DOI:
10.1039/C6RA24760A
(Paper)
RSC Adv., 2017,
7, 15342-15351
Polyaniline nanoflowers grafted onto nanodiamonds via a soft template-guided secondary nucleation process for high-performance glucose sensing†
Received
5th October 2016
, Accepted 21st February 2017
First published on 7th March 2017
Abstract
Superfine polyaniline (PANI) nanoflowers (NF) with protruded whiskers at the edge of the flowers were produced on the surface of nanodiamonds (NDs) using cetyltrimethylammonium bromide (CTAB) as a soft template and fine tuning the graft co-polymerization conditions. Typically, the amount of amino-functionalized ND (ND-NH2) and the soft template CTAB used in the graft polymerization plays an important role in directing the NF morphology. The correlation between the amount of ND-NH2 and morphology has been discussed in terms of secondary nucleation for PANI growth. More importantly, the ND-grafted PANI (NDx-g-PANI) NF exhibits a superior performance for glucose detection and possesses wider linear concentration range (1–30 mM), low detection limit (0.018 mM), and high sensitivity (2.03 μA mM−1) as compared to most of the enzyme glucose sensors fabricated with either PANI or carbon nanostructures. The results obtained from this study offer scope for future studies on the fabrication of new biosensors with other aniline derivatives and carbon nanostructures.
1. Introduction
Glucose is a universal nutrient that performs fundamental roles in energy supply, carbon storage, biosynthesis, and cell wall formation. Diabetes mellitus is a common metabolic disorder and the control of blood glucose is highly necessary to reduce the diabetes-related complications such as cardiovascular disease, chronic renal failure, nerve damage, and blindness.1,2 For the last 30 years, intensive studies have been conducted on the use of enzymes for bioelectrocatalysis towards the development of biosensors, bioreactors, and biofuel cells.3–5 Traditionally, enzymes are used to detect glucose via a process involving the reduction of the enzyme and simultaneous oxidation of the glucose molecules. However, there are few issues related to the electrochemistry of enzymes at bare electrodes; for example, enzymes have a short lifetime and lose their activity at bare electrodes.6 Moreover, there are electron transport issues associated with the long paths between the sites of a biocatalyst (enzyme) and the electrode. Thus, enzyme-based glucose sensors suffer from insufficient long-term stability, low sensitivity, and particularly complicated enzyme immobilization procedures. To resolve these problems, studies have been focused on the immobilization of enzymes at the electrode surface using different strategies.7,8 The strategies of enzyme immobilization on the electrodes include covalent binding of enzymes or wiring of enzymes to the electrode using a linker,9 sandwiching the enzymes between two layers of polymers,10 employing multicomponents to maintain enzyme activity,11 and entrapment in a polymer membrane/network.12 Covalent binding decreases the enzyme activity and sandwiching techniques result in non-uniform distribution of the enzyme. Among the various approaches for enzyme entrapment, the use of conductive polymers (CPs) is attractive.13–15 In the fabrication of biosensors, polyaniline (PANI) serves as a perfect matrix for the immobilization of enzymes16,17 and functions as a mediator to improve the signal application.18,19 The combination of carbon nanomaterials and PANI has been reported to be efficient in improving the sensitivity, selectivity, and stability of enzyme-based sensors.20,21 Carbon nanotube (CNT)/PANI nanocomposites exhibit improved catalytic properties for biomolecules such as hydrogen peroxide, oxygen, glucose, and ascorbic acid.22–25
Diamond has emerged as an electrode material because of its excellent electrochemical properties such as wide potential window,26,27 electrochemical stability, resistance to electrode fouling,28 and insensitivity to oxygen dissolution in aqueous solution.29 Diamond electrodes can be even used for the electrolysis of molecules with a high oxidation potential30 both in aqueous and non-aqueous media.31 Nanodiamond (ND) powder has found various applications such as intracellular imaging, drug delivery, biosensors, and biochips because of its biocompatibility, chemical inertness, chemical stability, wide potential window, and optical transparency.32,33 Moreover, undoped ND is an insulating material. The conductivity of ND can be improved by the introduction of impurities such as boron, nitrogen, phosphorous, and sulfur.34 These impurities may accumulate in the intercrystalline regions and promote sp2 carbon bonding in grain boundaries.35 However, doping is not a unique approach to make diamond conducting.
Surface modifications of ND have been facilitated due to the high surface area availability of unsaturated surface atoms. Surface modification of ND can induce a good dispersion of NDs in solvents and can modify its properties such as conductivity and electroactivity. Covalent surface modifications of diamond particles are generally used in the process of improving the properties of NDs as well to introduce functional groups. The functionalized surfaces of diamond are excellent for the self-assembly of proteins. Garrido et al.36 reported the modification of the surface layer of ND with amino groups for protein immobilization. A glucose sensor based on the covalent immobilization of glucose oxidase onto the surface of ND has been reported.37 Multilayer films of ND and hemoglobin were constructed via the layer by layer method, and the direct electron transfer was investigated.38 The immobilization of antibodies onto silica-functionalized ND and its efficiency towards bacterial binding were investigated.39 The use of polymer coatings on nanoparticles is very attractive. Li et al.40 reported the preparation of a diamond–polymer nanocomposite by grafting chains of poly(methyl methacrylate) through the atom transfer radical polymerization process. Polyethyleneimine-functionalized ND platforms (via covalent conjugation) were used as vehicles for gene delivery.41 Actis et al.42 reported the covalent linking of pyrrole units onto the boron-doped diamond surface through electropolymerization. The electrical conductivity of diamond was improved by polymerizing a layer of PANI on undoped ND.52 The results indicated that the resistance of the diamond electrode is greatly reduced after the deposition of PANI. However, note that the diamond/PANI composite reported in the earlier study43 consisted of a layer of PANI over diamond and the PANI chains were not chemically linked to the diamond surface. This bilayer configuration is not suited for sensor applications. Nanostructured conduction polymers (NCPs) have been widely utilized for chemical/biological sensors.44,45 Among the different types of NCPs, the topological features of nanoflowers (NF) have attracted the interest of scientists because of their high surface-to-volume ratio, which is beneficial for the enhancement of surface-based reactions. However, to date, no study has been reported on the utilization of flowers, such as CPs grafted on the ND backbone, towards enzyme immobilization and biosensor fabrication.
Herein, we report the preparation of ND-grafted PANI NF (NDx-g-PANI) NF through a soft template approach and their utilization towards the amperometric determination of glucose. The preparation conditions of NDx-g-PANI were optimized to obtain the desired NF morphology. Field emission scanning microscopy illustrates the formation of PANI NF on ND under the optimized conditions. The physicochemical characteristics of NDx-g-PANI-NF were evaluated through UV-visible spectroscopy, thermogravimetric analysis, and X-ray photoelectron spectroscopy. To the best of our knowledge, this is the first time that this NDx-g-PANI-NF electrode has been used for the sensitive electrochemical detection of glucose. The fabricated sensor exhibits excellent selectivity, fast response, wide linear range, and good reproducibility for glucose detection.
2. Materials and methods
2.1. Chemicals
Synthetic diamond (K. Diamond; USA) with an average size of 130 nm, which was supplied as a gift sample, was washed with a mixture of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) concentrated H2SO4 and HNO3.46 The characterization details of the supplied ND have been reported elsewhere.46,47 The reproducibility of the ND sample towards sensor applications has been demonstrated earlier.48,49 Aniline, ammonium persulfate (APS), and polyethylene glycol diamine (PEGDA) were obtained from Sigma-Aldrich, S. Korea. Human serum (from human male A plasma, USA origin, sterile-filtered) was obtained from Sigma-Aldrich, S. Korea. Cetyltrimethylammonium bromide (CTAB) was obtained from TCI (S. Korea). Reagent grade glucose oxidase (GOx) (E.C 1.1.3.4, 151 U mg−1, from Aspergillus niger; U: enzyme units) and glutaraldehyde were purchased from Sigma-Aldrich, Inc., USA. Double-distilled water (resistivity = ∼19 MΩ cm) was used throughout the experiments. Aqueous solutions of analytes were prepared in 0.1 M phosphate buffer saline (PBS; pH 7) fresh at the time of experiments. Indium tin oxide (ITO)-coated glass (specific resistance of 15 Ω, Corning Inc., USA) was first degreased with acetone and then washed in distilled water before use.
