
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biowaste chicken eggshell powder as a potential cure modifier for epoxy/anhydride systems: competitiveness with terpolymer-modified calcium carbonate at low loading levels†
Mohammad Reza Saeb*a,
Mehdi Ghaffarib,
Hadi Rastinc,
Hossein Ali Khonakdard,
Frank Simond,
Farhood Najafia,
Vahabodin Goodarzie,
Poornima Vijayan P.f,
Debora Pugliag,
Farzaneh Hassanpour Aslc and
Krzysztof Formelah
aDepartment of Resin and Additives, Institute for Color Science and Technology, P. O. Box 16765-654, Tehran, Iran. E-mail: saeb-mr@icrc.ac.ir; mrsaeb2008@gmail.com; Tel: +98 21 22956209
bDepartment of Polymer, Golestan University, P. O. Box 155, Gorgan, Golestan, Iran
cSchool of Chemical Engineering, College of Engineering, University of Tehran, P. O. Box: 11155-4563, Tehran, Iran
dLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Strasse 6, D-01069, Dresden, Germany
eApplied Biotechnology Research Center, Baqiyatallah University of Medical Sciences, P. O. Box 19945-546, Tehran, Iran
fCenter for Advanced Materials, Qatar University, P. O. Box 2713, Doha, Qatar
gMaterials Engineering Centre, University of Perugia, Department of Civil and Environmental Engineering, Strada di Pentima 4, 05100, Terni, Italy
hDepartment of Polymer Technology, Faculty of Chemistry, Gdansk University of Technology, G. Narutowicza Str. 11/12, 80-233 Gdansk, Poland
First published on 12th January 2017
Abstract
Biowaste chicken eggshell (ES) powder was applied as a potential cure modifier in epoxy/anhydride systems. Cure behaviour and kinetics of composites filled with very low content (0.1 wt% based on epoxy resin) of ES, calcium carbonate (CaCO3), and terpolymer-modified fillers, mES and mCaCO3, were discussed comparatively. Surface analysis was performed by X-ray photoelectron spectroscopy. Cure kinetics was investigated by differential (Friedman) and integral (Ozawa and Kissinger–Akahira–Sunose) isoconversional methods using dynamic differential scanning calorimetry (DSC) data. Overall, protein precursors naturally existing in the structure of pristine ES facilitated crosslinking of epoxy and hardener of anhydride with functional groups resulting from terpolymer attachment to CaCO3 particles. Accelerated/hindered cure was observed depending on the filler type and surface characteristics, as investigated via the autocatalytic/non-catalytic nature of reactions and comparison of activation energy values of four types of composites. An enhanced cure was identified for composites containing untreated ES, which could be inferred on account of the lower competitive cure of carboxyl groups in the terpolymer backbone with epoxy compared to peptide groups existing in microporous pristine ES. On the other hand, mCaCO3 revealed low values of activation energy compared to pristine CaCO3, but still of the same order as ground biowaste ES.
1. Introduction
Epoxy resins are versatile thermosetting polymers having unique characteristics such as high modulus, low cure shrinkage, good chemical and corrosion resistance and acceptable adhesion properties; hence, they have been widely served as structural adhesives, surface coatings, engineering composites, and electronic circuits.1,2 Despite above beneficial features, considerable brittleness was found to be the main drawback of epoxy resins, which brings about some disadvantages like low impact strength and poor fracture toughness. Thus, toughening of epoxy with various types of modifiers/additives has been the subject of several recent studies.3–5Diglycidyl ether of bisphenol-A (DGEBA) is the most common commercially available epoxy resin that can be cured by various kinds of hardeners; mostly by amine6–8 and anhydride curing agents.9 The cure reaction between epoxy and anhydride acids contains a two-stage reaction: (i) ring opening of epoxide that yields carboxylate anions followed by cross-linking of esterified constituent; (ii) etherification, which controls the overall degree of cure. In contrast to amine hardeners, easily curable with epoxy at ambient temperature, kinetics of epoxy/anhydride reaction at an identical condition is very slow, which comforts cure assessment of epoxy/anhydride composites containing fillers.10
Chicken eggshell (ES) is the by-product of food industry and causes a big waste disposal problem, but it has excellent mechanical properties, which gives reason to consider it as modifier for epoxy.11–13 Typically, ES contains CaCO3 (94%), MgCO3 (1%), Ca3(PO4)2 (1%) and organic materials (4%).14 This material is a biowaste used as fertilizer/soil conditioner,15 absorbent of heavy metals,16,17 as well as in the synthesis of hydroxyapatite bio-ceramic18 and in sorption of CO2.19,20 The abundant supply of ES makes it an alternative for mineral-based CaCO3 (chalk or calcite) in polymer composites.21 The unique hierarchical structure of ES provides a nano-scale porous and rough texture, which allows for an increased interaction with polymers.22 However, the most mysterious fact about ES is that porosities formed during incubation period in the structure of ES will remain often even after they are triturated; so that, micron-size ES could enhance properties of polymer composites somewhat more than that of nano-sized CaCO3.23
Surface modification of ES powder was found to enhance properties of resulting polymer composites. In case of thermoplastics, an improved adhesion with polypropylene (PP) was reported when ES was covered by stearic acid; so that composites containing 40 wt% treated ES enhanced Young's modulus by 200%, benefiting from an increased crystallinity of the PP matrix.24 Likewise, ES powders bearing isophthalic acid were found to cause 3–18% increase in modulus of PP composites compared to those containing unmodified ES or mineral-based micron-size CaCO3.21 Furthermore, ES functionalization with pimelic acid improves the nucleation of PP, dispersion of ES in the matrix, and impact strength of PP.25 ES was also applied as modifier of elastomers such as natural rubber, acrylonitrile butadiene rubber, styrene-butadiene rubber and epoxidized natural rubber.26 In case of thermosetting materials, enhancement of the mechanical properties of epoxy was reported upon addition of ES particles.11 Microanalysis of the fracture surface of three types of epoxy/ES composites containing natural grounded ES, NaOH-treated ES, and silane-modified ES, as well as fracture toughness and positron annihilation studies placed stress on the crucial role of protein molecules in achieving good interaction between ES and epoxy. However, due to presence of protein-based precursors in its structure, ES powder could be used directly as a cure modifier for epoxy, which has not been addressed so far.
In the light of above studies, the current work attempts to explore kinetics of cure of epoxy with anhydride in the presence of very low content of pristine and acid-treated ES and CaCO3 via dynamic differential scanning calorimetry (DSC) measurements. Study of cure reaction at very low concentrations (0.1–0.3 wt% of filler based on 100 parts by weight of epoxy resin) could help to reflect the competence of either pristine or functionalized particles in curing reactions. Attachment of reactive poly(N-vinyl-2-pyrrolidone-co-maleic acid-co-acrylic acid) to the ES and CaCO3 particles of the same size is responsible for two different curing behavior. Elemental surface analysis of untreated and acid-treated ES and CaCO3 was performed by X-ray photoelectron spectroscopy (XPS). Kinetic investigations were carried out using autocatalytic models; thereby the potential of all types of prepared composites to cure has been disclosed. Cure behavior and cure kinetics of all types of composites were thoroughly discussed applying differential and integral model-free approaches, where peculiar trends were observed before and after functionalization by terpolymer, despite comparable chemical composition of ES and CaCO3 particles.
2. Experimental
2.1. Materials
Epoxy resin used in the current work was a low-viscosity EPIKOTE (RIMR145) with epoxide equivalent weight of 170–210 g eq.−1. Methyltetrahydrophthalic anhydride (EPIKURE-RIMH145) was used as curing agent along with EPIKURE catalyst (RIMC145). Resin, curing agent and catalyst were all provided by Momentive (USA). The chemical structures of the used resin and hardener are illustrated in Fig. 1.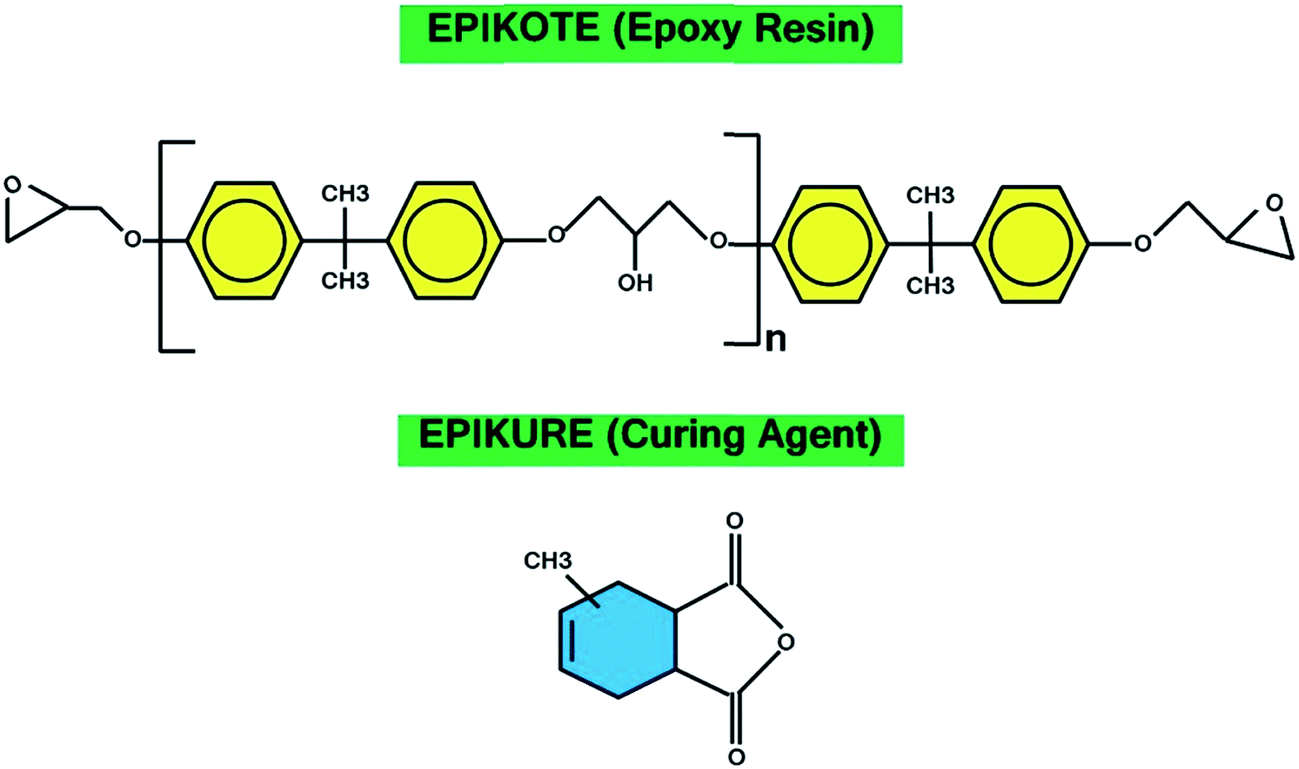 | ||
| Fig. 1 Chemical structure of the used resin and hardener. | ||
The micron-size precipitated CaCO3 under trade name of ALBAFIL® PCC was purchased from CARY Co., USA. Chicken ES was purchased from Fatim local farm. Prior to surface treatment, both types of particles were washed, dried and then grounded in a Retsch PM-400MA planetary ball mill with stabilized zirconia for 6 hours. The resulting powders were dried at 80 °C overnight and collected as pristine modifiers.
