
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica nanoparticles–nylon 6 composites: synthesis, characterization and potential use as sorbent†
E. M. Reyes-Gallardo,
R. Lucena and
S. Cárdenas *
*
Department of Analytical Chemistry, Institute of Fine Chemistry and Nanochemistry, Marie Curie Building, University of Córdoba, Campus of Rabanales, 14071 Córdoba, Spain. E-mail: qa1caarm@uco.es
First published on 12th January 2017
Abstract
Composite materials based on the combination of nanoparticles (NPs) and polymers have attracted much attention in recent years thanks to their positive characteristics. In this article an easy, cheap and green procedure for the synthesis of silica nanoparticles–nylon 6 composites is proposed. Several materials comprising different NPs–polymer ratios have been synthesized and characterized. The evaluation of their extraction performances indicated that the inclusion of NPs into the polymeric network increases their superficial area, boosting their sorbent capacity, because the NPs modify the normal stacking of the polymeric chains. Among the tested composite materials, the best one has been applied for the dispersive micro-solid phase extraction of selected estrogens (estrone, β-estradiol and estriol) from aqueous samples as a model analytical problem. The approach allows the determination of the target compounds by LC-MS/MS at the low μg L−1 range with acceptable precision (better than 13.9%, expressed as relative standard deviation). Finally, the potential use of the composite for the isolation of the targets from more complex samples has been outlined.
Introduction
Synthetic polymers have been applied for decades in extraction techniques thanks to their advantageous properties.1 Among these features, their chemical/mechanical stability, and their ability to interact with target analytes through different mechanisms can be highlighted. In addition, the physical/chemical architecture of synthetic polymers, in the so called molecularly imprinted polymers,2,3 can be adapted to the target analyte to achieve selective isolation from complex samples. The thermal stability of some polymers has allowed their use as extracting phase in solid phase microextraction (SPME), a research field where polymers have demonstrated prevalence over other materials.4–7In the same way, nanoparticles (NPs) have been extensively used in the microextraction context as supports or as extracting phases.8,9 Their high surface to volume ratio, a direct consequence of the nanometric size, guarantees a high extraction rate if aggregation is not an issue.10 Besides, the great variety of NPs, covering different interaction chemistries, and their potential functionalization increases even more the scope their applicability.11
Although both synthetic polymers and NPs present a great applicability, their combination in a synergic way to create composite materials opens a door to interesting applications.12 A deep revision of the scientific literature shows different alternative procedures for the synthesis of polymer nanocomposites.
The core shell approach consists of covering the nanoparticle surface with a polymeric layer in such a way that the resulting composite remains in the nanometric domain and it is characterized by a high superficial area. Magnetic NPs are used as the common support and the final magnetic nanocomposite is usually applied under the dispersive micro-solid phase extraction (dμSPE) format.13–16 The opposite approach, the coverage of the bulk polymer surface with NPs, was evaluated by our research group.17 The decoration of the polymer with magnetic NPs allowed the easy synthesis of a magnetic composite which resulted useful for the extraction of nitroaromatic compounds from water. The decoration of the surface resulted critical since an excessive coverage induced a reduction of the extraction surface.
Electrospun fibers containing NPs have attracted much attention in the last years due to the versatility of the resulting materials.18 These composites can be obtained in two ways, namely: (a) electrospinning a solution containing the polymer and the NPs or (b) loading the electrospun fiber after their synthesis with the NPs. Electrospun nanocomposites can be obtained in the form of fibers or films that can be applied in SPME,19,20 thin film microextraction21,22 and dμSPE.23
The inclusion of NPs inside the polymeric network has been also proposed for extraction purposes. The NPs may play two different roles. On the one hand, they can act as modifiers modifying the normal stacking of the polymer and therefore increasing the porosity of the composite.20 On the other hand, they can be the active extracting phase or they can complement the extraction capabilities of the polymer.24,25
Despite the potential of the above mentioned synthetic procedures, some of them are multistep protocols that require resources (energy, reagents and solvents) or special manifolds. Recently, our group have proposed a quick, easy, cheap and green method to synthesize nanocomposite playing with the switchable solubility of polymers.26,27 In this article, the procedure is extended to silica NPs and the role of these NPs in the extraction is evaluated using the determination of selected hormones as model analytical problem. In addition, the synthetic procedure has been modified to allow the synthesis of larger amount of material in a more reproducible way.