:1 (v/v) concentrated H2SO4 and HNO3.46 The characterization details of the supplied ND have been reported elsewhere.46,47 The reproducibility of the ND sample towards sensor applications has been demonstrated earlier.48,49 Aniline, ammonium persulfate (APS), and polyethylene glycol diamine (PEGDA) were obtained from Sigma-Aldrich, S. Korea. Human serum (from human male A plasma, USA origin, sterile-filtered) was obtained from Sigma-Aldrich, S. Korea. Cetyltrimethylammonium bromide (CTAB) was obtained from TCI (S. Korea). Reagent grade glucose oxidase (GOx) (E.C 1.1.3.4, 151 U mg−1, from Aspergillus niger; U: enzyme units) and glutaraldehyde were purchased from Sigma-Aldrich, Inc., USA. Double-distilled water (resistivity = ∼19 MΩ cm) was used throughout the experiments. Aqueous solutions of analytes were prepared in 0.1 M phosphate buffer saline (PBS; pH 7) fresh at the time of experiments. Indium tin oxide (ITO)-coated glass (specific resistance of 15 Ω, Corning Inc., USA) was first degreased with acetone and then washed in distilled water before use.
2.2. Preparation of NDx-g-PANI-NF
Two major stages were involved in the preparation of NDx-g-PANI-NF: (i) amine (–NH2) functionalization of ND to give ND-NH2 and (ii) surfactant (CTAP)-assisted graft polymerization of amine onto the ND-NH2 surface. ND-NH2 was prepared based on a similar procedure reported for the –NH2 functionalization of multi-walled carbon tubes.23,25 The calculated amount of ND-NH2 (x) (1, 5, and 10 mg) was dispersed in 42.7 mL of 1.0 M HCl containing 0.05 M CTAB and subjected to ultrasonication (BRANSON digital sonifier) for 1 h to obtain a homogeneous dispersion. To the ND-NH2 dispersion, 2.30 mL of aniline was added to achieve a concentration of 0.05 M aniline in the reaction system and stirred for 30 min. To the homogeneous dispersion, a precooled 5.0 mL solution of 0.25 M of APS was added dropwise and stirred for 6 h to obtain a green precipitate. The precipitate was dried at 60 °C for 6 h and stored in a refrigerator at 4 °C for 24 h. The product was identified as NDx-g-PANI, where x denotes the amount of ND-NH2 used in the polymerization process. For comparative purposes, a pristine PANI sample was prepared by maintaining similar experimental conditions as used for the preparation of NDx-g-PANI but in the absence of ND-NH2. Typically, 2.3 mL of aniline was added to 42.7 mL of homogeneous dispersion of CTAB (0.05 M) in 1.0 M HCl. Subsequently, 5.0 mL of 0.25 M aqueous APS in 1.0 M HCl was added dropwise and stirred for 6 h. The precipitate (pristine PANI) was collected and dried at 60 °C for 6 h.
2.2.1. Fabrication of NDx-g-PANI/GOx electrode. Pre-cleaned ITO was used for the fabrication of a glucose sensor electrode. Typically, about 20 mg of NDx-g-PANI was dispersed in a solution containing about 0.8 mL of water and 0.2 mL of Nafion solution in 5% ethanol and sonicated using a digital sonifier for 10 min to form a colloidal paste. About 50 μL of paste was applied onto the surface of the ITO (area = 1 cm2) and dried in a vacuum oven at 25 °C for 8 h. A solution of GOx (10 mg.mL−1) in 0.1 M PBS was prepared and stored at 4 °C. About 5 μL glutaraldehyde was added over the NDx-g-PANI layer coated on the ITO electrode and on top of that 20 μL GOx in 0.1 M PBS was added and allowed to dry at room temperature (25 °C). The NDx-g-PANI/GOx electrode was preserved in a refrigerator at 4 °C after being washed with a 0.1 M PBS solution.
2.3. Instrumentation
The morphology of NDx-g-PANI was examined via scanning electron microscopy (FESEM) (Hitachi, S-4200, Japan). Samples were mounted on the microscope and sputtered with a thin layer of platinum particles before viewing. FESEM images were obtained at the accelerating voltage of 15 kV. The oxidation states of PANI were evaluated by X-ray photoelectron spectroscopy (XPS) (ULVAC-PHI model Quantera SXM). A Fourier transform infrared spectrometer (FTIR) (Jasco, IR-610, USA) was used to obtain the spectra of the polymers and composites using KBr pellets. The thermal stability (TG-DTA) (Seiko, TMA 120, Japan) of the samples was determined at the heating rate of 10 °C min−1 under a nitrogen atmosphere over the temperature range of 30–800 °C. Ultraviolet-visible optical absorption spectra (Shimadzu, UV-2101, Japan) of the samples were obtained using quartz cuvettes (1 cm) at room temperature in the range of 200–800 nm. Chronoamperometry was performed via IVIUMSTAT and COMPACTSTAT (The Netherlands) instruments using the three-electrode cell set-up comprising Pt wire (auxiliary), Ag/AgClsatKCl (reference), and modified indium-doped tin oxide-coated glass plate (ITO) or glassy carbon (GC) (working) electrodes. The chronoamperometric response of NDx-g-PANI/GOx towards glucose was studied at the applied potential of +0.4 V under stirring. The current response was obtained as follows. After applying the potential (+0.4 V), the current was allowed to reach a stable value and small aliquots of glucose solutions were added at regular intervals, and then the response was obtained. The stock solution of glucose was prepared in 0.1 M PBS. The selectivity of NDx-g-PANI/GOx towards glucose was tested by performing experiments in the presence of generally known interfering substances.50
3. Results and discussion
3.1. Morphology
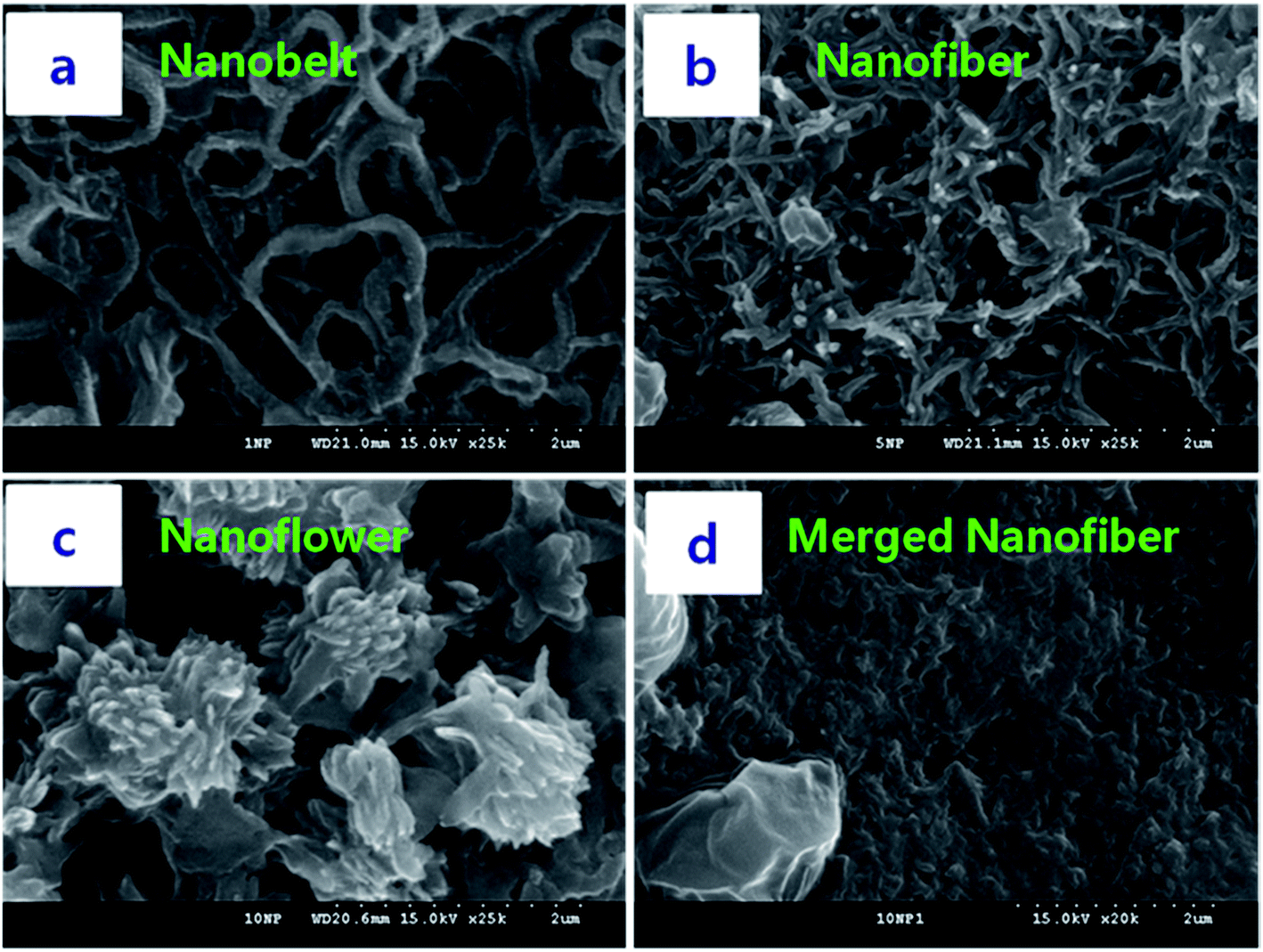
We optimized the conditions for NDx-g-PANI NF formation using different x values during graft polymerization and subsequently monitoring the morphology of the resultant product. Fig. 1 presents the FESEM images of various NDx-g-PANI materials obtained by changing x from 1 mg to 10 mg during graft polymerization. On examining the images shown in Fig. 1a–c, it can be noticed that x has a pronounced effect on the morphology of the resultant product. For comparison, the FESEM image of pristine PANI, prepared in the absence of ND-NH2, is presented Fig. 1d. Pristine PANI has an irregular fiber-like morphology (Fig. 1d) with an average diameter and length of ∼40 nm and ∼500 nm, respectively. The formation of PANI nanofibers during the CTAB-assisted polymerization of aniline has been documented earlier.51 The FESEM images in Fig. 1a–c show that the morphology varies from nanofibers to nanoplates to NF with an increase in x during graft polymerization. Typically, ND10-g-PANI exhibits a well-defined flower-like morphology with pronounced whiskers. In the case of ND1-g-PANI, a flattened fiber-like morphology is observed (Fig. 1a), which originates due to the secondary growth of PANI over the formed NDx-g-PANI in