Table 1 gives the specifications of the pristine CaCO3 and ES, as well as acid-modified CaCO3 (mCaCO3) and ES (mES). As can be seen, CaCO3 and ES particles have roughly the same size, while the surface area of ES is more than twice of that of CaCO3, which springs from nano-porous structure of natural ES.27 This table also confirms that a proper acid-modification (described later) assisted in reduction of particle size, particularly in case of mES. Chemicals used for surface treatment of CaCO3 and ES were tert-butyl hydroperoxide (TBHP), sodium formaldehyde sulfoxylate (SFS), maleic anhydride, acrylic acid, and vinylpyrrolidone, all commercial grades provided from Sigma-Aldrich and Merck and used without further purification. To have a more real judgment of the role of surface modification in cure reaction promotion, topology of pristine CaCO3 and ES are illustratively compared in Fig. 2. Both types of particles have a crystalline structure, but dendritical nature of ES is different from columnar carbonate structure.28,29 The outermost layer of ES contains hydroxyl groups, while protein molecules are attached to the innermost layer and grown to form huge poly(L-amino acids) with peptide repeating units and carboxyl end groups. These groups tend to react with anhydride hardener, which will be discussed in the upcoming sections.
| Modifier | Commercial grade (manufacturer) | Mean particle size (μm) | Surface area (m2 g−1) |
|---|---|---|---|
| CaCO3 | Cary Company | 700 | 1.95 |
| mCaCO3 | — | 660 | 2.40 |
| ES | (Fatim Local Farm) | 690 | 4.50 |
| mES | — | 570 | 5.50 |
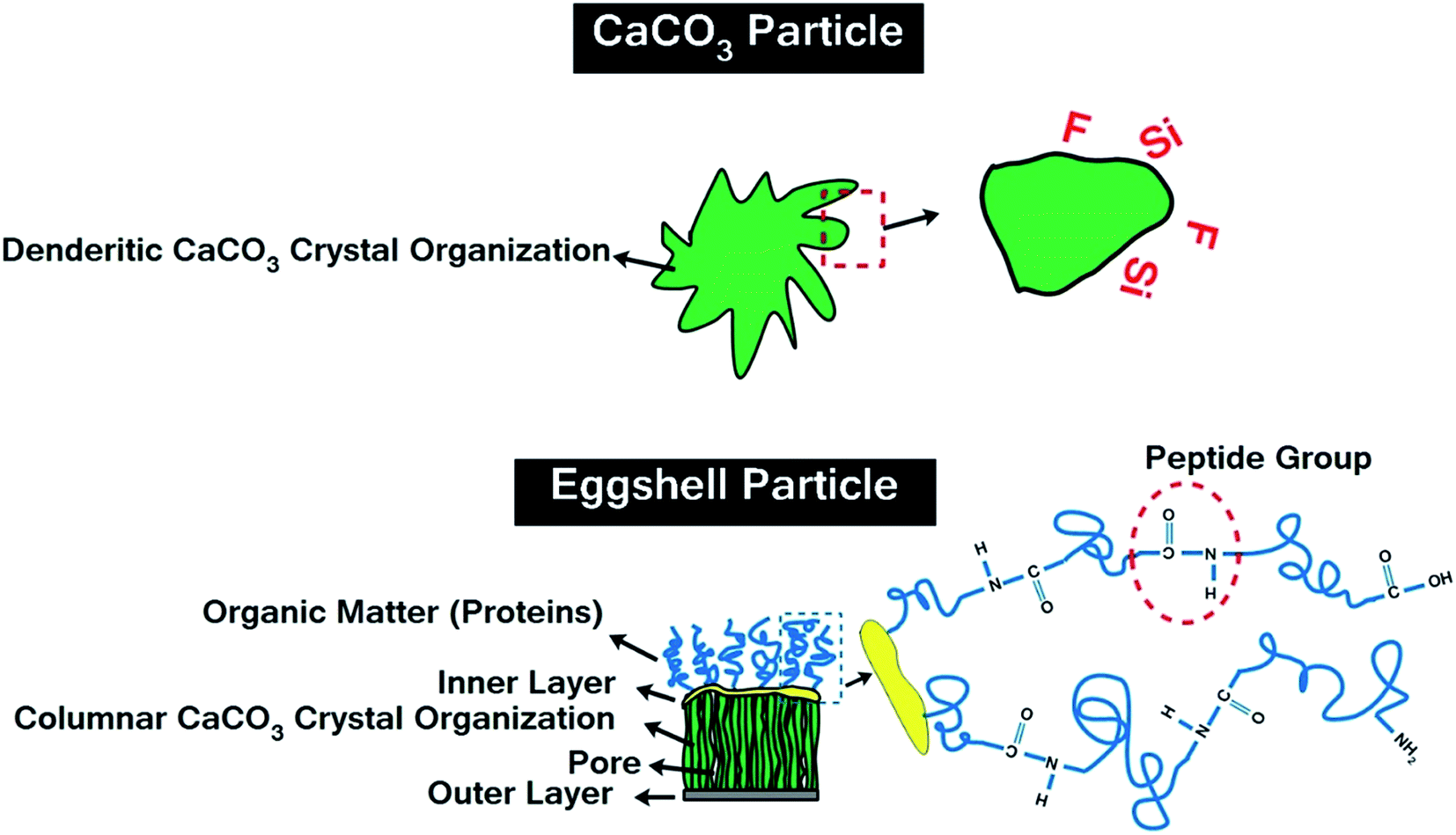 | ||
| Fig. 2 Topology of pristine CaCO3 and ES particles. | ||
2.2. Surface modification of CaCO3 and ES particles
A two-step preparation method was applied in the preparation of mCaCO3 and mES acid-modified particles. Firstly, a terpolymer precursor based on polycarboxylic acid containing three different monomers, namely vinylpyrrolidone, maleic anhydride, and acrylic acid, was prepared via aqueous radical polymerization by a redox initiator (TBHP/SFS). According to the illustration (Fig. 3), 9.8 g (0.1 mol) maleic anhydride, 7.2 g (0.1 mol) acrylic acid, and 23 g (0.21 mol) vinylpyrrolidone were dissolved in 60 ml water at 50 °C in a reactor equipped with condenser and mechanical stirrer. Then, 0.08 g TBHP and 0.09 g SFS were added to the mixture under stirring for 1 hour at 50 °C followed by further mixing at 75 °C for 2 hours. The resulting product was a clear solution containing 40 wt% solid terpolymer which was separated by filtration and used for surface modification of CaCO3 and ES particles. The resulting product was a clear solution containing solid terpolymer. To deposit terpolymer, ether was partly added to the transparent solution. Then, the aggregated solid terpolymer was dried in vacuum oven for 4 hours at 40 °C. The efficiency of terpolymer production was ca. 92%. In the second step, the synthesized terpolymer was grafted to filler surface. For instance, 40 g of terpolymer was dissolved in 450 g water, then 100 g precipitated CaCO3 was added to the solution while stirring at ambient temperature. The resulting mixture was stirred for further 12 hours, dried in vacuum oven at 80 °C, and collected without filtration to be grounded.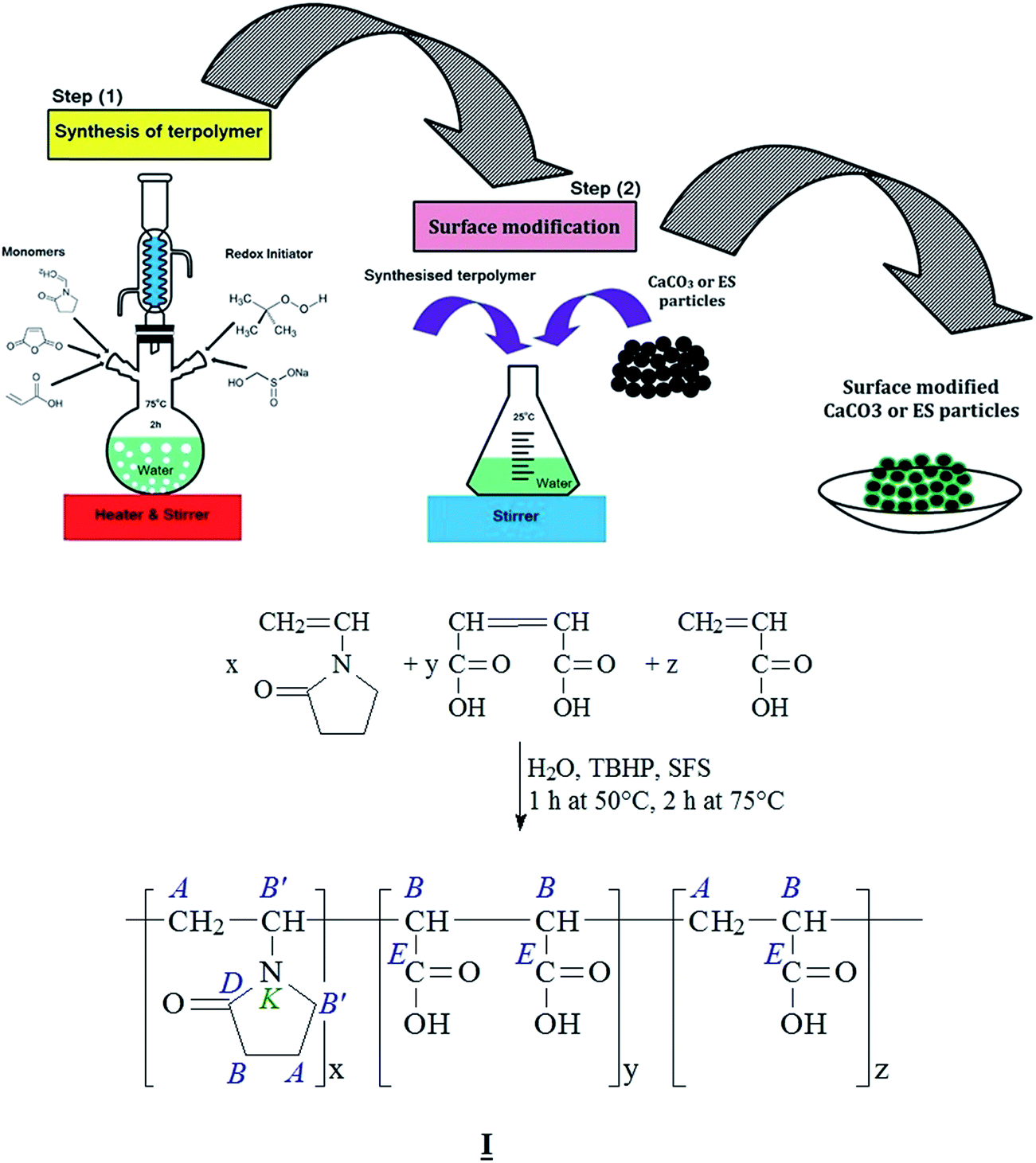 | ||
| Fig. 3 Schematic illustration of surface modification of CaCO3 and ES particles. | ||
2.3. Preparation of epoxy/CaCO3 and epoxy/ES composites
In order to prepare epoxy composites, the unmodified and copolymer-modified ES and CaCO3 were separately dispersed in the epoxy resin with the aid of sonication in pulse mode (50% duty cycle, i.e., pulse on, 2 s and pulse off, 2 s) for 1 hour in an oil bath maintained at 60 °C. The mixture was then degassed at 80 °C for 3 hours. Next, the neat and filled epoxy resins were mixed with the aforementioned curing agent and catalyst at ambient temperature in 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85:0.5 (resin:hardener:catalyst) ratio by weight and further mixed for 15 min. Samples containing very low concentration, i.e., 0.1 and 0.3 wt% of as-received CaCO3 and ES particles based on 100 parts by weight of epoxy, and 0.1 wt% in case of modified particles, were prepared to in an appropriate way reflect the potential of each type of filler to promote/obstruct curing reaction.