Experimental section
Reagents
All reagents were of analytical grade or better. Estrone (E1), β-estradiol (E2) and estriol (E3) were purchased from Sigma-Aldrich (Madrid, Spain). Stock standard solutions of the analytes were prepared at a concentration of 1000 mg L−1 in methanol (Carlo Erba Reagents, Italy) and stored at 4 °C. Working solutions were daily prepared by the appropriate dilution of the stock in methanol or Milli-Q water (Millipore Corp., Madrid, Spain) as required.Formic acid, nylon 6 (in the form of cubical pellets) and silica nanoparticles (10–20 nm particle size) were used for the synthesis of the nanocomposites. All these reagents were obtained from Sigma-Aldrich.
Saliva and urine samples were collected from volunteers who gave their informed consent prior to sampling.
Synthesis of the silica nanoparticles–nylon 6 composite
The composite is synthesized in an easy and rapid way. First of all, a defined amount of nylon 6 pellets (Table 1) is dissolved in 25 mL of formic acid under vigorous stirring in a magnetic stirrer (Velp Scientifica, Milan, Italy). In a second step, a weighted amount of silica nanoparticles (Table 1) are added to the solution. The resulted dispersion is stirred for 5 min to assess the homogeneous distribution of the nanoparticles. Finally, 25 mL of Milli-Q water are added dropwise into the solution inducing the gelation of the polymer around the nanoparticles.| Material | % nylon 6 | Amount of nylon 6 (g) | Amount of silica (g) | Carbon contenta (% ±SD) | Nitrogen contenta (% ±SD) |
|---|---|---|---|---|---|
| a Values obtained by elemental analysis. SD, standard deviation. | |||||
| A | 100 | 0.5 | 0 | 68.4 ± 0.3 | 13.3 ± 0.1 |
| B | 75 | 0.35 | 0.15 | 43.7 ± 0.8 | 8.83 ± 0.2 |
| C | 50 | 0.25 | 0.25 | 25 ± 5 | 5 ± 1 |
| D | 25 | 0.15 | 0.35 | 9 ± 3 | 1.9 ± 0.7 |
The obtained material is recovered by filtration under vacuum using filter paper as it is capable, due to its larger pores, to separate the composite from those silica NPs which have not been successfully introduced in the polymeric network. The material is washed with acetone and methanol and dried in an oven at 80 °C. The synthesis is shown in movie 1 (ESI†). Table 1 summarizes the different materials (A, B, C and D) that have been synthesized, based on different proportions of both components. Materials B, C and D are really composites (they combine silica NPs and nylon 6) while material A is just nylon subjected to the same synthetic procedure.
Characterization of the silica nanoparticles–nylon 6 composites
Micrographs of nylon 6, silica nanoparticles and their resulting composites were obtained by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) using a JEOL JSM 6300 and a Philips CM-10 microscopes, respectively. A EuroVector Elemental Analyser EA3000 (EuroVector SpA, Milan, Italy) was used for measuring the carbon and nitrogen content of the synthesized materials. Both the micrographs and elemental analysis were developed in the Central Service for Research Support (SCAI) of the University of Córdoba.Infrared spectra were acquired in a Bruker Tensor 37 FT-IR spectrometer, equipped with a diamond ATR cell (circular surface of 3 mm diameter and three internal reflections) and a deuterated triglycine sulfate (DTGS) detector. Spectra were collected between 4000 and 600 cm−1 at a 4 cm−1 resolution with 64 coadded scans each. Data collection was done using OPUS software (Bruker, Ettligen, Germany).