the initial stage. This is because PANI formation is nucleated on the surface of ND-NH2 since the surface of ND-NH2 acts as a heterogeneous nucleation site. Thus, the introduction of ND-NH2 into the mixture of 1 M HCl/0.05 M CTAB solution prior to the polymerization of aniline induces the secondary growth of PANI and results in different morphologies as compared to that of the nanofibers of pristine PANI (Fig. 1d). The FESEM image of ND5-g-PANI (Fig. 1b) reveals the presence of extensively interlinked and flattened fibers or belts. Obviously, the increase in –NH2 sites on the ND surface causes an increase in secondary nucleation sites, which results in cross-linking between them. Thus, the formation of NF in the case of ND10-g-PANI is ascribed to the extensive secondary nucleation and cross-linking reactions. When the number of –NH2 sites is higher, as in the case of 10 mg ND-NH2, the –NH2 sites nucleate and are converted into short PANI chains, which subsequently transform into whiskers to result in a NF (Scheme 1). Previous reports suggest morphological variations by these secondary nucleation processes.52–54
 |
| | Fig. 1 FESEM images of (a) ND1-g-PANI, (b) ND5-g-PANI, (c) ND10-g-PANI, and (d) PANI. | |
 |
| | Scheme 1 Morphological evolution during NDx-g-PANI formation with different amounts of ND-NH2(x): x = 1.0 mg (A), 5.0 mg (B), and 10.0 mg (C): (A) – nanofibers; (B) – nanoplates; and (C) – nanoflowers. | |
Herein, we proposed a plausible mechanism for the evolution of the NF morphology (Scheme 1C). First, the confined microenvironment derived from ND-NH2 allows the nucleation, growth, and assembly of PANI on the surface of ND. When the number of nucleating sites increases (as in the case of ND1-g-PANI), randomly oriented short fibers may be formed on the surface of ND (Fig. 1a, Scheme 1A). With a further increase in the number of ND-NH2 sites, the short fibers generated at the individual sites of the ND surface subsequently assemble and generate flattened fibers (Fig. 1a, Scheme 1B) through intermolecular interaction between the fibers to result in petal-like structures.55 It has been earlier reported that the introduction of nanocomponents such as carbon nanotubes or metal nanoparticles or even nanofibers of PANI greatly influences the morphology of the final product.56 A rough surface of PANI was observed when pristine ND was mixed with PANI.57 Depending on the shape and size of CTAB aggregates in the reaction system, nanostructured PANI materials such as nanowires,58,59 nanoparticles,60 and NF52,61 have been synthesized. In our study, we predicted the non-covalent functionalization of CTAB on the surface of ND-NH2.62,63 The CTAB molecules on the surface of ND-NH2 generate different assemblies depending on x. Thus, CTAB acts as a soft template to provide channels for PANI chains to grow into fibers, belts, or NF.
3.2. UV-visible spectroscopy
The UV-visible spectra of NDx-g-PANI and pristine PANI obtained in NMP show three characteristic absorption bands (Fig. 2) around the wavelengths of 330 nm, 430 nm, and >750 nm. The presence of these three absorption bands indicate that the PANI chains in NDx-g-PANI are in the emeraldine salt (ES) form.64 The peak at 330 nm is attributed to the π–π* transitions in the benzenoid rings. The other two absorption bands at around 430 and >750 nm are attributed to the polaron π* and π-polaron transitions, respectively.65 As reported in the literature, the UV-visible spectrum of PANI in NMP exhibits two characteristic absorptions, a broad band at around 630–650 nm (corresponding to the excitonic transitions of the organic acid-doped PANI chain) and a prominent peak at 320 nm (corresponding to the π–π*transition) of the emeraldine base (EB) form of PANI.65 It has been reported that the PANI ES salt, which mostly has an organic acid dopant, in NMP converts into the PANI EB form, whereas the de-doping of the PANI chains is predominant.66,67 It is known that the doping–dedoping of PANI chains with organic/inorganic acids is an acid–base type reaction. NMP is a basic polar solvent and can interact with the protons (H+) of organic acids. Thus, in a solution of organic acid-doped PANI in NMP, there are two competitive reactions: (1) PANI + acid dopant → PANI-H+ (doped PANI, PANI-ES) and (2) NMP + H+ (acid dopant) → NMP-H+ (hydrogen bonding). Thus, under the conditions where reaction (2) is predominant, PANI is dedoped to PANI-EB. However, the dedoping of PANI chains in PANI-ES is not the predominant process in the cases where NMP-hydrogen bonding (reaction 2) is hindered by another competing process, which can result in the co-existence of doped and undoped states for PANI chains or the predominant existence of the doped state of PANI (PANI-ES). It has also been reported that the UV-visible spectrum of PANI nanostructures (prepared in the presence of CTAB) exhibits three bands that are characteristics of PANI-ES.68 In this study, the three absorption bands that are characteristic of PANI-ES were observed in the UV-visible spectrum of NDx-g-PANI, which is prepared from an aqueous CTAB–HCl mixture. We attributed the following reason for the spectral features observed in this study. We employed a template (CTAB)-directed chemical oxidative polymerization for the synthesis of NDx-g-PANI and nanostructured pristine PANI. The surfactant CTAB, in addition to directing the morphology of the polymer, can also wrap-up the PANI chains through electrostatic interactions between the protonated ammonium sites of CTAB and Cl− ions in the doped PANI.69–71 As a consequence, the Cl− ions doped into the PANI chains are trapped within the PANI chains to result in PANI-ES. The ratio of the peak intensity at λ330nm and λ750nm is an indication of the level of PANI chains doping.72 It was observed that the doping was maximum for PANI and the value decreased with x in NDx-g-PANI (Table 1). The physicochemical properties such as doping level and conjugation length are highly dependent on the morphologies such as nanofibers, nanowires, nanorods, and NF, of the conductive polymers.73,74 It has been reported that the doping level of PANI nanostructures follow the trend nanofibers > nanorods > nanoparticles.75 The fiber-like morphology of pristine PANI (Fig. 1d) could be the reason for the higher doping level (Table 1). The enhanced doping level and conjugation length for pristine PANI are expected to originate from the high aspect ratio of nanofibers in pristine PANI (Fig. 1d) compared to that of platelet/NF morphologies for NDx-g-PANI (Fig. 1a–c).