:85:0.5 (resin:hardener:catalyst) ratio by weight and further mixed for 15 min. Samples containing very low concentration, i.e., 0.1 and 0.3 wt% of as-received CaCO3 and ES particles based on 100 parts by weight of epoxy, and 0.1 wt% in case of modified particles, were prepared to in an appropriate way reflect the potential of each type of filler to promote/obstruct curing reaction.
2.4. Measurements and characterizations
The chemical compositions of the surface of untreated and copolymer-modified CaCO3 and ES particles were studied by means of X-ray photoelectron spectroscopy (XPS). The XPS method is a very surface-sensitive analytical technique, which permits studying the elemental composition of materials surface as well as the chemical binding states of the elements. All XPS studies were carried out by means of an Axis Ultra photoelectron spectrometer (Kratos Analytical, Manchester, UK). The spectrometer was equipped with a monochromatic Al Kα (hν = 1486.6 eV) X-ray source of 300 W at 15 kV. The kinetic energy of photoelectrons was determined with a hemispheric analyzer set to pass energy of 160 eV for wide-scan spectra and 20 eV for high-resolution spectra. During all measurements, electrostatic charging of the sample was avoided by means of a low-energy electron source working in combination with a magnetic immersion lens. Later, all recorded peaks were shifted by the same value that was necessary to set the C 1s peak to 285.00 eV. Quantitative elemental compositions were determined from peak areas based on experimentally determined sensitivity factors and the spectrometer transmission function. Spectrum background was subtracted according to Shirley.30 The high-resolution spectra were deconvoluted by means of the Kratos spectra deconvolution software. Free parameters of component peaks were their binding energy (BE), height, full width at half maximum and the Gaussian–Lorentzian ratio. The powdery fillers were prepared as millimeter-thick films on a double-sided adhesive tape, which was mounted on the spectrometer's sample holder. Then, the samples were introduced in the entry airlock of the spectrometer's preparation chamber. The chamber was slowly evaporated. After transferring the samples in to the analysis chamber the XPS spectra were recorded at a typical base pressure in the range of about 10−8 mbar.Curing characteristics of ES and CaCO3-incorporated epoxy composites were assessed by nonisothermal differential scanning calorimetry (DSC) on a Q2000 DSC (TA Instruments, New Castle, DE, USA). Different samples of about 5 mg were sealed in aluminum pans and subjected to heating under nitrogen purge at 50 ml min−1. The curing was carried out at different heating rates, i.e., 5, 10, 15, and 20 °C min−1, over a temperature range of 0–300 °C. The reaction was expected to be accomplished when the recorded DSC curve leveled off to a baseline equivalent to that of empty chamber. The total area under the exothermic curve, based on the extrapolated baseline at the end of the reaction, was considered to calculate the total heat of reaction.
3. Results and discussions
3.1. Characterization of functionalized ES and CaCO3
The poly(N-vinyl-2-pyrrolidone-co-maleic acid-co-acrylic acid) copolymer molecule (I) contains a large number of carboxyl groups in its maleic and acrylic acid sequences. Some of these functional groups can decarboxylate the carbonate groups of ES and CaCO3 form stable salt pairs, where each of the Ca2+ ions can bridge two of the copolymer's carboxyl end groups and anchor the copolymer on the eggshell surface. Residual carboxyl end groups are suitable partners for the reaction with the epoxy resin, as shown in Fig. 3.The N-vinyl-2-pyrrolidone sequences in the copolymer can be considered as an inner amide with a nucleophilic reaction center on the oxygen atom. Although, the oxirane ring can be opened by a nucleophilic attack of a sterically accessible methylene group (–CH2–) in the oxirane group, the reactivity of the N-vinyl-2-pyrrolidone seems to be too low to get involved in the epoxy curing reactions. Nevertheless, the polarity of the N-vinyl-2-pyrrolidone units enables the formation of hydrogen bridges to the Brønsted acidic carboxyl groups and the polar groups formed during the curing of the epoxy resin.
Fig. 4 shows wide-scan and high-resolution element spectra of the modified and unmodified ES and CaCO3. The elemental surface compositions of CaCO3 and ES before and after modification are provided in Table S1 in ESI.†
 | ||
| Fig. 4 Wide-scan XPS spectra (left column), C 1s (middle column), and N 1s (right column) high-resolution XPS element spectra of unmodified CaCO3 (a), copolymer-modified CaCO3 (b), unmodified eggshell (c), and copolymer-modified eggshell (d). | ||
In addition, the unmodified eggshell powder also contained remnants of proteins and other biologically active substances, as considerable amount of nitrogen and traces of phosphorus (detected as P 2p peak at 133.4 eV and P 2s peak at 191 eV) and sulfur (detected as S 2p peak at 168.3 eV and S 2s peak at 232 eV) was observed in the spectra of ES samples. The copolymer (I) applied on the filler surfaces significantly increased the intensity of the C 1s and N 1s peaks, while the relative intensities of the oxygen contents ([O]:[C]|spec) considerably decreased. The copolymer wrapped vast areas of the particle surfaces. Photoelectrons escaped from the atoms of the substrate material were additionally scattered during their passage through the copolymer films and contribute less to the intensities of the elemental peaks.
High-resolution C 1s spectra were recorded to get more insights in the chemical changes induced by applying the copolymer film. It is expected that the C 1s spectrum of pure calcium carbonate shows only one component peak at 289.40 eV.31 In fact, highly dispersed calcium carbonate particles are excellent adsorbents, which immediately take up substances from atmosphere contaminating their surfaces. Hence, the C 1s spectra of the two unmodified filler samples, CaCO3 and ES, showed two intensive component peaks (A and G) and three further component peaks having lower intensities (Fig. 4a and c, middle column). The most intensive component peaks A at 285.00 eV showed the presence of saturated hydrocarbons (CxHy), which are the main component of the non-specifically adsorbed surface contaminations mentioned above. The other intensive component peaks G resulted from the carbonate ions (CO32−) of the calcium carbonate. Their binding energy values of 289.44 eV and 289.36 eV excellently agreed with the literature data.26 Component peaks C (at 286.61 eV) presented C–O bonds of alcohol (C–OH) and/or ether (C–O–C) groups. In the CaCO3 sample alcohol-sided carbon atoms of carboxylate ester groups (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–C) also contribute to component peak C. The corresponding carbonyl carbon atoms of the carboxylate ester groups (OC–O–C) were identified as component peak E (at 288.15 eV). Carbonyl carbon atoms observed as component peak D in the C 1s spectrum of the eggshell sample had a slightly lower binding energy of 287.83 eV. It indicated the presence of carboxylate amide groups (OC–NH–C), which are constituents of remnants of proteins. The corresponding amine-sided carbon atoms (OC–NH–C) contributed to component peak B found at 285.81 eV. In addition, the component peaks B of both of the two samples also showed carbon atoms in α-position to the strongly electronegative carbonyl carbon atoms. The high-resolution N 1s spectrum (Fig. 4c, right column) of the eggshell sample showed only one component peak K. Its binding energy value (400.16 eV) is typical for nitrogen atoms of carboxylate amide groups (OC–NH–C) but also for amino groups (C–NR2). The absence of a component peak showing protonated amino groups (C–N+R2H) usually observed at binding energy values higher than 401 eV indicated the predominant presence of amide (or protein) groups.
C–O–C) also contribute to component peak C. The corresponding carbonyl carbon atoms of the carboxylate ester groups (OC–O–C) were identified as component peak E (at 288.15 eV). Carbonyl carbon atoms observed as component peak D in the C 1s spectrum of the eggshell sample had a slightly lower binding energy of 287.83 eV. It indicated the presence of carboxylate amide groups (OC–NH–C), which are constituents of remnants of proteins. The corresponding amine-sided carbon atoms (OC–NH–C) contributed to component peak B found at 285.81 eV. In addition, the component peaks B of both of the two samples also showed carbon atoms in α-position to the strongly electronegative carbonyl carbon atoms. The high-resolution N 1s spectrum (Fig. 4c, right column) of the eggshell sample showed only one component peak K. Its binding energy value (400.16 eV) is typical for nitrogen atoms of carboxylate amide groups (OC–NH–C) but also for amino groups (C–NR2). The absence of a component peak showing protonated amino groups (C–N+R2H) usually observed at binding energy values higher than 401 eV indicated the predominant presence of amide (or protein) groups.
The application of the copolymer (I) on the filler surfaces significantly affected the shape of the C 1s spectra (Fig. 4b and d, middle column). The intensities of component peaks G characterizing the substrate material seemed to be strongly decreased. As shown in Fig. 3, the component peaks resulting from the deconvolution of the C 1s spectra confirmed the chemical structure of the applied copolymer (I).
