
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of N-heterocycles and enimino carbocycles by tandem dehydrative coupling–reductive cyclization of halo-sec-amides and dehydrative cyclization of olefinic sec-amides†‡
Pei-Qiang
Huang
 *,
Ying-Hong
Huang
and
Shu-Ren
Wang
*,
Ying-Hong
Huang
and
Shu-Ren
Wang
Department of Chemistry, Fujian Provincial Key Laboratory of Chemical Biology, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn
First published on 16th December 2016
Abstract
We report two efficient and versatile alkenylative cyclization methods for the one-pot synthesis of substituted pyrrolidine, piperidine, indolizidine, and quinolizidine ring systems, and enimino carbocycles, respectively. The first method consists of amide activation (Tf2O) induced dehydrative coupling of halogenated secondary amides with alkenes and the NaBH4 reduction triggered tandem cyclization reaction, while the second one features the Tf2O-promoted novel modes of extended Bischler–Napieralski cyclization reactions of olefinic secondary amides. Taking advantage of triflic anhydride (Tf2O) as the amide activating reagent, the hitherto failed two-step process for the construction of the quinolizidine ring system via intramolecular vinylogous Bischler–Napieralski cyclization has been realized in one pot. The first method was applied to the protecting-group-free one-pot synthesis of the cytotoxic natural product caulophyllumine B (5) and its bioactive derivatives, and to the synthesis of δ-coniceine and 6-styrylpiperidin-2-one. When ethyl vinyl ether and enamides were used as functionalized alkenes, saturated 1,3-amino-ether/amido products 4A1–4A3 were obtained in 73%–74% yields.
Introduction
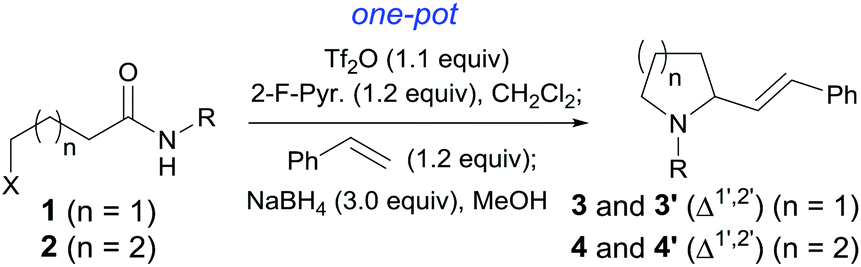
Efficiency is essential to modern organic synthesis.1 A tandem reaction2 is a valuable tactic towards this goal. In this regard, tertiary amide-based tandem reactions have been proven to be a powerful strategy to access molecule complexity.3 In comparison, this chemistry has been less explored for secondary amides.4 In connection with our endeavor to develop amide-based C–C bond forming reactions,5–7 very recently, we have disclosed a highly chemoselective, intermolecular C–H alkyliminylation and acylation of alkenes with secondary amides,7a–c which provides direct access to α,β-unsaturated ketimines (1-aza-1,3-dienes, enimines) and α,β-enones. As a continuation of this work, the tandem dehydrative alkenylation–reductive cyclization of secondary halogenated amides 1/2 to give 2-alkenylpyrrolidines 3 and 2-alkenylpiperidines 4 was envisioned (Scheme 1). | ||
| Scheme 1 Designed tandem dehydrative coupling–reductive cyclization reaction of halogenated secondary amides. | ||
