Hussain, K. Takahashi and D. J. Searles, J. Phys. Chem. A, 2016, 120, 2009 CrossRef CAS PubMed.
- T. Hussain, M. S. Islam, G. S. Rao, P. Panigrahi, D. Gupta and R. Ahuja, Nanotechnology, 2015, 26, 275401 CrossRef CAS PubMed.
- T. Hussain, S. Chakraborty, T. W. Kang, B. Johansson and R. Ahuja, ChemPhysChem, 2014, 16, 634 CrossRef PubMed.
- T. Hussain, A. De Sarkar and R. Ahuja, Int. J. Hydrogen Energy, 2014, 39, 2560 CrossRef CAS.
- K. Lee, E. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- H. Lee, J. Ihm, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115412 CrossRef.
- A. Bhattacharya, S. Bhattacharya, C. Majumdar and G. P. Das, J. Phys. Chem. C, 2010, 114, 10297 CAS.
- C.-G. Zhang, R. Zhang, Z.-X. Wang, Z. Zhou, S. B. Zhang and Z. Chen, Chemistry, 2009, 15, 5910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24890j |
| This journal is © The Royal Society of Chemistry 2017 |