For the CaCO3 sample, the intensity of the component peak D showing the carbonyl carbon atoms of the inner amide groups of the pyrrolidone sequences, is in well agreement with the [N]:[C]|spec ratio, which was separately determined from the corresponding wide-scan spectrum. Compared to the binding energy values of component peaks B (ca. 286.15 eV) – resulting from photoelectrons escaped from carbon atoms in α-position to the carbonyl carbon atoms of the carboxyl acid groups (C–COOH) the carbon atoms of the amine-like C–N bonds (component peaks B′) – were shifted to lower values of 285.15 eV. Their intensities were twice as those of the component peaks D, which indicated that the protein residuals did not significantly contribute to the C 1s spectrum of the modified eggshell sample.
It is assumed that the copolymer also wrapped areas of the eggshell surface, which were covered with the remains of proteins. Component peaks E, representing the carbonyl carbon atoms of the carboxyl acid groups (COOH), were found at binding energy values of 288.59 and 289.01 eV. These values are slightly lower than the binding energy values which are expected for carboxylc acids (289.2–289.3 eV (ref. 32)). Thus, it can be assumed that the copolymer's carboxyl groups were present in their dissociated form as carboxylate groups (⊖O–CO ↔ OC–O⊖).
As mentioned above, the negatively charged carboxylate groups strongly interact with the Ca2+ ions of the filler materials. Regarding the detection of carboxylate groups in the C 1s spectra, the Ca 2p spectra showed also widened peaks (Fig. 5), which were separated into two component peaks P and Q. While the component peaks P in the Ca 2p3/2 spectra (346.9 eV) result from CaCO3, the slightly shifted component peaks Q (347.9 eV) shows calcium ions involved in salt-like interactions with the copolymer's carboxylate sequences. The areas of the component peaks Q equalled the areas of the component peaks E observed in the C 1s spectra of the copolymer-modified filler samples. The corresponding N 1s spectra showed only a single component peak K at ca. 400 eV, which is attributed to the nitrogen atoms in the vinyl 2-pyrrolidone sequences (Fig. 4b and d, right column) of the copolymer (I).
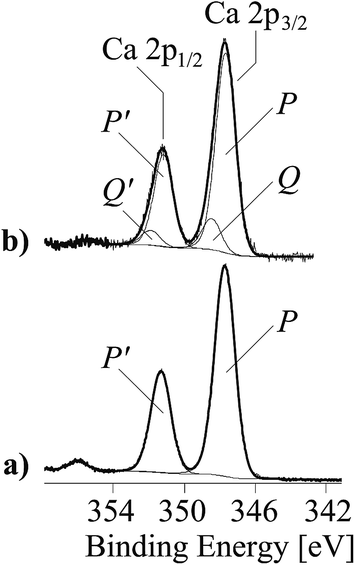 | ||
| Fig. 5 Ca 2p high-resolution XPS element spectra (spectra are composed of the Ca 2p3/2 and Ca 2p1/2 peaks) of unmodified CaCO3 (a), copolymer-modified CaCO3 (b). | ||
3.2. Nonisothermal cure analysis
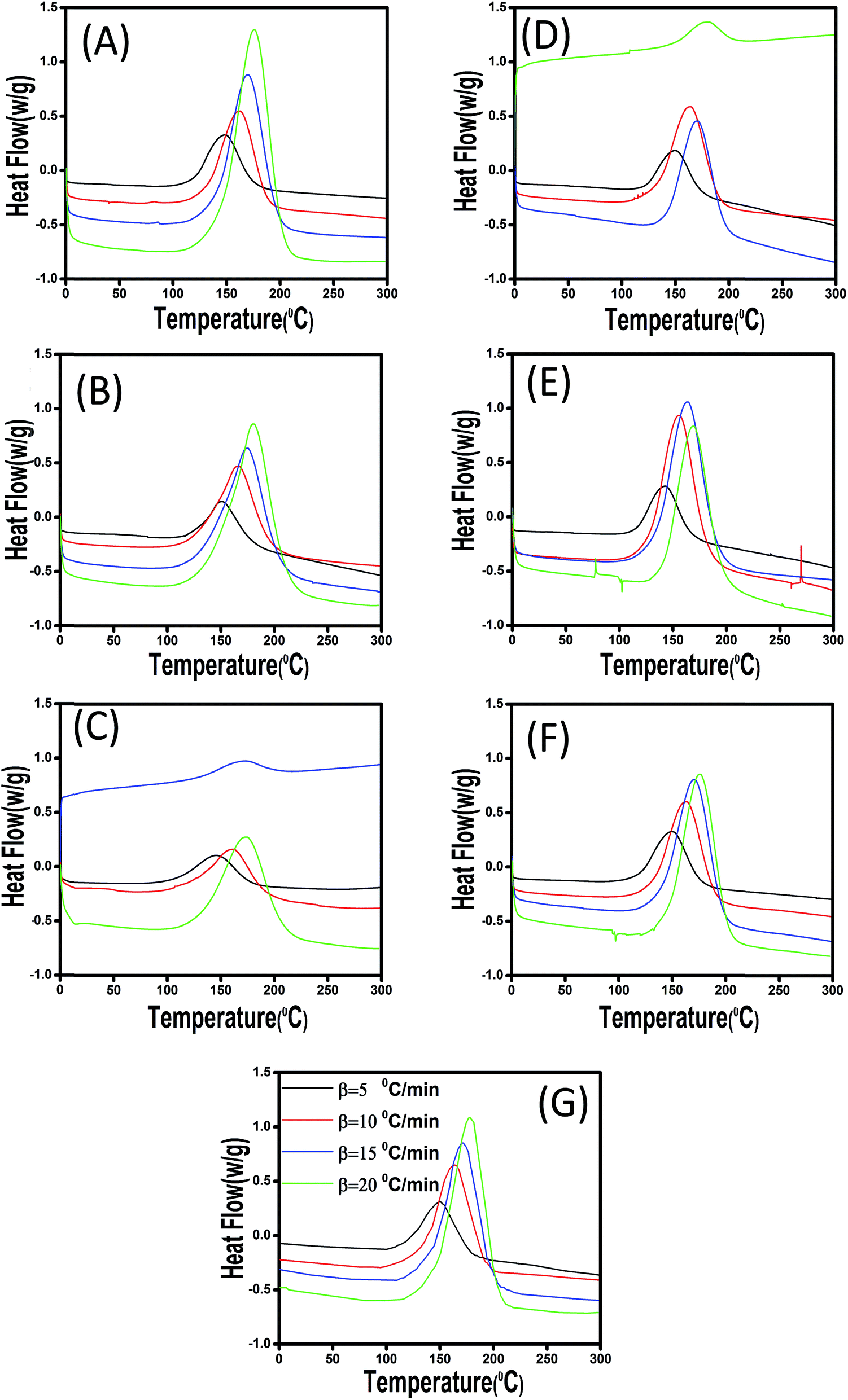 | ||
| Fig. 6 Dynamic DSC thermograms of epoxy/anhydride and its nanocomposites containing (A) 0.1-CaCO3, (B) 0.3-CaCO3, (C) 0.1-mCaCO3, (D) 0.1-ES, (E) 0.3-ES, (F) 0.1-mES, and (G) unfilled samples at different heating rates of 5, 10, 15, and 20 °C min−1. | ||
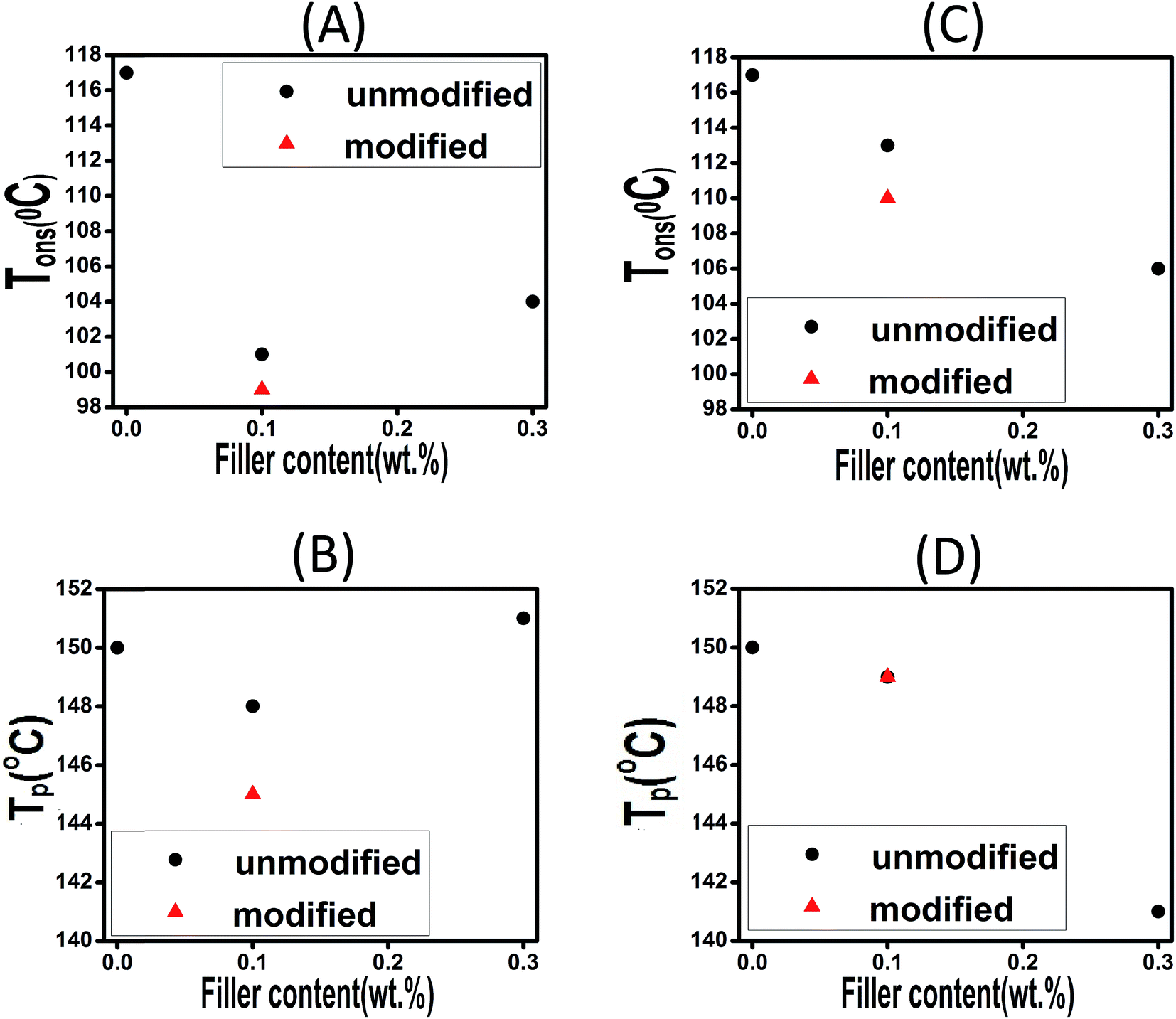 | ||
| Fig. 7 Effect of filler loading on the Tons and TP of composites filled with CaCO3 (A and B) and ES (C and D). | ||
The plots confirm that Tp and Tons are both dropped as a consequence of introduction of 0.1 wt% of CaCO3, while a very limited rise can be seen at 0.3 wt% CaCO3 loading. This contradict behavior can be explained in this way that at very low filler concentration the functional groups anchored to the filler surface compensate steric hindrance effect, while for higher loading level of 0.3 wt% the steric hindrance effect can overcome the effect caused by functional groups. In case of ES-incorporated samples, Tp and Tons continuously decreased with ES loading, which was attributed to the amine and carbonyl functional groups of the proteins present on ES surface able to react with epoxide groups. This behavior was observed in all experiments at different heating rates. Furthermore, in case of modified CaCO3, one can observe that Tp and Tons take lower values in comparison with the corresponding samples filled with the same content of pristine filler. This can be ascribed to the presence of more functional groups on the surface of fillers after chemical modification, which is discussed in details later. Of note, however, samples having modified and pristine ES particles have nearly the same Tp values indicating that modification of ES does not accelerate epoxy–anhydride cure reaction that much as functionalized CaCO3.