The superficial area of the composites were measured using a Quantachrome® ASiQwin™-Automated Gas Sorption Data based on the nitrogen adsorption/desorption measures at −196 °C. The specific surface area values were calculated according to the BET (Brunauer–Emmett–Teller) equation.
Sample extraction procedure
The extraction unit, which is described elsewhere,26 consists of a 10 mL-syringe which is adapted to a pipette tip section where a cotton bead is located as frit. 40 mg of the composite is placed between the cotton bead and the syringe plunger (see Fig. 1).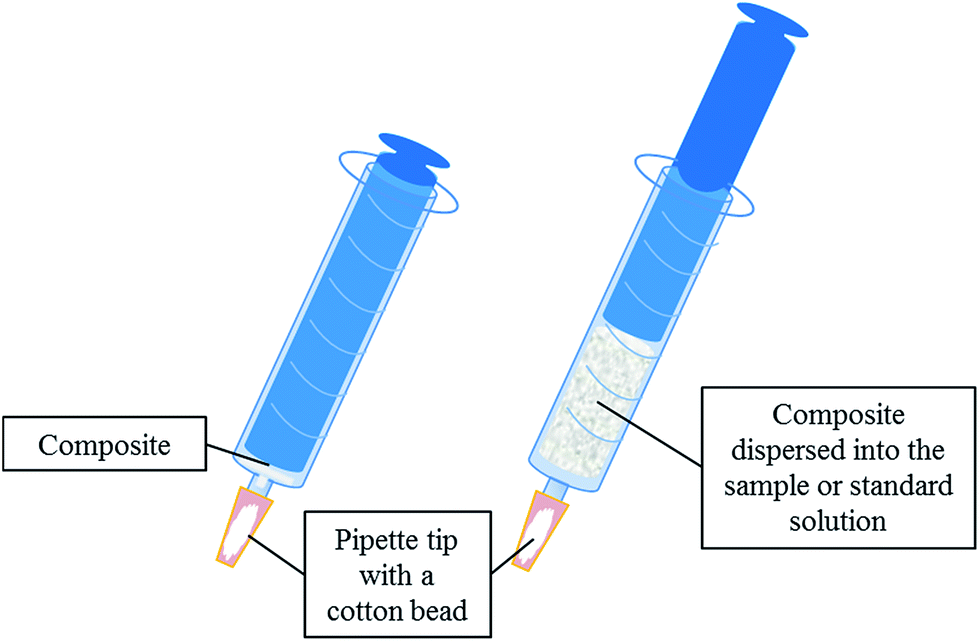 | ||
| Fig. 1 Schematic diagram of the extraction device used in this work. | ||
The extraction procedure involves several steps. First of all, the sorbent is conditioned by means of 1 mL of methanol followed by 1 mL of milli-Q water. Secondly, 10 mL of the sample or standard (containing the analytes in the range from 0.5 to 200 μg L−1) is drawn into the syringe inducing the composite dispersion. The sample is aspirated and ejected twice to increase the interaction between the analytes and the sorbent. Thirdly, a washing step is performed using 1 mL of milli-Q water.
Finally, the analytes are eluted with 500 μL of methanol. The eluate is evaporated to dryness in a vacuum concentrator (Eppendorf, Germany) and the final residue is redissolved in 50 μL of mobile phase before the final LC analysis (ESI†).