 |
| | Fig. 2 UV-visible spectra of (a) ND1-g-PANI, (b) ND5-g-PANI, (c) ND10-g-PANI, and (d) PANI. | |
Table 1 Investigation of doping of NDx-g-PANI and PANI from the intensity ratio
| Sample |
Intensity ratioa |
| At the wavelengths between 330 nm and 750 nm. |
| PANI |
1.065 |
| ND1-g-PANI |
0.778 |
| ND5-g-PANI |
0.759 |
| ND10-g-PANI |
0.718 |
3.3. X-ray photoelectron spectroscopy
The N1s core level spectra of NDx-g-PANI and pristine PANI were deconvoluted into Gaussian component peaks76,77 (Fig. 3). The core level N1s spectra were fitted into three main chemical states due to the different chemical environments possible for PANI. The chemical states were imine (–NH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), amine (–NH–), and protonated amine and imine (Table 2).78 The results revealed that the PANI chains in pristine PANI and NDx-g-PANI existed in the emeraldine form containing both benzenoid amine (–NH) and quinoid amine (N–) units. However, there are differences in the doping level of PANI chains (the contribution of –N+ species to the total nitrogen content) between PANI and NDx-g-PANI. Pristine PANI is highly doped with the doping level of 48%. On the contrary, the doping level of PANI is much lower in NDx-g-PANI. This suggests that the inclusion of ND into the PANI chains decreases the doping level.
), amine (–NH–), and protonated amine and imine (Table 2).78 The results revealed that the PANI chains in pristine PANI and NDx-g-PANI existed in the emeraldine form containing both benzenoid amine (–NH) and quinoid amine (N–) units. However, there are differences in the doping level of PANI chains (the contribution of –N+ species to the total nitrogen content) between PANI and NDx-g-PANI. Pristine PANI is highly doped with the doping level of 48%. On the contrary, the doping level of PANI is much lower in NDx-g-PANI. This suggests that the inclusion of ND into the PANI chains decreases the doping level.
 |
| | Fig. 3 XPS spectra of (a) ND1-g-PANI, (b) ND5-g-PANI, (c) ND10-g-PANI, and (d) PANI. | |
Table 2 Deconvolution of the XPS peaks and proportion of nitrogen chemical states
| Electrode |
Proportion (%) of nitrogen chemical states |
| N–(imine) |
–NH–(amine) |
N+ (protonated amine) |
| PANI |
0.335 |
0.182 |
0.481 |
| ND1-g-PANI |
0.158 |
0.808 |
0.033 |
| ND5-g-PANI |
0.444 |
0.413 |
0.141 |
| ND10-g-PANI |
0.57 |
0.377 |
0.047 |
3.4. Fourier transform infrared spectroscopy
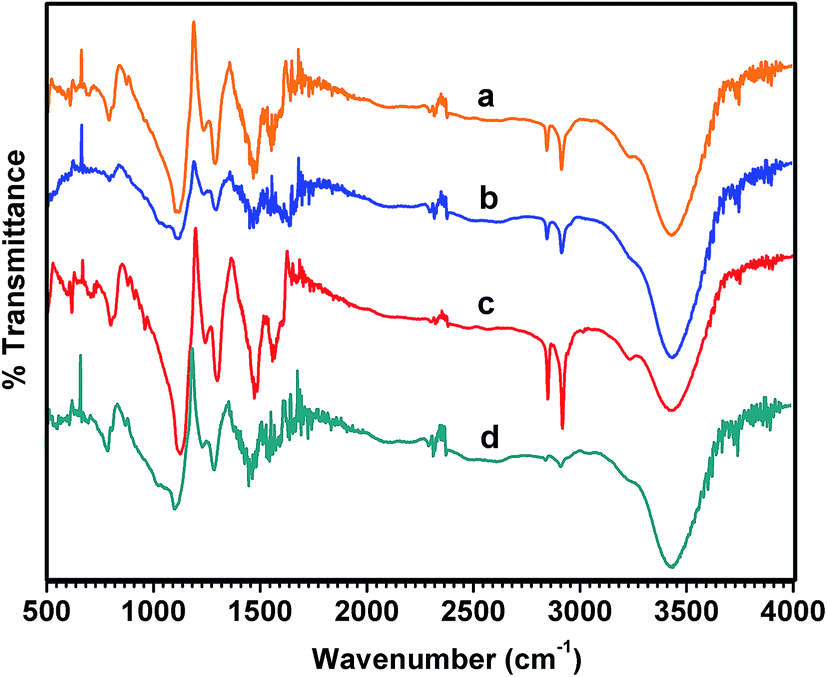
Fig. 4 shows the FTIR spectra of the pristine PANI and NDx-g-PANI samples prepared with different amounts of ND. The bands at around 1600 cm−1 and 1500 cm−1 are attributed to the CN stretching mode and CC stretching vibration of quinoid (Q) and benzenoid (B), respectively. The peaks were assigned based on earlier literature.79–81 There was a shift in the band position of the Q and B group stretching vibrations between PANI and NDx-g-PANI. The bands at around 1300 cm−1 and 1120 cm−1 in Fig. 4 are attributed to the C–N stretching of the B group and in-plane bending vibration of the C–H aromatic rings (NQN, QN–B, and Q–NH–B). The band at 830 nm is attributed to the C–H out of plane bending of the 1–4, disubstituted ring structures (para–para coupling) and likely signifies the head to tail coupling of the monomeric structural units during aniline polymerization. The intensity ratio of Q to B is near unity for pristine PANI, suggesting the emeraldine salt-like form. However, the Q/B intensity ratio is shockingly low for NDx-g-PANI, suggesting a low level doping. These results are in accordance with the XPS results (Table 2).