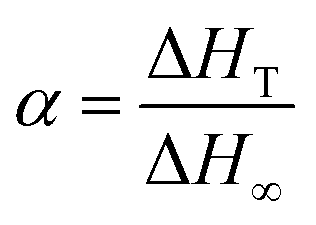 | (1) |
The mechanisms of the thermoset epoxy curing are generally classified into two major kinetics reactions, namely an nth order and an autocatalytic reaction (more information on how to come to this equations is given in ESI†):
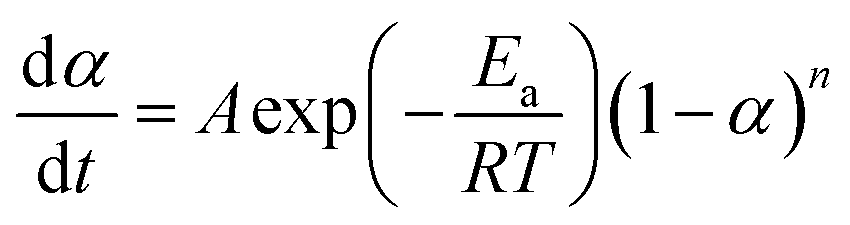 | (2) |
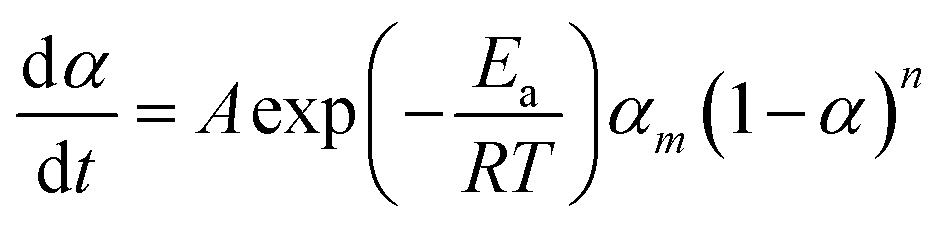 | (3) |
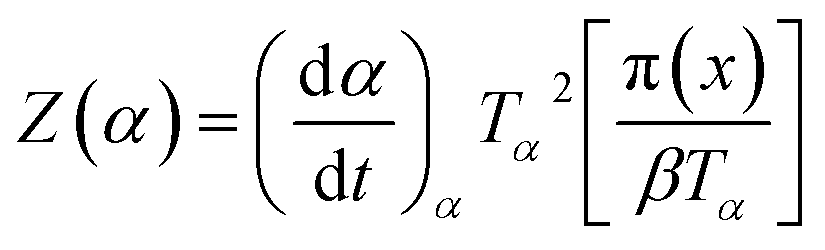 | (4) |
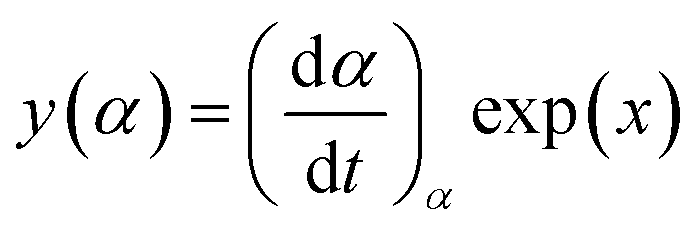 | (5) |
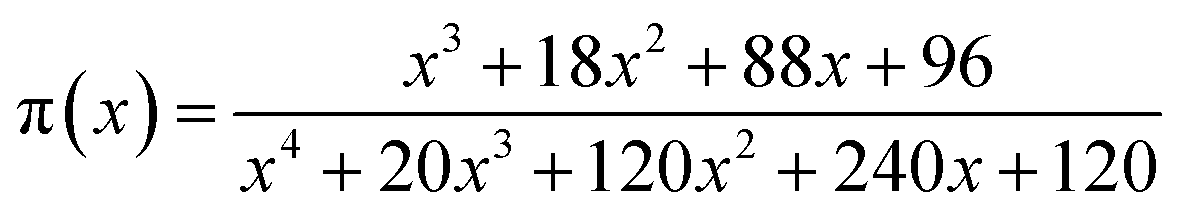 | (6) |
It has been recognized that the term in the brackets in eqn (4) has a negligible effect on the shape of the Z(α) function.33 Thus, the values of Z(α) can be determined for each value of α by multiplying the experimental values of (dα/dt)α and Tα2. In this work, the value of Ea (calculated by Ozawa method, the approach discussed in the following section) was used to measure both the y(α) and Z(α) functions using eqn (4) and (5). Fig. 8 shows the variation of y(α) and Z(α) functions for the sample containing 0.1 wt% ES at heating rate of 5 °C min−1. The apexes αm and αp of the curves are determined accordingly, where the best match between the experimental and theoretical Z(α) master plots donated the kinetics model. As typically shown in Fig. 8, the values of αm were lower than that of 0.63 obtained for αp. In agreement with the outcomes of Malek on curing kinetics of epoxy under nonisothermal conditions, curing systems studied in this work can be described well with the two-parameter autocatalytic kinetic model.
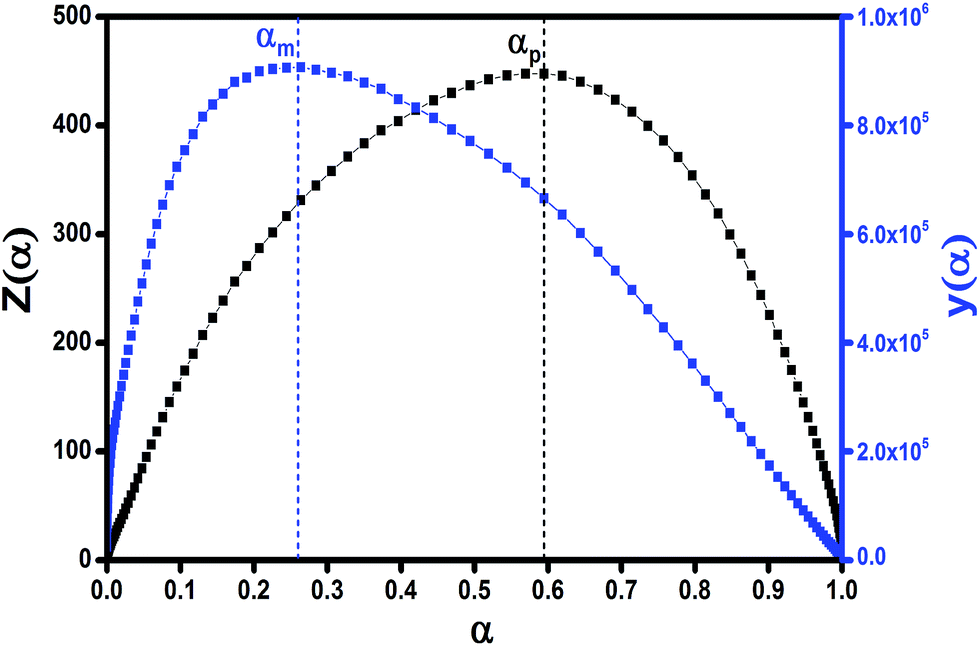 | ||
| Fig. 8 Distribution fashion of y(α) and Z(α) functions calculated by DSC data for the composite containing 0.1 wt% ES at heating rate of 5 °C min−1. | ||
With converting eqn (3), and defining p = αm/(1 − αm), we can obtain the following expression:
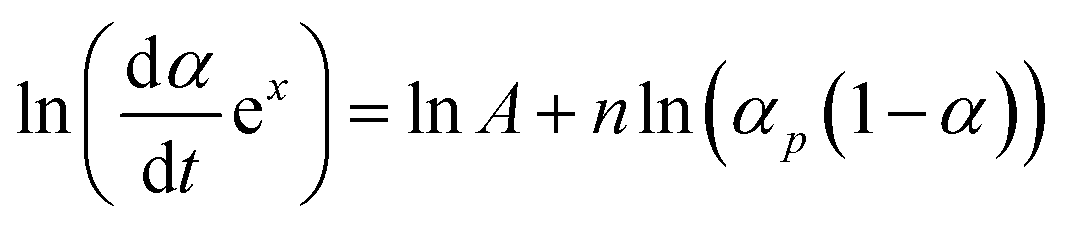 | (7) |
The DSC data is then converted to plot 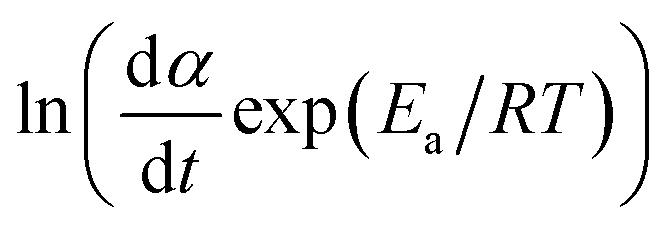 against ln(αp(1 − α)).
against ln(αp(1 − α)).
The reaction orders m and n are then obtained from the slope of the resulting curve in the linear region and the relation m = pn, respectively.