2-Substituted pyrrolidines and piperidines are salient structural features found in a number of alkaloids and medicinal agents.8 Among them some possess a 2-alkenyl pyrrolidine9/piperidine10 motif. For example, caulophyllumine B (5, Fig. 1) was isolated from Caulophyllum thalictroides (L.) Michx (Berberidaceae), an indigenous perennial plant found in north-eastern North America.10a This piperidine alkaloid has been shown to exhibit cytotoxic activity on the human cancer cell lines of the lung, breast and ovary.10b,c Using caulophyllumine B (5) as a leading compound, several piperidine alkene-alkaloids represented by the general structure 6 have found to possess higher growth inhibition activity than the standard drug cisplatin.10c Alkaloid himbacine (9) is a potent and selective antagonist of the muscarinic M2 receptor, which constitutes an attractive lead compound of a drug for the treatment of Alzheimer's disease.10d In addition to their important biological profile, 2-substituted pyrrolidines and piperidines also serve as key intermediates for the synthesis of aza-bicyclic pyrrolizidine,9a,11 indolizidine9a,12 and quinolizidine12 alkaloids such as δ-coniceine13a (7) and epiquinamide,13b,c (8). The latter was isolated from the skin of Ecuadorian frog Epipedobates tricolor.13b
 | ||
| Fig. 1 Representative bioactive alkaloids and medicinal agents related to the present investigation. | ||
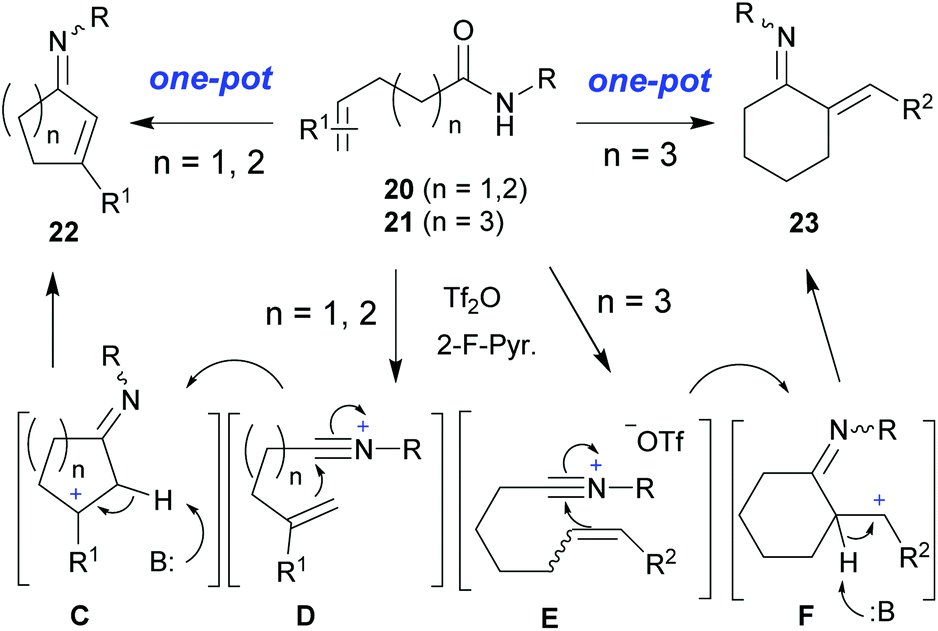
On the other hand, the Bischler–Napieralski (B–N) reaction14 has been known over one century, and the P2O5 or POCl3-mediated dehydrative cyclization of the secondary amide group onto an internal cyclohexenyl moiety (extended Bischler–Napieralski cyclization) (Scheme 2, eqn (a))15a or onto a styryl terminator (vinylogous Bischler–Napieralski cyclization) (Scheme 2, eqn (b))15b have been reported since 1950s. However, to date, this type of cyclization reactions are restricted to the synthesis of heterocycles 13, 17, and 19,15,16 and suffer from limited substrate scope and cyclization mode. Even the modern version utilizing PPSE (polyphosphoric acid trimethylsilyl ester) as a more efficient dehydrative cyclization agent, it has been reported that all attempts to undertake the dehydrative cyclization of 15 to 18 failed.16 In addition, the harsh reaction conditions and hazardous solvent (benzene or CCl4) used in these methods render them with low functional group tolerance and make them environmentally harmful. In this regard, Gawley and Chemburker have noted that the substrate containing a benzyl ether [14: R = (CH2)2OBn] or an ethyl chloride moiety [14: R = (CH2)2Cl] was destroyed under the reaction conditions.16a,b Thus development of novel modes of extended Bischler–Napieralski-type cyclizations (cf.Scheme 3) and tandem reactions that allow access of diverse nitrogenous carbocycles (cf.Scheme 1) is highly desirable.