Results and discussion
Characterization of the synthesized materials
Nylon 6 presents a chemical structure where two different polarity domains can be highlighted, namely: the polar amide group and the non-polar hydrocarbon backbone. Both domains may interact with target analytes favouring their extraction from the samples. Nylon 6 is usually marketed as pellets (or as granular solid) which presents a low surface to volume ratio due to its uniform surface as it can be observed in Fig. 2. This aspect hinders the extraction capacity of the polymer. | ||
| Fig. 2 SEM micrographs of one nylon 6 pellet at different magnifications. | ||
The synthetic procedure proposed in this article improves this aspect providing a rougher surface due to two different mechanisms: (a) the slow gelation of the nylon produced by the dropwise addition of water into the solution of nylon 6 in formic acid and (b) the introduction of silica NPs into the polymeric network. In this sense, the micrographs of a nylon pellet (Fig. 3A), material A (Fig. 3B) and material B (Fig. 3C) confirm this hypothesis. The superficial structures of the pellet and material A are dramatically different although both are just nylon. In addition, the inclusion of NPs in the polymeric network provides to the solid B with a spongier surface as it can be deduced from the superficial area values shown in the Fig. 3. The values for nylon 6 pellets were negligible (lower than the sensitivity of the technique).
 | ||
| Fig. 3 SEM micrographs of a nylon pellet at 800 magnifications (A), material A at 800 magnifications (B) and material B at 1000 magnifications (C). Surface area (m2 g−1) of B and C materials is also shown. | ||
TEM micrographs of the different synthesized materials were obtained. For simplicity, Fig. 4 only presents the TEM images obtained for material A (Fig. 4A), material B (Fig. 4B) and silica NPs (Fig. 4C). Fig. 4A shows a dark spot that can be ascribed to the polymeric network of nylon 6. Besides, the presence of NPs into the polymeric network can be easily observed in Fig. 4B. Finally, Fig. 4C illustrates how the NPs are easily dispersed in the absence of the polymeric network which confirms, comparing with Fig. 4B, the efficient inclusion of the NPs in the polymer.
 | ||
| Fig. 4 TEM micrographs of material A (A), material B (B) and silica NPs (C). | ||
The elemental analysis of the synthesized materials, which is presented in Table 1, demonstrated, as it was expected, that the carbon and nitrogen content decreases when silica NPs are incorporated to form the composites. However, the precision of the measurements are lower for materials C and D. This lower precision, that we assume is not related to the elemental analysis itself, may indicate that when the amount of NPs is increased their inclusion in the polymeric network (due to the lower amount of nylon) is less reproducible.
ATR-FTIR spectra of the composites and the silica NPs were recorded and compared, as it can be seen in Fig. 5. In the silica NPs spectrum, a strong absorbing region at 1200–1000 cm−1 appears, being characteristic of the Si–O–H and Si–O–Si bonds. On the other hand, nylon 6 (material A) spectrum shows a characteristic band around 1642 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the amide group. Furthermore, another band around 1546 cm−1 is observed, which can be assigned to the N–H deformation band of the amide. Finally, in the other composites spectra, the representative bands of both compounds can be noticed. It was also observed that the intensity of the characteristic bands of nylon 6 decreased from solid B to D as well as the characteristic band of silica NPs increased, what verify the composition of the different sorbents.
O stretching of the amide group. Furthermore, another band around 1546 cm−1 is observed, which can be assigned to the N–H deformation band of the amide. Finally, in the other composites spectra, the representative bands of both compounds can be noticed. It was also observed that the intensity of the characteristic bands of nylon 6 decreased from solid B to D as well as the characteristic band of silica NPs increased, what verify the composition of the different sorbents.