 |
| | Fig. 4 Fourier transform infrared spectra of (a) ND1-g-PANI, (b) ND5-g-PANI, (c) ND10-g-PANI, and (d) PANI. | |
3.5. Thermogravimetric analysis
The thermogravimetric curves of NDx-g-PANI and PANI are shown in Fig. 5. NDx-g-PANI and pristine PANI demonstrate a three-stage weight loss. The initial weight loss at around 100 °C is mainly attributed to the elimination of residual water, and the second stage weight loss from 220 °C is due to the removal of dopant molecules. The third stage weight loss starting at around 380 °C is caused by the decomposition of the PANI chains.82 Table 3 shows the %weight loss at each stage and the residual weight (%) at 600 °C. The residual weight% at 600 °C increases with an increase in ND content in the composite, which is attributed to the fact that ND inclusion in NDx-g-PANI alters the thermal stability of the PANI backbone.83
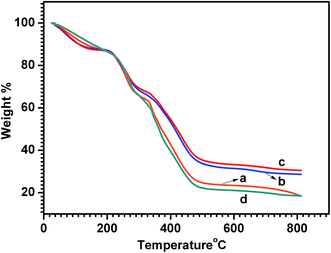 |
| | Fig. 5 TGA curves of (a) ND1-g-PANI, (b) ND5-g-PANI, (c) ND10-g-PANI, and (d) PANI. | |
Table 3 Weight loss and residual weight (%) of NDx-g-PANI and PANI
| Samples |
Percentage (%) weight losses |
Residual weight (%) at 600 °C |
| Stage-I |
Stage-II |
Stage-III |
| PANI |
10.7 |
24.2 |
51.6 |
18.7 |
| ND1-g-PANI |
11.6 |
22.4 |
43.2 |
22.7 |
| ND5-g-PANI |
10.9 |
20.9 |
35.3 |
32.9 |
| ND10-g-PANI |
11.1 |
19.3 |
34.8 |
34.8 |
3.6. Amperometric determination of glucose by NDx-g-PANI
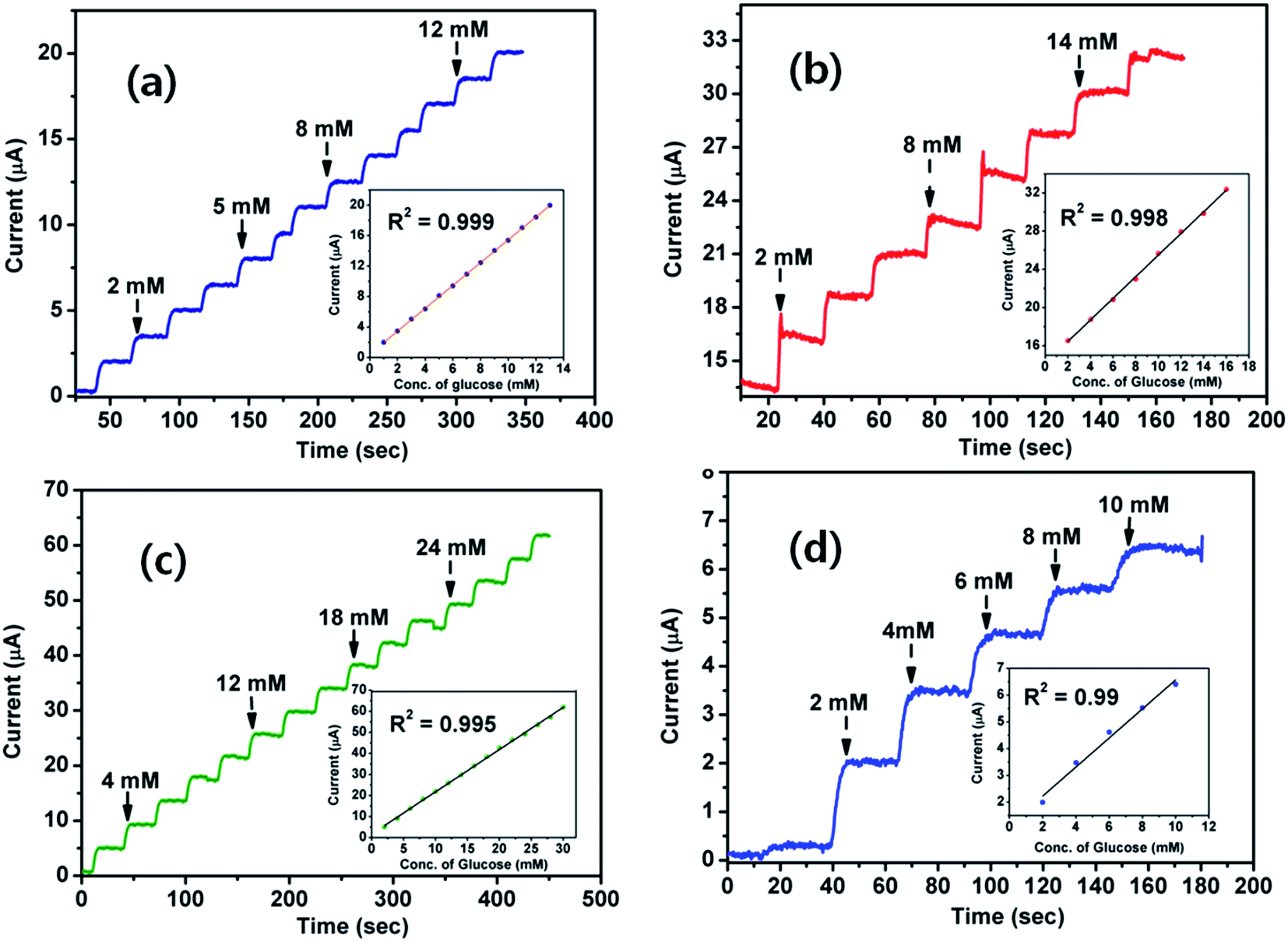
The electrochemical properties and interfacial characteristics of ND1-g-PANI, ND5-g-PANI, ND10-g-PANI, and pristine PANI were investigated using cyclic voltammetry and electrochemical impedance measurements, respectively, and the details are presented in the ESI SI-1 and 2.† As is known, the applied potential has a definite influence on the sensing performances of an electrochemical sensor. To determine the optimum potential for amperometric glucose measurements, amperometric studies (Fig. SI-3†) were conducted by varying the applied potential between +0.25 V and +0.65 V. Typically, the potential at the ND10-g-PANI/GOx-modified electrode was varied and the transduced current values were obtained for a 1 mM glucose solution in 0.1 M PBS. The glucose oxidation current was stable and maximum at +0.40 V (Fig. SI-3†). Hence, the operating potential of +0.40 V was chosen for further amperometric glucose measurements. Fig. 6a–d depict the amperometric responses upon the successive addition of glucose at the applied potential of +0.40 V for the enzymatic glucose biosensors ND1-g-PANI/GOx, ND5-g-PANI/GOx, ND10-g-PANI/GOx, and pristine PANI/GOx, respectively. The ND10x-g-PANI/GOx biosensor exhibits a much higher linear current response for glucose in the concentration range of 1–30 mM, with the correlation coefficient of 0.995 (Fig. 6, inset). This biosensor also exhibits a lower detection limit (0.018 mM) than that reported for multilayer films of chitosan/MWNT/GOx (21 μM).84 The sensitivity of the glucose sensor increases from 1.13 μA mM−1 (ND1-g-PANI/GOx) to 1.49 μA mM−1 (ND5-g-PANI/GOx) and then to 2.03 μA mM−1 (ND10-g-PANI/GOx). The sensitivity of (ND10-g-PANI/GOx) is much higher than that of chitosan/MWNT/GOx (0.45 μA mM−1),84 Nafion/GOx/CNTs (0.33 μA mM−1),85 and GOx–Ru/CF microelectrode (0.43 nA mM−1).86 The increased sensitivity with ND clearly demonstrates its compatibility with GOx for the sensitive determination of glucose. Table 4 shows the comparison between the glucose biosensor performances and various other PANI-based biosensors reported in the literature. Table 4 reveals two striking pieces of information: (i) the biosensor based on ND10-g-PANI NF exhibits much-improved sensitivity (2.03 μA mM−1), wider linear concentration range (1–30 mM), and lower detection limit (0.018 mM) and (ii) the ND10-g-PANI NF-based glucose sensor shows superior sensor performances than many PANI and carbon nanostructure (e.g. MWNT)-based biosensors (Table 4).