 | ||
| Scheme 2 Known dehydrative cyclization of olefinic amides. | ||
 | ||
| Scheme 3 Designed novel modes of extended Bischler–Napieralski-type cyclization reactions. | ||
We anticipated that a new mode of dehydrative olefinic amide cyclization would provide direct access to enimino carbocycles 22 and 23 (Scheme 3), which could in turn serve as valuable building blocks for the synthesis of related alkaloids and medicinal agents such as VPC01091 (10, Fig. 1) and flavagline. VPC01091![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17a is a drug candidate developed by the scientists of Abbott Laboratories for the treatment of multiple sclerosis.17a Flavagline derivative 11 exhibited cytotoxicity on human cancer cell lines and the neuroprotection effect on culture models of Parkinson's disease and cisplatin-induced neurotoxicity.17b
17a is a drug candidate developed by the scientists of Abbott Laboratories for the treatment of multiple sclerosis.17a Flavagline derivative 11 exhibited cytotoxicity on human cancer cell lines and the neuroprotection effect on culture models of Parkinson's disease and cisplatin-induced neurotoxicity.17b
Investigations along these two lines (cf.Schemes 1 and 3) have been undertaken and the results are reported herein.
Results and discussion
We first investigated the tandem intermolecular dehydrative alkenylation–reductive cyclization of halo amides (Scheme 1). For this purpose, 5-chloro-N-(2,6-dimethylphenyl)pentanamide 2a was selected as a phenotype substrate, and the reaction conditions were optimized on the basis of those we previously established for the coupling of alkenes with amides.7a–c Thus, chloro-amide 2a was successively treated with trifluoromethanesulfonic anhydride (Tf2O, 1.1 equiv.) and 2-fluoropyridine18,6i–k (2-F-Pyr., 1.2 equiv.) in CH2Cl2 (0 °C, 10 min), and styrene (1.2 equiv.) (Scheme 4). After the disappearance of the starting material as indicated by TLC monitoring, NaBH4 and MeOH were added. The mixture was stirred at 0 °C for 30 min, and at RT for 3 hours. As shown in Scheme 4, when 1.5 equiv. of NaBH4 was used, 1,2-disubstituted piperidine 4a and uncyclized intermediate chloro-enimine 24 were obtained in 15% and 71% yields, respectively. With the amount of NaBH4 increased to 2.0 equiv., the yield of 4a increased to 35% and that of 24 decreased to 46%. On further increasing the amount of NaBH4 to 3.0 equiv., the yield of 4a increased to 88% and only a trace amount of 24 was observed. Thus 3.0 equiv. was determined to be the optimized amount for NaBH4 (cf.Table 1, 4a). | ||
| Scheme 4 The tandem dehydrative coupling–reductive cyclization reaction of chloro-amide 2a. | ||

With the optimized reaction conditions in hand, the reaction was extended to a series of substituted styrenes as well as 1-vinylnaphthalene and 1H-indene. As can be seen from Table 1, the reactions produced the corresponding 2-substituted piperidines 4b–i in 90–98% yields. These results implicated that the reaction tolerated styrylic alkenes bearing either an electron-donating or an electron-withdrawing group, and these groups have little effects on the yield. Besides styrene and its derivatives, gem-dialkyl alkenes also reacted smoothly. Thus, the reaction of methylenecyclohexane afforded the corresponding product 4j in 90% yield. The reaction also tolerated ester and α,β-unsaturated ester group bearing alkenes (4k and 4l). Interestingly, the reaction of ethyl vinyl ether unexpectedly yielded saturated 2-ethoxyethylpiperidine 4A1 in 74% yield. Similarly, the reactions of enamides 25b and 25c, aza-analogues of the enol form of acetaldehyde, produced the saturated 1,3-amino-amido products 4A2 and 4A3 in 73% and 74% yields, respectively. It is worth mentioning that 1,3-diamine is an important structural motif found in many natural products, pharmaceuticals,10a,19 and chiral ligands.