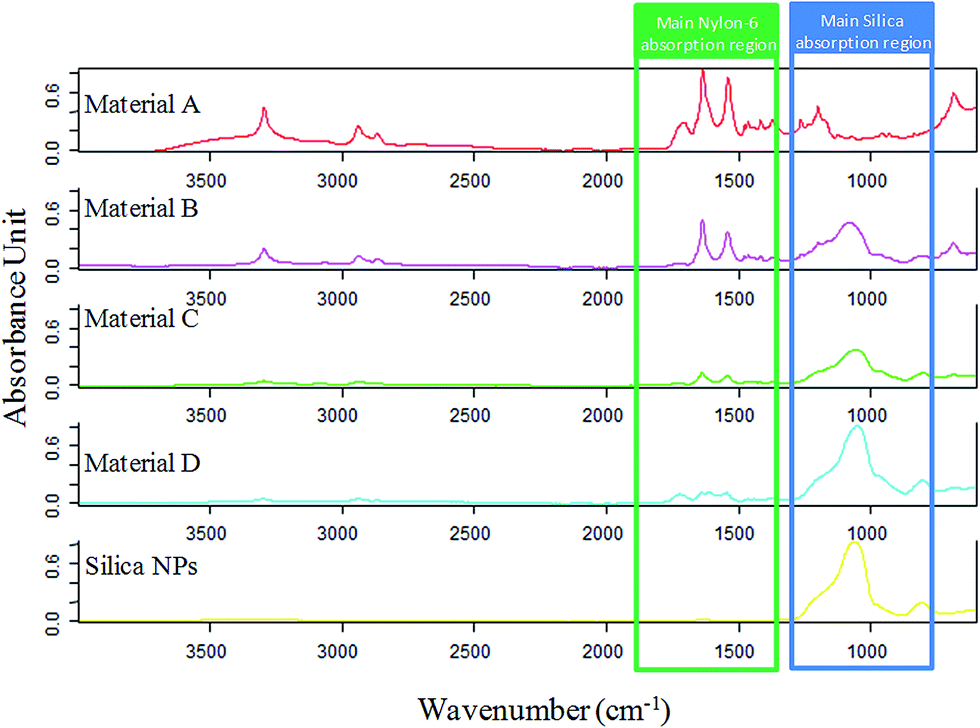 | ||
| Fig. 5 FT-IR spectra of the synthesized materials. The main absorption region of the components of the composites are highlighted in order to clearly observe how the ratio of each component varies. | ||
Study of the extraction performance of the synthesized materials
As it has been explained, several materials comprising different proportions of nylon 6 and silica NPs (Table 1) were synthesized by the proposed method. All these materials were preliminary evaluated to know their sorption abilities. For this purpose, 1 mL of an aqueous standard solution containing the analytes at 1 mg L−1 was extracted with 5 mg of each material. The retained analytes were eluted with 500 μL of methanol which was further analyzed. The chromatograms obtained for this test, which are shown in Fig. 6, lead to several conclusions. | ||
| Fig. 6 Obtained chromatograms for the extraction of 1 mL of aqueous standards containing the analytes [estrone (E1), β-estradiol (E2) and estriol (E3)] at the same concentration (1 mg L−1) by different materials (see Table 1). | ||
On the one hand, the synthetic procedure enhances the extraction performance as it can be concluded by comparing material A and nylon 6 pellets results and considering that both materials have the same composition. On the other hand, materials B and C provided the best results in terms of extraction as a consequence of the introduction of nanoparticles in the polymeric network. However, the results for material C present a lower precision probably due to the synthesis (see Table 1 where the precision obtained for several batches is lower for material C than for material B). Material D presents a slightly lower performance due to the lower content of nylon 6 in this composite. In the light of these results, material B was considered for further studies.
In syringe d-μSPE optimization
The extraction of the target analytes can be influenced by several factors such as the sample pH, the ionic strength, the amount of composite and the volume of sample. All these variables were studied in depth using aqueous standards spiked with the analytes at a concentration of 25 μg L−1. The elution step was carried out using 500 μL of methanol.First of all, pH was studied in the range from 3 to 10, showing a negligible effect on the extraction of the analytes. This behavior is in accordance with the octanol partition coefficient of these compounds which is almost constant in this range. Secondly, ionic strength was evaluated in the range from 0 to 300 g L−1 using sodium chloride as model electrolyte. The extraction recoveries slightly increased with the ionic strength up to 100 g L−1, decreasing at higher concentrations due to an increase on the sample viscosity. Therefore, pH and ionic strength adjustment is not required for sample extraction which simplifies the overall procedure.