The kinetic parameters are obtained using the multiple heating rates Ozawa method assuming that the activation energy Ea is not dependent on the conversion in the course of curing reaction. Assuming the maximum conversion being independent of the heating rate, the accurate Ea can be determined.34,35 In the present study, the reactions were conducted at four different heating rates: 5, 10, 15, and 20 °C min−1. Ozawa suggests that Ea can be calculated correlating the Tp with heating rate β as follows:
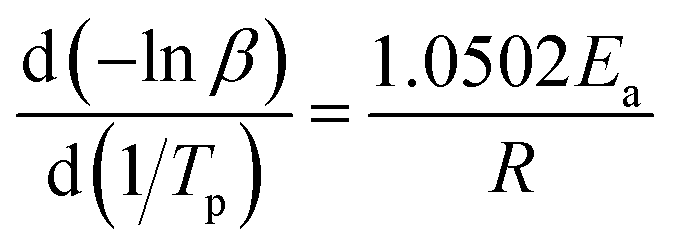 | (8) |
The transformed form of eqn (8) is the below relation:
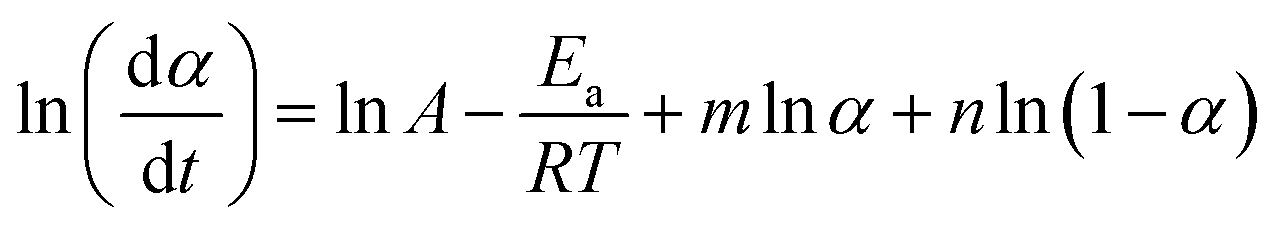 | (9) |
Changing α to (1 − α) we obtain:
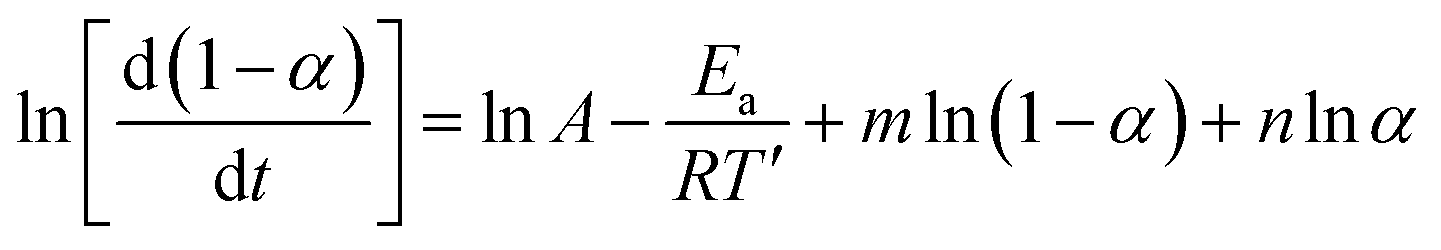 | (10) |
Subtracting eqn (10) from (9) we obtain:
 | (11) |
On the other hand, when we add the aforementioned relations together we get:
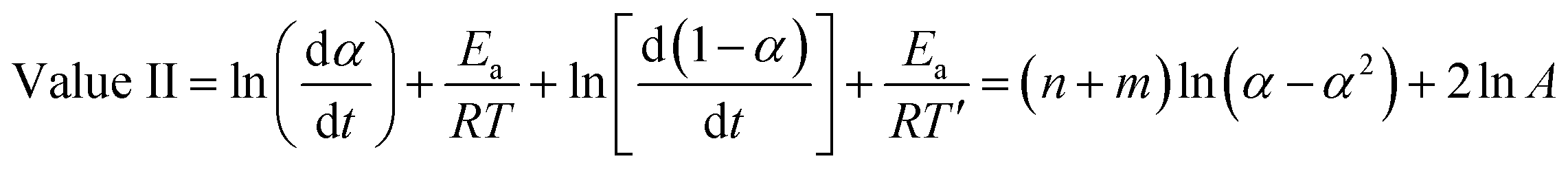 | (12) |
The value of (n − m) was determined from the slope of the plot of value I against ln[(1 − α)/α], while values of (n + m) and 2lnA were obtained from the slope and intercept of the plot of value II against ln(α − α2).36
The Friedman's model is based on the assumption that the activation energy Ea is a function of α, means that it changes throughout the reaction.37 The Friedman's model can be derived directly from eqn (4), as follows:
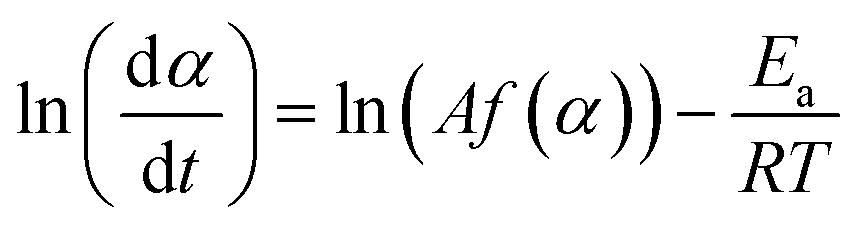 | (13) |
The plot ln(dα/dt)–1/T at an identical α from a series of dynamic DSC tests performed at different heating rates results in a line with a slope of −Ea/R and an intercept of ln(Af(α)).
By fitting linear regression lines a series of Ea and Af(α) values are obtained for each value of α.
In the case of Friedman's method, we can also apply eqn (8) and (14) to obtain kinetics triplet which is shown in Table 2.
| Model | Friedman | Malek | Ozawa | KAS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ea (kJ mol−1) | n | m | lnA (s−1) |
Ea (kJ mol−1) | n | m | lnA (s−1) |
Ea (kJ mol−1) | n | m | lnA (s−1) |
Ea (kJ mol−1) | n | m | lnA (s−1) |
|
| Unfilled | — | 1.04 | 0.57 | 13.6 | — | 0.83 | 0.38 | 16.1 | 75.3 | 1.05 | 0.4 | 16.4 | 65.0 | 1.04 | 0.57 | 13.6 |
| 0.1-ES | — | 1.06 | 0.34 | 17.4 | — | 0.86 | 0.3 | 14.1 | 72.6 | 1.03 | 0.38 | 15.7 | 68.4 | 1.01 | 0.4 | 14.6 |
| 0.3-ES | — | 1.09 | 0.4 | 15.9 | — | 0.87 | 0.25 | 15.1 | 75 | 1.1 | 0.38 | 16.9 | 71.2 | 1.08 | 0.41 | 15.8 |
| 0.1-mES | — | 1.17 | 0.31 | 19 | — | 0.98 | 0.27 | 15.9 | 79.9 | 1.15 | 0.34 | 17.9 | 74.5 | 1.13 | 0.38 | 16.3 |
| 0.1-CaCO3 | — | 1.24 | 0.41 | 17.9 | — | 0.99 | 0.28 | 15.4 | 68.5 | 1.2 | 0.46 | 15.9 | 76.7 | 1.22 | 0.43 | 17.1 |
| 0.3-CaCO3 | 1.05 | 0.44 | 14.4 | — | 0.8 | 0.46 | 14.3 | 68.5 | 1.05 | 0.44 | 14.5 | 82.1 | 1.11 | 0.34 | 18.3 | |
| 0.1-mCaCO3 | — | 1.12 | 0.17 | 16.3 | — | 0.98 | 0.12 | 15.9 | 74.4 | 1.12 | 0.17 | 16.1 | 71.8 | 1.1 | 0.2 | 15.4 |
The kinetics of curing reactions of the epoxy composites may be nth order or autocatalytic, depending on the curing agent and/or filler type, filler content, and filler topology.38,39 For nth order reaction, ln[Af(α)] values may be written as:
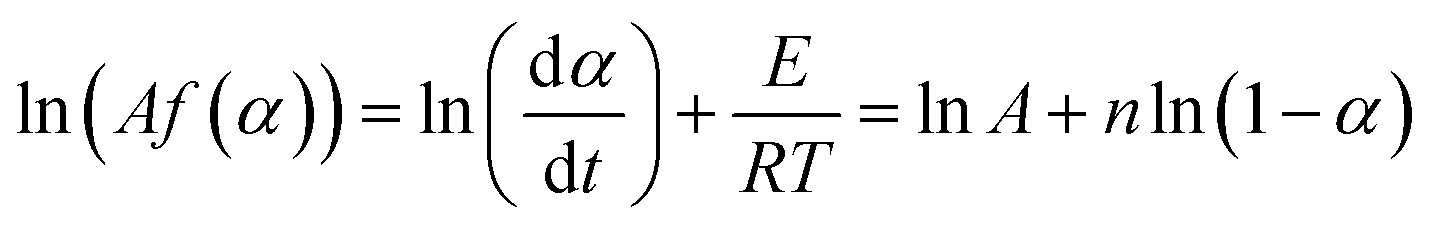 | (14) |
Friedman suggested that the relationship between ln[Af(α)] and ln(1 − α) should yield a beeline of which the slope corresponding to the order “n” of the reaction can be obtained. For an autocatalytic process, the Friedman plot frequently shows a maximum of ln(1 − α) roughly around −0.51 to −0.22, which is equivalent to α of about 0.2–0.4.40 Typical plots on Ozawa model described through eqn (8)–(14) are provided in the ESI (Fig. S2 and S3†).
Fig. 9 shows Friedman plots of the composite filled with 0.1 wt% of ES. Since ln[Af(α)] is not linearly related to the ln(1 − α), and show a maximum in the conversion range mentioned above, it can deduced that curing reactions of this system has an autocatalytic nature.