A plausible mechanism for the formation of saturated 1,3-amino ether/amides when employing a vinyl ether or an enamide in lieu of an alkene as a nucleophile is depicted in Scheme 5. A vinyl ether or an enamide reacts with a nitrilium ion A, generated in situ from a chloro-amide and Tf2O/2-F-Pyr., to give an oxonium–iminium intermediate B-4A1 or N-acyliminium–iminium intermediates B-4A2, B-4A3. Bis-reduction of the two electrophilic sites in B-4A1, B-4A2, B-4A3 followed by tandem cyclization produces the 1,3-amino ether/amides 4A.
 | ||
| Scheme 5 A plausible mechanism for the formation of 1,3-amino ether/amides. | ||
Encouraged by these results, the effects of the N-substituent, ω-leaving group, and chain length were examined. The reaction of 4-chloro-N-(2,6-dimethylphenyl)butanamide 1a, a one-carbon lower homologue of 2a afforded (E)-2-styrylpyrrolidine derivative 3a in an unexpected lower yield (80%, Table 2, entry 1). The reaction of 4-bromo-N-(2,6-dimethylphenyl)butanamide gave 3a in the same yield (80%, entry 2) as that for its chloro analogue (entry 1). Thus the chloro and bromo amides displayed comparable reactivity in the tandem reaction. To our delight, the tandem reaction of N-n-butylamide 2b with styrene produced the desired piperidine derivative 4m in 70% yield along with the corresponding side chain-saturated product 4m′ in 10% yield (entry 3). The reaction of its lower homologue 1c afforded the (E)-2-styrylpyrrolidine derivative 3b in 52% yield along with its saturated analogue 3b′ in 12% yield (entry 4). Similar yields (56% and 50%) were obtained with N-allyl and N-benzyl chloro-amides, where the yields of the side products bearing saturated substituents were higher (21% and 27%, entries 5 and 6). The successful incorporation of the well-known N-protecting groups such as N-ally and N-benzyl groups will allow further elaboration via ring closing metathesis (RCM) and N-deprotection.









A tandem reaction on amido ester 26 was also attempted. The reductive addition with styrene gave α-styryl amino ester 27 in 70% yield along with 12% of its saturated analogue 27′ (Scheme 6). Treatment of 27 with NaH at 80 °C yielded 6-styrylpiperidin-2-one 28 in 80% yield.
 | ||
| Scheme 6 The stepwise dehydrative coupling–lactamization of amido ester 26. | ||
To demonstrate the synthetic value of this method, the synthesis of some simple targets were envisaged. Caulophyllumine B (5) and its analogues (cf.6 in Fig. 1) appeared to be ideal targets for our methodology. To this end, amide 2d and p-methoxystyrene was subjected to the standard tandem reaction conditions, which provided compound 4n (Scheme 7). The caulophyllumine analogue 4n has been shown to exhibit higher growth inhibition activity than the standard drug cisplatin.10c In addition, this compound has served as the immediate intermediate in a racemic synthesis of caulophyllumine B (5).20a Although this approach is more efficient than the reported ones,20 in view of developing procedure-economical synthesis,21 an even more efficient one-pot access to racemic caulophyllumine B (5) was envisioned. According to the above mentioned plausible mechanism, the reaction medium is acidic. Thus we anticipated that if we use an acid cleavable O-protecting group such as TBS and THP, it would be possible to perform an in situ O-deprotection by the in situ generated acid. Indeed, subjecting chloro-amide 2d to the reaction with p-TBSO-styrene under slightly modified tandem reaction conditions (40 °C, 12 h) yielded 5 directly in 53% yield (Scheme 7). Under the same conditions, the reaction of p-THPO-styrene produced 5 in 57% yield. Significantly, the direct use of unprotected p-hydroxystyrene also afforded 5 in an appreciable yield of 48%.