Sorbent amount, which defines the maximum sorption capacity, was evaluated at four different levels, namely: 5, 10, 20 and 50 mg using 5 mL of aqueous standard. The results show that the peak areas for all the analytes increased with sorbent amount until 20 mg, decreasing slightly for 50 mg. This final diminution of the signal can be assigned to the worse dispersion of the sorbent in such conditions. Finally, the sample volume was evaluated maintaining the sorbent amount/sample volume ratio constant and equal to the previous optimized value (20 mg per 5 mL) in order to work in the best dispersibility conditions. As it was expected (data not shown) the peak areas for all the analytes increased linearly with the sample volume. Therefore, 10 mL of standard was selected as optimum value.
In order to improve the enrichment of the analytes, an evaporation/redissolution of the eluates was implemented. The best values were obtained when the analytes were eluted from the composite with 500 μL of solvent, this solution being evaporated to dryness and redissolved in 50 μL of mobile phase. In such conditions, the enrichment factors resulted to be 80.2, 88.2 and 43.4 for E1, E2 and E3 respectively.
Analytical figures of merit and sample analysis
Once the procedure was optimized, it was characterized in terms of linearity, precision, sensitivity and extraction efficiency. For each analyte, a calibration graph was constructed by extracting in duplicate seven aqueous standards containing all the analytes at different concentrations. The calibration graphs were built by the representation of the two values obtained for each concentration level and were used for sample analysis. The analytical figures of merit, which are summarized in Table 2, show good linearity, limits of quantification in the low μg L−1 range and acceptable precision (lower than 13.9%, expressed as relative standard deviation, evaluated in septuplicate at 10 μg L−1).Finally, the selectivity of the composite for the extraction of the target compounds from more complex samples, like urine and saliva, was outlined. Direct infusion MS was selected for monitoring the selectivity, since it is more affected by the sample matrix. Urine and saliva samples were spiked with the analytes at 10 μg L−1 levels and were analyzed by direct infusion MS before and after the proposed sample treatment. The analysis of the raw samples provided no signal revealing a high ion suppression effect. However, after the sample treatment the characteristics transition of the analytes are again detected thanks to the clean-up ability of the extraction technique. Although these results are promising about the potential of the composite in biofluids extraction, the quantitative evaluation of the results (data not shown) demonstrated that the intensities of the transitions are lower in biological samples than in aqueous standards even when the composite is used. This resulted especially marked for E2 and E3 extraction. This suggests that the extraction procedure should be further improved for bioanalytical applications.
The proposed method has been compared (see Table 3) with other counterparts published in the literature for the determination of the same compounds in several samples.28–34 Our method provided similar28,29 or even better31 sensitivity than several approaches. However, these methods have been proposed for urine analysis which is not affordable by our method due to selectivity issues. In addition, it should be remarked that two LC-DAD methods provided similar LOQ than our proposal. This fact could be ascribed to a better extraction efficiency of the SBSE and cloud point extraction which are well developed techniques. In addition, those methods proposed for the analysis of environmental waters32–34 are characterized by a better sensitivity due to the large volume of sample that is processed during the extraction technique.