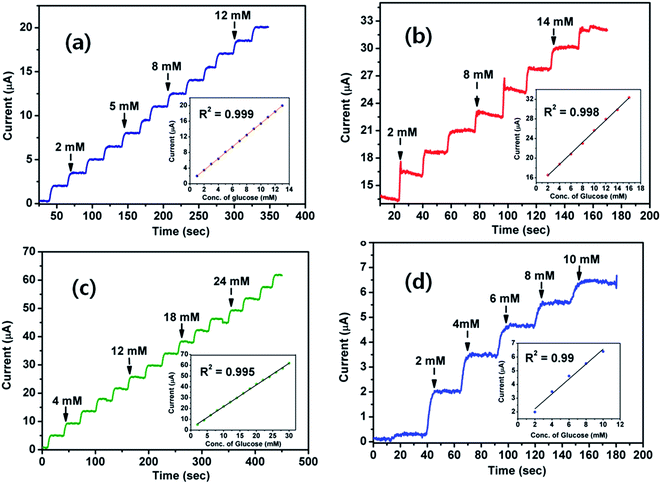 |
| | Fig. 6 Amperometric current responses of the (a) ND1-g-PANI/GOx, (b) ND5-g-PANI/GOx, (c) ND10-g-PANI/GOx, and (d) PANI/GOx biosensors at +400 mV in 0.1 M PBS (pH 7.0). Inset: the calibration curve. | |
Table 4 Comparison of glucose biosensor performances
| Material |
Detection limit |
Linear range |
Sensitivity |
Ref. |
| PANI/GOx |
0.062 mM |
1–10 mM |
0.54 μA mM−1 |
This work |
| ND1-g-PANI/GOx |
0.035 mM |
1–13 mM |
1.13 μA mM−1 |
This work |
| ND5-g-PANI/GOx |
0.02 mM |
1–16 mM |
1.49 μA mM−1 |
This work |
| ND10-g-PANI/GOx |
0.018 mM |
1–30 mM |
2.03 μA mM−1 |
This work |
| NiHCF/PANI/graphene |
0.5 μM |
0.001–0.765 mM |
487.33 μA mM−1 cm−2 |
88 |
| AuNPs/PANI-NF/GOx |
0.5 μM |
0.5–16 mM |
— |
89 |
| PANI/PANAA-GOx |
1 μM |
0.005–5 mM |
16 mA M−1 cm−2 |
90 |
| PANI/MWCNTs |
0.02 mM |
0.5–12 mM |
0.94 μA mM−1 |
91 |
| Nafion-silica/MWCNTs-g-PANI/GOx |
0.1 μM |
1–10 mM |
5.01 μA mM−1 |
25 |
| BNNTs-PANI-Pt |
0.18 μM |
0.01–8 mM |
19.02 mA M−1 cm−2 |
92 |
| PANI/gold nanorod/GOx |
5.8 μM |
17.6 μM−1 mM |
13.8 μA mM−1 cm−2 |
93 |
| PANI/PB |
0.4 μM |
2–1600 μM |
99.4 μA mM−1 cm−2 |
94 |
| Cyt c/Au NPs/PANI-NS |
0.01 mM |
0.01–3.2 mM |
63.1 μA mM−1 cm−2 |
95 |
| Pt NPs/PANI hydrogel heterostructure |
0.7 μM |
0.01–8 mM |
96.1 μA mM−1 cm−2 |
96 |
| GOx–PANI–PecNPs |
43.5 μM |
0.06–4 mM |
79.49 μA mM−1 cm−2 |
97 |
| GOx-graphene/PANI/Au NPs/GCE |
0.6 μM |
4.0 μM −1.12 mM |
— |
98 |
| GOx/n-TiO2/PANI/GCE |
18 μM |
0.02–6.0 mM |
6.31 μA mM−1 cm−2 |
99 |
3.7. Interferences
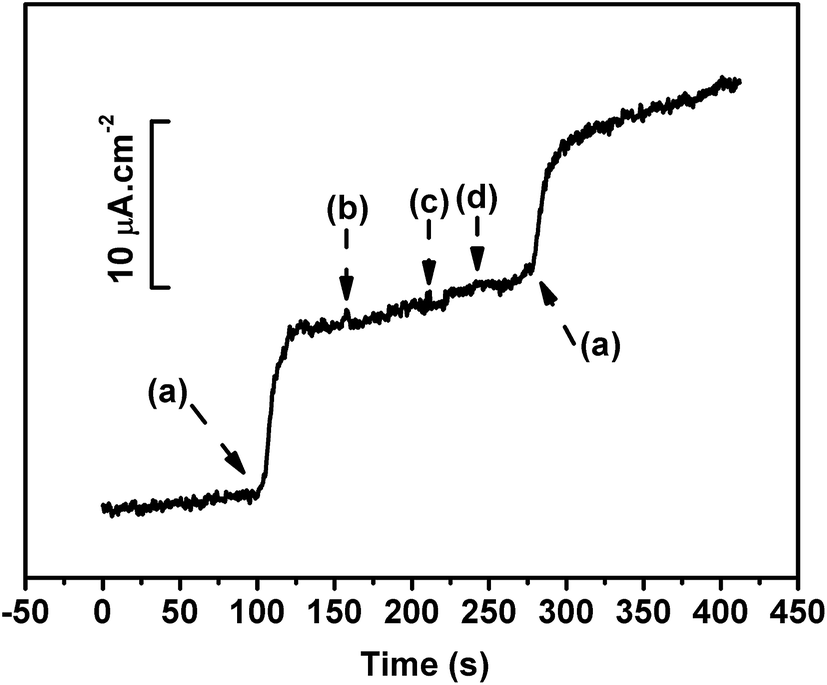
In the present study, an applied potential of +0.40 V was used to monitor the glucose concentration. Other electroactive species such as ascorbic acid (AA), dopamine (DA), and acetaminophen (AP) present in the sensing solution had higher electron transfer rates than glucose. As a result, the transduced net current would contain an interference signal. Thus, the concentration of glucose would be overestimated. Hence, it was essential to eliminate the current contributions from interfering substances. Nafion, which has ion-exchange capabilities, can minimize the interferences caused by these electroactive species.87 Fig. 7 represents the amperometric response of the ND10-g-PANI/GOx biosensor for 5 mM of glucose (a) with ascorbic acid (2 mM) (b), dopamine (2 mM) (c), and acetaminophen (2 mM) (d). It can be seen from Fig. 7 that the spiking interference solution did not alter the current response of the ND10-g-PANI/GOx biosensor. Thus, the ND10-g-PANI/GOx biosensor exhibits high selectivity towards sensing glucose.
 |
| | Fig. 7 Effect of interfering signals of glucose (5 mM) (a), ascorbic acid (2 mM) (b), dopamine (2 mM) (c), and acetaminophen (2 mM) (d) at ND10-g-PANI/GOx. | |
3.8. Real sample analysis
The reliability and practical use of ND10-g-PANI/GOx were tested by estimating the glucose concentration in a human serum sample and comparing the value with a commercial glucose meter (CareCens™N, GM5051A, i-SENS, Inc., Korea). Table 5 presents the glucose levels separately determined using the fabricated ND10-g-PANI/GOx sensor and glucose meter. The glucose levels obtained at ND10-g-PANI/GOx agree well with the glucose values observed from the glucose meter. The recovery values for the added glucose concentrations were between 98.3 and 100.21%. With the root mean square deviation (R.S.D) value of ∼3.0% (n = 3) for the recovery, ND10-g-PANI/GOx was proven to be useful for the detection of glucose in human serum samples.
Table 5 Real sample analysis
| Sample (serum + added [Glu]) |
Reference valuesa (mM) |
Found valuesb (mM) |
RSDc (%) |
Recovery (%) |
| Determined using a commercial glucose meter (CareCens™ N, GM5051A, i-SENS, Inc., Korea). Determined using the ND10-g-PANI/GOx biosensor. Number of measurements = 3. |
| Serum + 0 mM |
4.44 |
4.44 |
2.81 |
100 |
| Serum + 1 mM |
5.43 |
5.38 |
4.02 |
99.07 |
| Serum + 5 mM |
9.41 |
9.43 |
2.35 |
100.21 |
| Serum + 8 mM |
12.36 |
12.15 |
3.57 |
98.3 |
3.9. Reproducibility, repeatability, and stability
The reproducibility, repeatability, and lifetime (stability) of the fabricated sensor were evaluated and the details are presented in SI-3.† The results revealed that the sensor has satisfactory reproducibility (RSD = 1.2%, n = 6), repeatability (RSD = 1.42%, n = 4), and good stability for 30 days (retaining 97% of its original current responses).
4. Conclusions
Nanoflowers (NF) of PANI chains were grafted onto the surface of nanodiamonds (ND) by optimizing the binding sites on the ND surface to augment secondary nucleation for PANI chains. The NF of PANI on ND caused the effective immobilization of glucose oxidase (the enzyme) and resulted in high performances (wide concentration range and low detection limit) for glucose detection. Our studies demonstrated a simple flower-like modification strategy on the surface of ND and also opened-up further avenues for the development of efficient biosensing platforms for other target molecules.
Acknowledgements
This work was supported by the Priority Research Centers Program through a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (2009-0093819) and a funded research project (NRF- 2014R1A1A4A03004026). The authors gratefully acknowledge Dr. Miklos Verios, Institute for Solid State Physics and Optics, Higher Research Center for Physics, Hungarian Academy of Science, Hungary, for providing the ND sample.