 | ||
| Scheme 7 The one-pot racemic syntheses of caulophyllumine B (5) and 4n. | ||
Both approaches to caulophyllumine B (5) are highly efficient which merit comments. In the first approach, by judicious selection of the O-protecting group, three reactions took place sequentially in one pot. The fact that the O-deprotection was achieved by taking advantage of TfOH generated in situ from amide activation, without the need to use any additional reagent, implicates that in addition to being procedure-economical, the method also features a symbiotic catalysis.22 Using O-unprotected 4-hydroxystyrene as a nucleophile, the second approach is not only procedure-economical, but also a protecting group-free synthesis, which are key elements of modern green synthesis.2a
We next addressed the construction of the indolizidine ring system by the established method. The synthesis started from 2e, which was subjected to the tandem reaction to produce 4o in 53% yield along with its saturated analogue 4o′ in 19% yield (Scheme 8). Treatment of 4o with the Grubbs second generation catalyst and Ti(OiPr)4 in CH2Cl2 at reflux yielded 3,5,6,7,8,8a-hexahydroindolizine (29) in 82% yield. Catalytic hydrogenation of the latter produced the indolizidine alkaloid δ-coniceine (7)13a in 93% yield. Hexahydroindolizine 29 can also be converted to indolizidinediol by dihydroxylation.23
 | ||
| Scheme 8 The synthesis of racemic δ-coniceine (7). | ||
Alternatively, a more efficient direct entry to the indolizidine ring system was also envisaged. Indeed, tandem dehrdracyclization–reductive cyclization of 1f led to 3e in 78% yield (Scheme 9). In view of the synthesis of 1-benzylideneoctahydro-2H-quinolizine 4p as a potential synthetic intermediate of quinolizidine alkaloids12 such as epiquinamide (8),13 we needed to challenge the previously reported unsuccessful vinylogous Bischler–Napieralski reaction (cf.Scheme 1).16 To this end, compound 2f was prepared and subjected to the standard tandem reaction conditions, which, to our delight, afforded the desired cyclization product 4p in 81% yield. In view of the failure in the previous attempts for the PPSE-promoted dehydrative cyclization of 15 [R = (CH2)3CO2Et] to give 18 (Scheme 1),16 this result is significant. Gawley and Chemburker attributed the failure of dehydrative cyclization of 15 to the inability of the intermediate generated from 15 to achieve the correct orbital overlap geometry. On the other hand, by incorporating a p-methoxy substituent, Angelastro and co-workers have accomplished the six-membered ring formation of 16via a vinylogous Bischlep–Napieralski cyclization.16c On the basis of the experimental results and in combination with semiempirical and ab initio molecular orbital calculations, they suggested that the overall reaction is under kinetic rather than thermodynamic control.16c
 | ||
| Scheme 9 One-pot construction of indolizidine and quinolizidine ring systems. | ||
Our results might implicate that the activation of secondary amides 1f/2f by the Tf2O/2-F-Pyr. system generates more reactive O-triflylimidate salt H, which can either undergo intramolecular cyclization with the styryl moiety to give intermediate I directly or generate more efficiently the highly reactive nitrilium intermediate A-1f/A-2f thus ensuing a successful cyclization reaction to take place.
We next turned our attention to explore novel modes of dehydrative cyclization of olefinic amides as those displayed in Scheme 3. To this end, compound 20a was prepared by Michael addition24 and treated with Tf2O/2-F-Pyr. The reaction proceeded smoothly to produce the expected cyclization product 22a in 89% yield (Scheme 10). Similarly, the dehydrative cyclization of 20b afforded 1-imino-2-cyclopentene (22b) in 87% yield. Possessing two parochial functionalities, 1-imino-2-cyclohexenes and 1-imino-2-cyclopentenes such as 22a and 22b could serve both as a versatile platform for enantiomeric synthesis and as building blocks for the synthesis of related natural products and medicinally relevant molecules such as VPC01091 (10). It is worth mentioning that although several methods for the synthesis of fused and poly-substituted 1-imino-2-cyclopentene derivatives have been reported,25 a flexible method for direct synthesis of properly substituted and thus synthetically useful 1-imino-2-cyclopentenes from simple starting materials is rare.26 Our approach thus provides an attractive transition-metal-free alternative, which is potentially applicable to the synthesis of related natural products. To demonstrate the versatility of this method, olefinic amide 20c was subjected to dehydrative cyclization and acidic hydrolysis to give 3-phenylcyclohex-2-en-one 32 directly in 81% yield. Alternatively, subjecting 20c to dehydrative cyclization and reduction with NaBH4 to give 3-butylamino-phenylcyclohex-1-ene (33) directly in 80% yield.