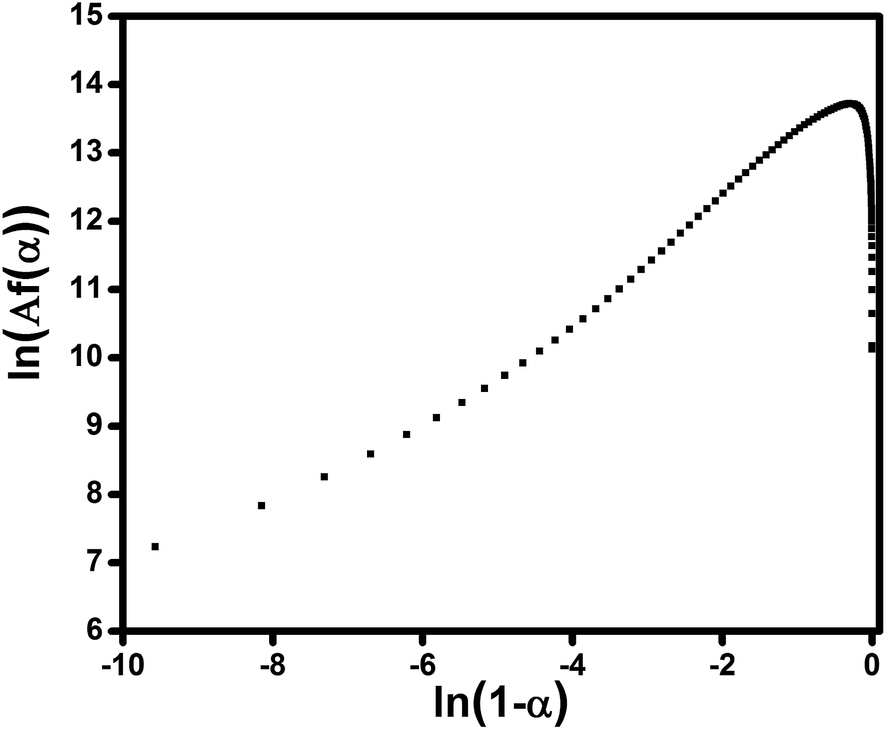 | ||
| Fig. 9 Plots of ln[Af(α)] vs. ln(1 − α) for the sample containing 0.1 wt% ES studied under heating rate of 5 °C min−1 to calculate the activation energy using Ozawa method. | ||
The Kissinger–Akahira–Sunose (KAS) method is performed to determine the activation energy prior to identifying other kinetic parameters:
 | (15) |
In KAS model, Tα is the temperature at a specified conversion α, extracted from the DSC curve at a given heating rate. The Eα of the curing reaction at various α values is obtained from the linear plots of ln(β/Tα2) vs. 1/Tα (see Fig. S6 in the ESI†).
Since KAS and Friedman models lack the assumption of constant Ea, the kinetic equations of the cure reaction calculated by these methods should better fit the actual values on dynamic DSC compared to the Malek and Ozawa approaches. Fig. 10 shows the variation of Eα values obtained by the Friedman method at various α values for the studied epoxy/anhydride systems. As a whole, the results suggest that the curing mechanism in these systems is complicated and depends obviously on the type and functionalization of particles. At low conversions, an overall fall in the Ea value can be explained on account of autocatalytic nature of reaction between epoxy and methyltetrahydrophthalic anhydride. The low viscosity of system at the assigned interval permits for movement of chain segments leading to facilitation of curing reaction. At later stages, however, as the cure reaction proceeds, the free volume decreases and the system undergoes gelation (liquid-to-rubber) and vitrification (rubber-to-glass) transitions. Intensive cross-linking that takes place at high conversions causes a reduction in molecular mobility, then the cure changes from kinetic-controlled to diffusion-controlled mechanism.19 The rise in Ea at higher conversions for the system is a vivid evidence for this event.20
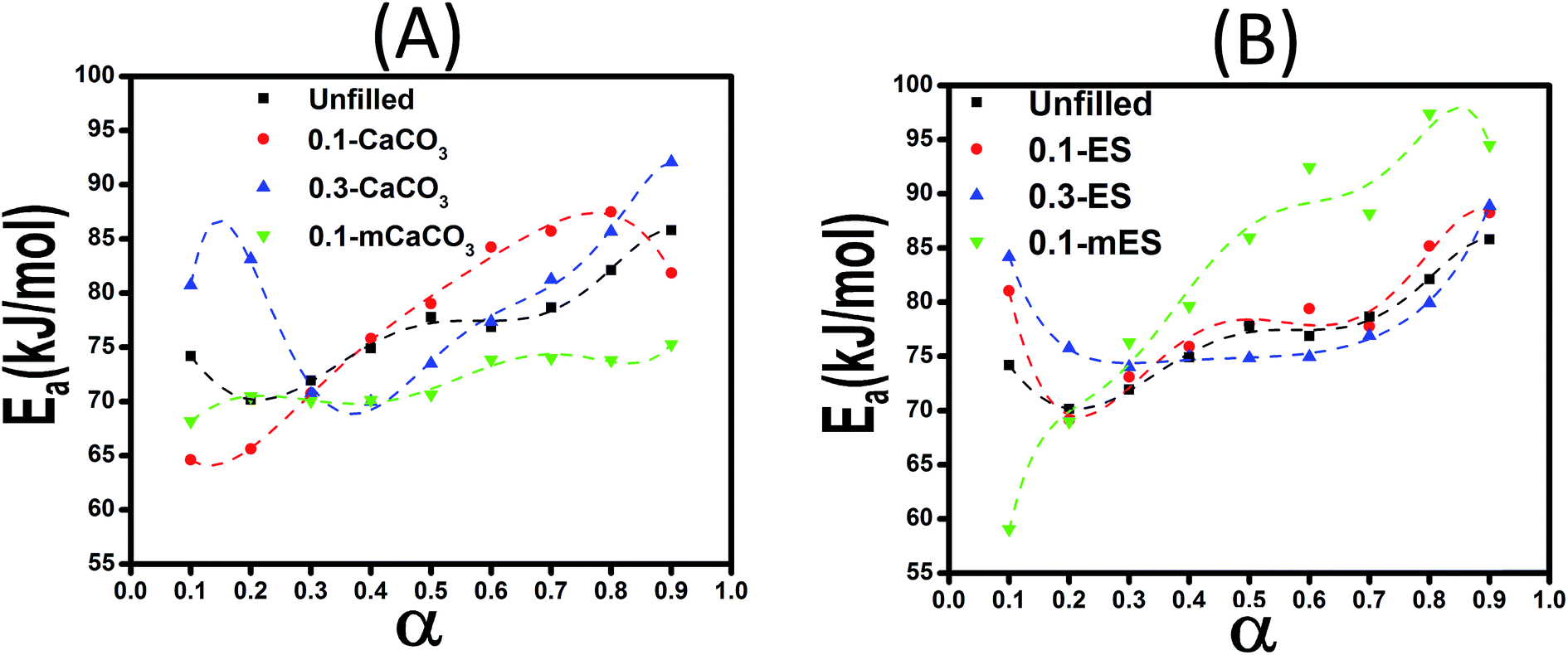 | ||
| Fig. 10 Variation of activation energy vs. conversion calculated using Friedman method for systems containing CaCO3 (A) and ES (B) particles. | ||
Comparison of Ea values of samples containing ES particles (Fig. 10B) suggests that addition of 0.1 wt% ES particle effectively facilitates the epoxy–anhydride cure reaction, while the same amount of acid-modified ES (mES) cannot catalyze the cure reaction. In agreement with this, Ozawa model suggests a higher activation energy of 79.98 kJ mol−1 for 0.1-mES sample compared to value of 72.58 kJ mol−1 for 0.1-ES sample (Table 2). A similar trend with somewhat more positive effect can be seen in case of 0.3-ES sample. Of note, the presence of ES biofiller poses very limited hindrance effect on the cure reaction between epoxy and anhydride. This is while even nanofillers like carbon nanotubes with a comparatively very higher price frequently harden the curing reaction at high conversions.10 Likewise, addition of CaCO3 makes difficult the interaction between resin and hardener (Fig. 10A).
Despite very similar chemical composition of two types of fillers used in this work, there might be a reason behind this contradictive behavior, which necessitates further explanations. Functionalization of particles of each type leads to such an opposite trends in activation energy variation. The presence of a functional group on the surface of a filler allows for an effective dispersion of particles in the system leading to a viscosity upturn.41 Moreover, the functional groups anchored to the surface of modified filler can open epoxide rings as a secondary hardener. Although the increase in viscosity retards the cross-linking reaction, the presence of functional group may speed up this event at higher conversions. In case of samples containing modified fillers, the higher surface area of ES particles (based on BET results in Table 1) enables more copolymer entities to attach the particle surface giving the epoxy groups more access to carboxyl groups, which has a retardation effect on cross-linking. On the contrary, less number of terpolymer chains attached to CaCO3 particles can catalyze the cure reaction with the aid of functional groups with less interference, which compensates the retardation of CaCO3. Thus, chemical functionalization of ES is expected to somewhat neutralize the power of peptide groups naturally attached to the surface of porous ES particles. Fig. 11 provides the schematic illustration for the proposed mechanism with contradictive effect of acid functionalization to ES and CaCO3 particles.
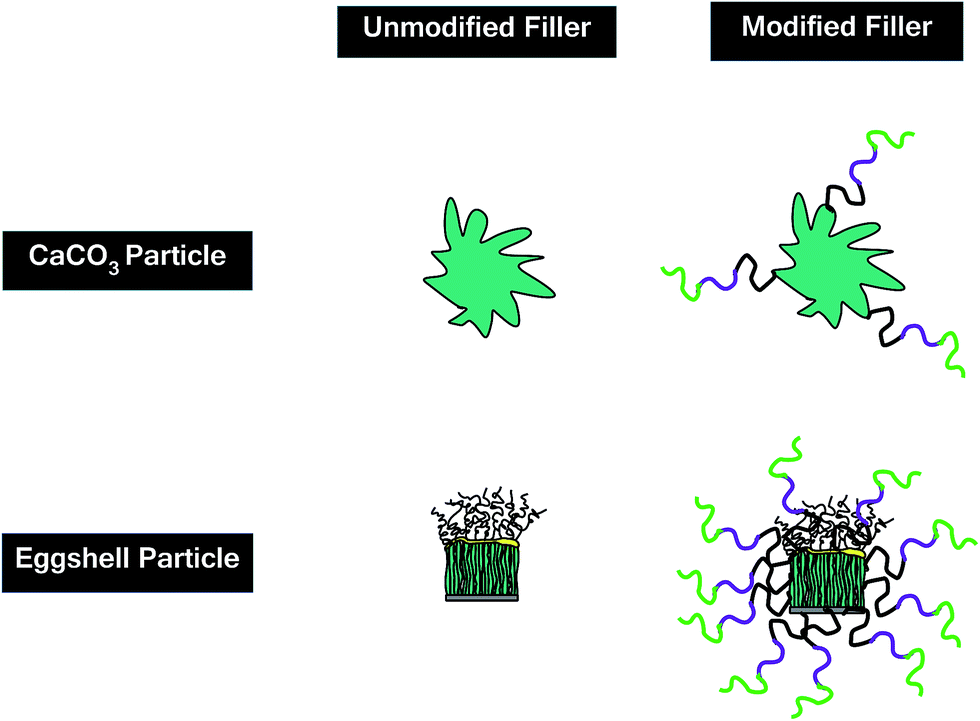 | ||
| Fig. 11 Proposed surface topology for the modified ES and CaCO3. | ||
From a mathematical point of view, the kinetic parameters of the studied epoxy/anhydride systems can be compared using data in Table 2. The overall order of cure reaction (m + n) is greater than 1 for all studied systems indicating non-elementary nature of cure in such systems. In view of activation energy data, despite its simplicity, the Ozawa model provides a quantitative comparison for assessing the role of filler type, filler content, and filler functionalization. Fig. 12 provides a general framework to assess the reliability of the used models in the current study. Overall, the Ozawa, KAS and Friedman, i.e., eqn (15) and (13), models are fitted better to the experimental data than the Malek or Friedman based on polynomial fitting. At early stages of cure reaction, before gelation and vitrification take place, the reaction proceeds in the liquid phase and it is cogently controlled by the chemical kinetics. Therefore, the kinetic behavior can be predicted well by the used models. As the reaction progresses, a deviation appears due to the onset of gelation and vitrification, where the mobility of reactive groups is hindered, the rate of reaction is controlled by diffusion of molecules rather than by kinetic factors, hence, the values predicted by the model are differ somewhat by the experimental data. Obviously, however, the polynomial-based Friedman and Malek kinetic models fail to predict cure behavior at late stages of reaction.