| Extraction | Instrument | Sensitivitya | RSDb | Ref. |
|---|---|---|---|---|
| a Sensitivity expressed as limit of detection (LOD) or limit of quantification (LOQ).b RSD, relative standard deviation. | ||||
| Stir bar sorptive extraction | LC-DAD | LOQ (μg L−1): 3.0 (E1), 3 (E2) | <7.6% (E1), <4.9% (E2) | 28 |
| Cloud point extraction | LC-DAD | LOD (μg L−1): 0.2 (E1), 0.1 (E2) | <2.36 (E1), <3.86% (E2) | 29 |
| Magnetic solid phase extraction | LC-UV | LOQ (μg L−1): 1.0 (E1), 0.8 (E2), 0.9 (E3) | <4.6% (E1), <4.0% (E2), <5.3% (E3) | 30 |
| Solid phase extraction | LC-FD | LOD (μg L−1): 2.7 (E2), 8.3 (E3) | <3.3% (E2), <1.3% (E3) | 31 |
| Solid phase extraction | LC-MS/MS | LOD (ng L−1): 15 (E1), 30 (E2), 30 (E3) | <6% (E1), <16.0% (E2), <15% (E3) | 32 |
| Immuno solid phase extraction | LC-MS | LOD (ng L−1): 0.07 (E1), 0.18 (E2) | <5% (E1 and E2) | 33 |
| Stir bar sorptive extraction | LC-UV | LOD (μg L−1): 0.29 (E1), 0.28 (E2) | <5.8% (E1), <4.5% (E2) | 34 |
| In-syringe dispersive SPE | LC-MS/MS | LOQ (μg L−1): 0.5 (E1), 1.0 (E2), 1.0 (E3) | <6.9% (E1), <2.8% (E2), <13.9% (E3) | This work |
Conclusions
In this article, different composites comprising nylon 6 and silica NPs have been synthesized, characterized and their extraction abilities towards selected hormones have been evaluated. Several advantages can be highlighted. First of all, the positive effect of the synthetic procedure on the extraction performance has been demonstrated. This effect relies on two facts, namely: (a) the solubilization/precipitation of nylon due to a solvent changeover infers a different superficial structure to the polymer and (b) the inclusion of silica NPs on the polymeric network enhances the extraction of the sorbent. In short, the composite presents better extraction performance than the raw polymer. Second, the synthesis is easy and relatively cheap.The optimum composite has shown good extraction behaviour towards the target analytes in aqueous standards and their potential application to bioanalytical samples is promising according to the direct infusion profiles obtained for real samples. However, the quantitative results indicated a matrix effect even when the extraction procedure is applied. Therefore a further optimization should be done in order to develop an analytical method for the determination of these hormones in saliva or urine.
Although the composite has demonstrated a great potential for the treatment of aqueous based samples, the chemical nature of nylon opens a door for processing non-aqueous ones. In fact, the amide group which does not prevail in a polar environment may be exploited in a hydrophobic one.
Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (grant CTQ2014-52939R) is gratefully acknowledged. E. M. Reyes Gallardo expresses her gratitude for the predoctoral grant (refs FPU12/02670) from the Spanish Ministry of Education. The authors would like to thank the Central Service for Research Support (SCAI) of the University of Córdoba for the service provided to obtain the micrographs and elemental analysis of the synthesized material.Notes and references
- C. W. Huck and G. K. Bonn, J. Chromatogr. A, 2000, 885, 51–72 CrossRef CAS PubMed.
- A. Martín-Esteban, Trends Anal. Chem., 2013, 45, 169–181 CrossRef.
- W. J. Cheong, S. H. Yang and F. Ali, J. Sep. Sci., 2013, 36, 609–628 CrossRef CAS PubMed.
- A. Spietelun, M. Pilarczyk, A. Kloskowski and J. Namieśnik, Chem. Soc. Rev., 2010, 39, 4524–4537 RSC.
- L. Lin, H. Chen, H. Wei, F. Wang and J.-M. Lin, Analyst, 2011, 136, 4260–4267 RSC.
- F. Zhao, Y. Meng and J. L. Anderson, J. Chromatogr. A, 2008, 1208, 1–9 CrossRef CAS PubMed.
- M. Zhang and J. Huang, RSC Adv., 2016, 6, 94098–94104 RSC.
- R. Lucena, B. M. Simonet, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2011, 1218, 620–637 CrossRef CAS PubMed.
- M. L. Castillo-García, M. P. Aguilar-Caballos and A. Gómez-Hens, Trends Anal. Chem., 2016, 82, 385–393 CrossRef.