References
- S. Arimori, C. J. Ward and T. D. James, Tetrahedron Lett., 2002, 43, 303–305 CrossRef CAS.
- H. Cao, D. I. Diaz, N. Dicesare, J. R Lakowicz and M. D. Heagy, Org. Lett., 2002, 4, 1503–1505 CrossRef CAS PubMed.
- O. Schaetzle, F. Barrière and U. Schröder, Energy Environ. Sci., 2009, 2, 96–99 CAS.
- E. Simon, C. M. Halliwell, C. S. Toh, A. E. Cass and P. N. Bartlett, Bioelectrochemistry, 2002, 55, 13–15 CrossRef CAS PubMed.
- N. L. Akers, C. M. Moore and S. D. Minteer, Electrochim. Acta, 2005, 50, 2521–2525 CrossRef CAS.
- Z. Wen, B. Ye and X. Zhou, Bioelectrochemistry, 1997, 9, 641 CAS.
- S. Calabrese Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867 CrossRef.
- S. Komathi, N. Muthuchamy, K. P. Lee and A. I. Gopalan, Biosens. Bioelectron., 2016, 84, 64–71 CrossRef CAS PubMed.
- T. Kuwahara, T. Homma, M. Kondo and M. Shimomura, Synth. Met., 2009, 159, 1859–1864 CrossRef CAS.
- X. B. Yan, X. J. Chen, B. K. Tay and K. A. Khor, Electrochem. Commun., 2007, 9, 1269–1275 CrossRef CAS.
- L. Xu, Y. Zhu, L. Tang, X. Yang and C. Li, Electroanalysis, 2007, 19, 717–722 CrossRef CAS.
- M. Zhao, X. Wu and C. Cai, J. Phys. Chem. C, 2009, 113, 4987–4996 CAS.
- J. Lu, I. Do, L. T. Drzal, R. M. Worden and I. Lee, ACS Nano, 2008, 2, 1825–1832 CrossRef CAS PubMed.
- J. Y. Chiu, C. M. Yu, M. J. Yen and L. C. Chen, Biosens. Bioelectron., 2009, 24, 2015 CrossRef CAS PubMed.
- M. Yuan, N. Konstantopoulos, J. Lee, L. Hansen, Z. W. Li, M. Karin and S. E. Shoelson, Science, 2001, 293, 1673–1677 CrossRef CAS PubMed.
- H. Liu, J. Kameoka, D. A. Czaplewski and H. G. Craighead, Nano Lett., 2004, 4, 671–675 CrossRef CAS.
- Z. Wang, S. Liu, P. Wu and C. Cai, Anal. Chem., 2009, 81, 1638–1645 CrossRef CAS PubMed.
- A. I. Gopalan, N. Muthuchamy, S. Komathi and K. P. Lee, Biosens. Bioelectron., 2016, 84, 53–63 CrossRef CAS PubMed.
- N. Muthuchamy, K. P. Lee and A. I. Gopalan, Biosens. Bioelectron., 2017, 89, 390–399 CrossRef CAS PubMed.
- M. Ding, Y. Tang, P. Gou, M. J. Reber and A. Star, Adv. Mater., 2011, 23, 536–540 CrossRef CAS PubMed.
- Y. Zhang, X. Yang and Z. Gao, RSC Adv., 2015, 5, 21675 RSC.
- P. Santhosh, K. M. Manesh, A. Gopalan and K. P. Lee, Sens. Actuators, B, 2007, 125, 92–99 CrossRef CAS.
- P. Santhosh, A. Gopalan and K. P. Lee, J. Catal., 2006, 238, 177–185 CrossRef CAS.
- P. Santhosh, K. M. Manesh, A. Gopalan and K. P. Lee, Anal. Chim. Acta, 2006, 575, 32–38 CrossRef CAS PubMed.
- A. I. Gopalan, K. P. Lee, D. Ragupathy, S. H. Lee and J. W. Lee, Biomaterials, 2009, 30, 5999–6005 CrossRef CAS PubMed.
- L. F. Li, D. Totir, B. Miller, G. Chottiner, A. Argoitia, J. C. Angus and D. Scherson, J. Am. Chem. Soc., 1997, 119, 7875–7876 CrossRef CAS.
- N. Vinokur, B. Miller, Y. Avyigal and R. Kalish, J. Electrochem. Soc., 1996, 143, L238 CrossRef CAS.
- D. R. Dunphy, S. B. Mendes, S. S. Saavedra and N. R. Armstrong, Anal. Chem., 1997, 71, 3086 CrossRef PubMed.
- T. Yano, E. Popa, D. A. Tryk, K. Hashimoto and A. Fujishima, J. Electrochem. Soc., 1999, 146, 1081 CrossRef CAS.
- J. Xu, Q. Chen and G. M. Swain, Anal. Chem., 1998, 70, 3146–3154 CrossRef CAS PubMed.
- Q. Chen, M. C. Granger, T. E. Lister and G. M. Swain, J. Electrochem. Soc., 1997, 144, 3086 Search PubMed.
- C. C. Fu, H. Y. Lee, K. Chen, T. S. Lim, H. Y. Wu, P. K. Lin, P. K. Wei, P. H. Tsao, H. C. Chang and W. Fann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 727–732 CrossRef CAS PubMed.
- R. Lam, M. Chen, E. Pierstorff, H. Huang, E. Osawa and D. Ho, ACS Nano, 2008, 10, 2095–2102 CrossRef PubMed.
- J. Wang, G. Chen, M. P. Chatrathi, A. Fujishima, D. A. Tryk and D. Shin, Anal. Chem., 2003, 75, 935–939 CrossRef CAS PubMed.
- Q. Chen, D. M. Gruen, A. R. Krauss, T. D. Corrigan, M. Witek and G. M. Swain, J. Electrochem. Soc., 2001, 148, E44–E51 CrossRef CAS.
- A. Härtl, E. Schmich, J. A. Garrido, J. Hernando, S. C. Catharino, S. Walter, P. Feulner, A. Kromka, D. Steinmüller and M. Stutzmann, Nat. Mater., 2004, 3, 736–742 CrossRef PubMed.
- W. Zhao, J. J. Xu, Q. Q. Qiu and H. Y. Chen, Biosens. Bioelectron., 2006, 22, 649–655 CrossRef CAS PubMed.
- J. T. Zhu, C. G. Shi, J. J. Xu and H. Y. Chen, Bioelectrochemistry, 2007, 71, 243–248 CrossRef CAS PubMed.
- T. S. Huang, Y. Tzeng, Y. K. Liu, Y. C. Chen, K. R. Walker, R. Guntupalli and C. Liu, Diamond Relat. Mater., 2004, 13, 1098–1102 CrossRef CAS.
- L. Li, J. L. Davidson and C. M. Lukehart, Carbon, 2006, 44, 2308 CrossRef CAS.
- X. Q. Zhang, M. Chen, R. Lam, X. Xu, E. Osawa and D. Ho, ACS Nano, 2009, 3, 2609–2616 CrossRef CAS PubMed.
- P. Actis, M. Manesse, C. Nunes-Kirchner, G. Wittstock, Y. Coffinier, R. Boukherroub and S. Szunerits, Phys. Chem. Chem. Phys., 2006, 8, 4924–4931 RSC.
- X. Y. Zhao, J. B. Zang, Y. H. Wang, L. Y. Bian and J. K. Yu, Electrochem. Commun., 2009, 11, 1297–1300 CrossRef CAS.
- L. Li, Y. Wang, L. Pan, Y. Shi, W. Cheng, Y. Shi and G. Yu, Nano Lett., 2015, 15, 1146–1151 CrossRef CAS PubMed.
- P. Santhosh, K. M. Manesh, S. Uthayakumar, A. I. Gopalan and K. P. Lee, Biosens. Bioelectron., 2009, 24, 2008–2009 CrossRef CAS PubMed.
- A. V. Karmenyan, E. Perevedentseva, M. Veres and C. L. Cheng, Plasmonics, 2013, 8, 325–333 CrossRef CAS.
- P. Csikvari, A. Somogyi, M. Veres, G. Hárs and A. Tóth, Diamond Relat. Mater., 2009, 18, 1459–1465 CrossRef CAS.