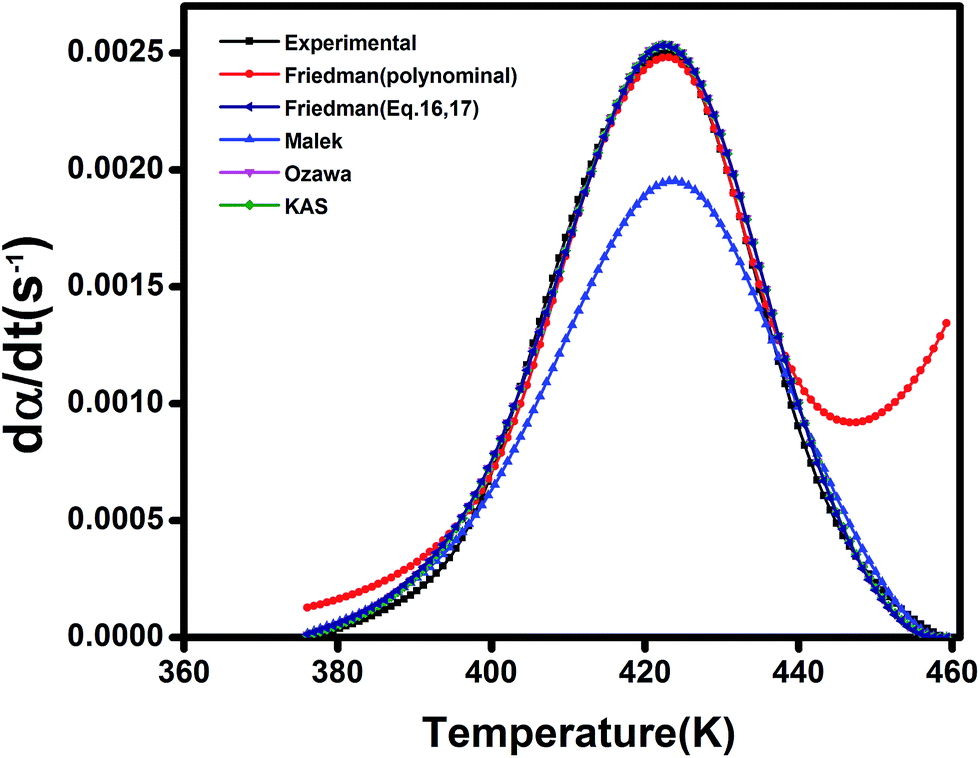 | ||
| Fig. 12 Comparison of experimental data with the kinetic models for sample containing 0.1 wt% ES measures at heating rate of 5 °C min−1. | ||
4. Conclusions
XPS analysis of pristine and terpolymer-modified particles of each type showed that the intensity of the C 1s and N 1s peaks and relative intensities of the oxygen contents ([O]:[C]|spec) strongly changed by chemical functionalization. The results suggest that terpolymer wrapped vast areas of the particle surfaces. Photoelectrons escaped from the atoms of the substrate material were additionally scattered during their passage through the copolymer films and contributed less to the intensities of the elemental peaks. The attachment of peptide groups to the surface of pristine ES has facilitated cure kinetics, as recognized by lesser activation energy values compared to CaCO3. Ozawa and KAS methods were in better agreement with the experimental data than Malek or Friedman method. The peculiar behavior of nanocomposites containing terpolymer-modified fillers, however, was explained mechanistically; so that functionalized ES, unlike modified CaCO3, hindered the cure reaction of epoxy with anhydride. In case of pristine fillers, the higher specific surface area of ES particle resulting from its porous structure enabled ES to receive more terpolymer entities by utilizing more carboxyl groups. On the other hand, functionalization of ES provided less amount of residual carboxyl group in ES particles to reach epoxy leading to a reduction in the number of effective contacts between epoxy and anhydride, compared to the treated CaCO3 particles.5. Conflict of interest
The authors of this work confirm that they have no competing interests.Acknowledgements
The authors of this work would like to express their special thanks to Leibniz-Institut für Polymerforschung, Dresden, Germany for Technical and Financial supports.References
- D. Ratna, Handbook of thermoset resins, iSmithers, 2009 Search PubMed.
- V. Fombuena, L. Bernardi, O. Fenollar, T. Boronat and R. Balart, Mater. Des., 2014, 57, 168–174 CrossRef CAS.
- T. Vidil, F. Tournilhac, S. Musso, A. Robisson and L. Leibler, Prog. Polym. Sci., 2016, 62, 126–179 CrossRef CAS.
- J. J. Chruściel and E. Leśniak, Prog. Polym. Sci., 2015, 41, 67–121 CrossRef.
- L.-J. Cui, Y.-B. Wang, W.-J. Xiu, W.-Y. Wang, L.-H. Xu, X.-B. Xu, Y. Meng, L.-Y. Li, J. Gao, L.-T. Chen and H.-Z. Geng, Mater. Des., 2013, 49, 279–284 CrossRef CAS.
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- N. A. S. John and G. A. George, Prog. Polym. Sci., 1994, 19, 755–795 CrossRef.
- H. He, K. Li, J. Wang, G. Sun, Y. Li and J. Wang, Mater. Des., 2011, 32, 4521–4527 CrossRef CAS.
- V. Trappe, W. Burchard and B. Steinmann, Makromol. Chem., Macromol. Symp., 1991, 45, 63–74 CrossRef CAS.
- M. R. Saeb, E. Bakhshandeh, H. A. Khonakdar, E. Mäder, C. Scheffler and G. Heinrich, Sci. World J., 2013, 2013 Search PubMed.
- G. Ji, H. Zhu, C. Qi and M. Zeng, Polym. Eng. Sci., 2009, 49, 1383–1388 CAS.
- M. M. Rahman, A. N. Netravali, B. J. Tiimob and V. K. Rangari, ACS Sustainable Chem. Eng., 2014, 2, 2329–2337 CrossRef CAS.
- D. Cree and A. Rutter, ACS Sustainable Chem. Eng., 2015, 3, 941–949 CrossRef CAS.
- S. H. Katz and W. W. Weaver, Encyclopedia of food and culture, Scribner, New York, 2003 Search PubMed.
- A. G. J. Tacon, Agric. Wastes, 1982, 4, 335–343 CrossRef CAS.
- M. Pettinato, S. Chakraborty, H. A. Arafat and V. Calabro, Ecotoxicol. Environ. Saf., 2015, 121, 57–62 CrossRef CAS PubMed.
- H. J. Park, S. W. Jeong, J. K. Yang, B. G. Kim and S. M. Lee, J. Environ. Sci., 2007, 19, 1436–1441 CrossRef CAS.
- S.-C. Wu, H.-C. Hsu, S.-K. Hsu, Y.-C. Chang and W.-F. Ho, Ceram. Int., 2015, 41, 10718–10724 CrossRef CAS.
- T. Witoon, Ceram. Int., 2011, 37, 3291–3298 CrossRef CAS.
- M. Mohammadi, P. Lahijani and A. R. Mohamed, Chem. Eng. J., 2014, 243, 455–464 CrossRef CAS.
- R. Kumar, J. S. Dhaliwal, G. S. Kapur and Shashikant, Polym. Compos., 2014, 35, 708–714 CrossRef CAS.
- J. Zhou, S. Wang, F. Nie, L. Feng, G. Zhu and L. Jiang, Nano Res., 2011, 4, 171–179 CrossRef CAS.
- H. R. Dakhel, e-Polym., 2008, 8, 1618–1626 Search PubMed.
- T. Ghabeer, R. Dweiri and S. Al-Khateeb, J. Reinf. Plast. Compos., 2013, 32, 402–409 CrossRef.
- Z. Lin, Z. Zhang and K. Mai, J. Appl. Polym. Sci., 2012, 125, 61–66 CrossRef CAS.
- P. Intharapat, A. Kongnoo and K. Kateungngan, J. Polym. Environ., 2013, 21, 245–258 CrossRef CAS.
- M. R. Saeb, H. Ramezani-Dakhel, H. A. Khonakdar, G. Heinrich and U. Wagenknecht, J. Appl. Polym. Sci., 2013, 127, 4241–4250 CrossRef CAS.
- A. B. Rodríguez-Navarro, P. Marie, Y. Nys, M. T. Hincke and J. Gautron, J. Struct. Biol., 2015, 190, 291–303 CrossRef PubMed.
- M. S. Fernandez, K. Passalacqua, J. I. Arias and J. L. Arias, J. Struct. Biol., 2004, 148, 1–10 CrossRef CAS PubMed.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- W. J. Landis and J. R. Martin, J. Vac. Sci. Technol., A, 1984, 2, 1108–1111 CAS.
- G. Beamson and D. Briggs, J. Chem. Educ., 1993, 70, A25 Search PubMed.
- J. Málek, Thermochim. Acta, 1995, 267, 61–73 CrossRef.
- D. D. Shin and H. T. Hahn, Composites, Part A, 2000, 31, 991–999 CrossRef.
- J. Gao, D. Kong and S. Li, Polym. Compos., 2010, 31, 60–67 CrossRef CAS.
- T. Zhou, M. Gu, Y. Jin and J. Wang, Polymer, 2005, 46, 6174–6181 CrossRef CAS.
- S. Vyazovkin and C. A. Wight, Thermochim. Acta, 1999, 340–341, 53–68 CrossRef CAS.
- C. Li, C. Zuo, H. Fan, M. Yu and B. Li, Thermochim. Acta, 2012, 545, 75–81 CrossRef CAS.
- S. H. Ryu, J. H. Sin and A. M. Shanmugharaj, Eur. Polym. J., 2014, 52, 88–97 CrossRef CAS.
- Y. Bai, P. Yang, S. Zhang, Y. Li and Y. Gu, J. Therm. Anal. Calorim., 2015, 120, 1755–1764 CrossRef CAS.
- M. Abdalla, D. Dean, P. Robinson and E. Nyairo, Polymer, 2008, 49, 3310–3317 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24772e |
| This journal is © The Royal Society of Chemistry 2017 |