- C. Carrillo-Carrión, R. Lucena, S. Cárdenas and M. Valcárcel, Analyst, 2007, 132, 551–559 RSC.
- A. H. Lu, E. L Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- E. M. Reyes-Gallardo, R. Lucena, S. Cárdenas and M. Valcárcel, Bioanalysis, 2015, 7, 1723–1730 CrossRef CAS PubMed.
- Z. Huang and H. K. Lee, J. Chromatogr. A, 2015, 1414, 41–50 CrossRef CAS PubMed.
- H. Bagheri, R. Daliri and A. Roostaie, Anal. Chim. Acta, 2013, 794, 38–46 CrossRef CAS PubMed.
- A. Mehdinia, S. Dadkhah, T. B. Kayyal and A. Jabbari, J. Chromatogr. A, 2014, 1364, 12–19 CrossRef CAS PubMed.
- M. C. Alcudia-León, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2016, 1455, 57–64 CrossRef PubMed.
- E. M. Reyes-Gallardo, G. Lasarte-Aragonés, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2013, 1271, 50–55 CrossRef CAS PubMed.
- E. M. Reyes-Gallardo, R. Lucena and S. Cárdenas, Trends Anal. Chem., 2016, 84, 3–11 CrossRef.
- H. Bagheri and A. Roostaie, J. Chromatogr. A, 2014, 1324, 11–20 CrossRef CAS PubMed.
- H. Bagheri and A. Roostaie, J. Chromatogr. A, 2015, 1375, 8–16 CrossRef CAS PubMed.
- K. Yoshimatsu, L. Ye, J. Lindberg and I. S. Chronakis, Biosens. Bioelectron., 2008, 23, 1208–1215 CrossRef CAS PubMed.
- X. M. He, G. T. Zhu, J. Yin, Q. Zhao, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2014, 1351, 29–36 CrossRef CAS PubMed.
- X. M. He, G. T. Zhu, H. B. Zheng, S. N. Xu, B. F. Yuan and Y. Q. Feng, Talanta, 2015, 140, 29–35 CrossRef CAS PubMed.
- S. Tong, X. Zhou, C. Zhou, Y. Li, W. Li, W. Zhou and Q. Jia, Analyst, 2013, 138, 1549–1557 RSC.
- B. Fresco-Cala, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2016, 1468, 55–63 CrossRef CAS PubMed.
- E. M. Reyes-Gallardo, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2014, 1345, 43–49 CrossRef CAS PubMed.
- E. M. Reyes-Gallardo, R. Lucena, S. Cárdenas and M. Valcárcel, Microchem. J., 2016, 124, 751–756 CrossRef CAS.
- C. Almeida and J. M. F. Nogueira, J. Pharm. Biomed. Anal., 2006, 41, 1303–1311 CrossRef CAS PubMed.
- Y. Zou, Y. Li, H. Jin, H. Tang, D. Zou, M. Liu and Y. Yang, Anal. Biochem., 2012, 421, 378–384 CrossRef CAS PubMed.
- S. Wang, R. Wang, X. Wu, Y. Wang, C. Xue, J. Wu, J. Hong, J. Liu and X. Zhou, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 905, 105–112 CrossRef CAS PubMed.
- L. Mao, C. Sun, H. Zhang, Y. Li and D. Wu, Anal. Chim. Acta, 2004, 522, 241–246 CrossRef CAS.
- M. Pedrouzo, F. Borrull, E. Pocurull and R. M. Marcé, Talanta, 2009, 78, 1327–1331 CrossRef CAS PubMed.
- P. L. Ferguson, C. R. Iden, A. E. McElroy and B. J. Brownawell, Anal. Chem., 2001, 73, 3890–3895 CrossRef CAS PubMed.
- C. Hu, M. He, B. Chen, C. Zhong and B. Hu, J. Chromatogr. A, 2013, 1310, 21–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24739c |
| This journal is © The Royal Society of Chemistry 2017 |