- A. I. Gopalan, K. P. Lee and S. Komathi, Biosens. Bioelectron., 2010, 26, 1638–1643 CrossRef CAS PubMed.
- A. I. Gopalan, S. Komathi, G. S. Anand and K. P. Lee, Biosens. Bioelectron., 2013, 46, 136–141 CrossRef CAS PubMed.
- T. J. Ohara, R. Rajagopalan and A. Heller, Anal. Chem., 1994, 66, 2451 CrossRef CAS PubMed.
- D. Zhou, Y. Li, J. Wang, P. Xu and X. Han, Mater. Lett., 2011, 65, 3601–3604 CrossRef CAS.
- C. Zhou, J. Han and R. Guo, Macromolecules, 2008, 41, 6473–6479 CrossRef CAS.
- S. Sadasivan, D. Khushalani and S. Mann, Chem. Mater., 2005, 17, 2765–2770 CrossRef CAS.
- G. Sai-Anand, A. I. Gopalan, S. W. Kang and K. P. Lee, IEEE Electron Device Lett., 2013, 34, 1065–1067 CrossRef CAS.
- W. Chen, R. B. Rakhi and H. N. Alshareef, J. Phys. Chem. C, 2013, 117, 15009–15019 CAS.
- M. Hosseini, M. M. Momeni and M. Faraji, J. Mater. Sci., 2010, 45, 2365 CrossRef CAS.
- I. Kovalenko, D. G. Bucknall and G. Yushin, Adv. Funct. Mater., 2010, 20, 3979 CrossRef CAS.
- A. D. Carswell, E. A. O'Rea and B. P. Grady, J. Am. Chem. Soc., 2003, 125, 14793–14800 CrossRef CAS PubMed.
- H. Mao, X. Lu, D. Chao, L. Cui and W. Zhang, Mater. Lett., 2008, 62, 998–1001 CrossRef CAS.
- L. S. Jan and M. A. Siddig, Chinese J. Poly. Scien., 2011, 29, 181–190 CrossRef CAS.
- T. Wang, W. Zhong, X. Ning, Y. Wang and W. Yang, J. Colloid Interface Sci., 2009, 334, 108–112 CrossRef CAS PubMed.
- Q. Xu and X. Zhao, J. Mater. Chem., 2012, 22, 16416–16421 RSC.
- X. Zhang, S. Wang, M. Liu, J. Hui, B. Yang, L. Tao and Y. Wei, Toxicol. Res., 2013, 2, 335–342 RSC.
- W. Yin and E. Ruckenstein, Synth. Met., 2000, 108, 39–46 CrossRef CAS.
- P. Jiménez, P. Castell, R. Sainz, A. Ansón, M. Martínez, A. Benito and W. K. Maser, J. Phys. Chem. B, 2010, 114, 1579–1585 CrossRef PubMed.
- J. De Albuquerque, L. Mattoso, R. Faria, J. Masters and A. MacDiarmid, Synth. Metal, 2004, 146, 1–10 CrossRef CAS.
- S. Dutt and P. Siril, Mater. Lett., 2014, 124, 50–53 CrossRef CAS.
- X. Zhu, K. Hou, C. Chen, W. Zhang, H. Sun, G. Zhang and Z. Gao, High Perform. Polym., 2015, 27, 207–216 CrossRef CAS.
- T. Hashimoto, R. Iwase, A. Murakami and T. Yamaoka, Polym. Degrad. Stab., 2009, 94, 1349–1353 CrossRef CAS.
- V. Chaudhary and A. Kaur, RSC Adv., 2016, 6, 95349–95357 RSC.
- S. Palaniappan and V. Nivasu, New J. Chem., 2002, 26, 1490–1494 RSC.
- S. Shreepathi and R. Holze, Chem. Mater., 2005, 17, 4078–4085 CrossRef CAS.
- S. Virji, J. Huang, R. B. Kaner and B. H. Weiller, Nano Lett., 2004, 4, 491–496 CrossRef CAS.
- J. Jang, J. Ha and J. Cho, Adv. Mater., 2007, 19, 1772–1775 CrossRef CAS.
- S. Cho, O. S. Kwon, S. A. You and J. Jang, J. Mater. Chem. A, 2013, 1, 5679–5688 CAS.
- F. J. Liu, L. M. Huang, T. C. Wen, C. F. Li, S. L. Huang and A. Gopalan, Synth. Met., 2008, 158, 767 CrossRef CAS.
- F. J. Liu, L. M. Huang, T. C. Wen and A. Gopalan, Synth. Met., 2007, 157, 651 CrossRef CAS.
- E. T. Kang, K. G. Neoh, S. H. Khor, K. L. Tan and B. T. G. Tan, Polymer, 1990, 31, 202–207 CrossRef CAS.
- A. Dey, S. De, A. De and S. K. De, Nanotech., 2004, 15, 1277 CrossRef CAS.
- V. Rajendran, A. Gopalan, T. Vasudevan and T. C. Wen, J. Electrochem. Soc., 2000, 147, 3014–3020 CrossRef CAS.
- L. Zhang, Z. D. Zujovic, H. Peng, G. A. Bowmaker, P. A. Kilmartin and J. Travas-Sejdic, Macromolecules, 2008, 41, 8877–8884 CrossRef CAS.
- D. Lee and K. Char, Polym. Degrad. Stab., 2002, 75, 555–560 CrossRef CAS.
- Y. He, Q. Cheng, V. Pavlinek, C. Li and P. Saha, Inter. J. Poly. Anal. Ch., 2008, 13, 25 CrossRef CAS.
- B. Y. Wu, S. H. Hou, M. Yu, X. Qin, S. Li and Q. Chen, Mater Sci Eng C Mater Biol Appl., 2009, 29, 346 CrossRef CAS.
- Y. C. Tsai, S. C. Li and J. M. Chen, Langmuir, 2005, 21, 3653 CrossRef CAS PubMed.
- T. Kohma, D. Oyamatsu and S. Kuwabata, Electrochem. Commun., 2007, 9, 1012 CrossRef CAS.
- K. M. Manesh, H. T. Kim, P. Santhosh, A. I. Gopalan and K. P. Lee, Biosens. Bioelectron., 2008, 23, 771 CrossRef CAS PubMed.
- Y. Kong, Y. Sha, Y. Tao, Y. Qin, H. Xue and M. Lu, J. Electrochem. Soc., 2014, 161, B269–B274 CrossRef.
- Y. Xian, Y. Hu, F. Liu, Y. Xian, H. Wang and L. Jin, Biosens. Bioelectron., 2006, 21, 1996–2000 CrossRef CAS PubMed.
- D. Shan, S. Wang, Y. He and H. Xue, Mater. Sci. Eng. C., 2008, 28, 213–217 CrossRef CAS.
- Q. Sheng and J. Zheng, Biosens. Bioelectron., 2009, 24, 1621–1628 CrossRef CAS PubMed.
- J. Wu and L. Yin, ACS Appl. Mater. Interfaces, 2011, 3, 4354–4362 CAS.
- U. Tamer, A. I. Seçkin, E. Temur and H. Torul, Int. J. Electrochem., 2011, 2011, 869742 Search PubMed.
- X. Chen, Z. Chen, R. Tian, W. Yan and C. Yao, Anal. Chim. Acta, 2012, 723, 94–100 CrossRef CAS PubMed.
- C. Xiang, Y. Zou, S. Qiu, L. Sun, F. Xu and H. Zhou, Talanta, 2013, 110, 96–100 CrossRef CAS PubMed.
- D. Zhai, B. Liu, Y. Shi, L. Pan, Y. Wang, W. Li, R. Zhang and G. Yu, ACS Nano, 2013, 7, 3540–3546 CrossRef CAS PubMed.
- B. Thakur, C. A. Amarnath and S. N. Sawant, RSC Adv., 2014, 4, 40917–40923 RSC.
- Q. Xu, S. X. Gu, L. Jin, Y. E. Zhou, Z. Yang, W. Wang and X. Hu, Sens Actuators B, 2014, 190, 562–569 CrossRef CAS.
- W. Tang, L. Li and X. Zeng, Talanta, 2015, 131, 417–423 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24760a |
| ‡ Authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *bc
*bc