 | ||
| Scheme 10 Novel modes of dehydrative cyclization of olefinic amides (20a–c). | ||
Finally, another mode of dehydrative cyclization was investigated, for this purpose 21 was prepared and subjected to the Tf2O/2-F-Pyr.-mediated dehydrative cyclization to yield the expected cyclization product 23 in 76% yield (Scheme 11). It is worth mentioning that although N-acyl/tosyl-1-aza-dienes are widely used in aza-[4 + 2]-cycloaddition reactions for the synthesis of heterocycles,27 known methods for the synthesis of this type of bis-exo-cyclic 1-aza-dienes (enimines) either require the use of the corresponding enones as starting materials28 or formed in situ as reactive intermediates (o-quinone methide imines, aza-o-quinone methides).29 Our metal-free direct synthesis of enimino cyclohexane 23 from either E or Z olefinic amides is straightforward and high yielding.
 | ||
| Scheme 11 A novel mode of dehydrative cyclization of olefinic amide (21). | ||
Conclusions
In summary, we have developed, on one hand, a tandem reductive alkenylation–cyclization reaction starting from halogenated secondary amides and terminal alkenes,30 and on the other hand, new cyclization modes of olefinic secondary amides to give two kinds of enimino carbocycles. Employing Tf2O as an amide activating reagent, the reactions were run under mild conditions, and the intermediates generated were more reactive than those using other activating reagents, allowing the hitherto failed vinylogous Bischler–Napieralski cyclization leading to the quinolizidine ring system. These methods provide efficient one-pot access to α-vinylic substituted pyrrolidines, piperidines, and 8-benzylideneindolizidine and 1-benzylidenequinolizidine, as well as to enimino carbocycles from easily available starting materials. The efficacy of the method was demonstrated by the one-pot synthesis of caulophyllumine B (5) and its bioactive derivatives 4n. The enimino carbocycles could serve as versatile scaffolds for many transformations such as asymmetric reduction/nucleophilic addition, aza-cycloaddition. Because the Tf2O-activated Vilsmeier-type intermolecular cross-coupling reaction of arenes with tertiary N,N-dimethylformamide has been reported,31 it is expectable that the current method can be extended to the intermolecular coupling of alkenes with tertiary amides. In addition, the enantioselective version32 of these reactions will be explored. The results of the investigations along these lines will be reported in due course.Experimental section
General method
Melting points were determined on Büchi M560 Automatic Melting Point apparatus and are uncorrected. Infrared spectra were recorded with a Nicolet Avatar 360 FT-IR spectrometer using film/KBr pellet techniques. 1H NMR and 13C NMR spectra were recorded on a Bruker spectrometer at 400 and 100 MHz (or at 500 and 125 MHz). Chemical shifts (δ) are reported in ppm and respectively referenced to internal standard Me4Si or solvent signals (Me4Si, 0 ppm for 1H NMR and CDCl3, 77.0 ppm for 13C NMR). Mass spectra were recorded on Bruker Dalton Esquire 3000 plus LC-MS apparatus (ESI direct injection). HRMS spectra were recorded on 7.0T FT-MS apparatus.General procedure A: tandem dehydrative coupling–reductive cyclization reaction of halogenated secondary amides
Into a dry 10 mL round-bottom flask equipped with a magnetic stirring bar were added successively a halo-amide (0.5 mmol, 1.0 equiv.), 2 mL of anhydrous CH2Cl2 and 2-fluoropyridine (0.6 mmol, 1.2 equiv.) under an argon atmosphere. After being cooled to 0 °C, trifluoromethanesulfonic anhydride (Tf2O) (155 mg, 93 μL, 0.55 mmol, 1.1 equiv.) was added dropwise via a syringe and the reaction was stirred for 10 min. To the resulting mixture, an alkene (0.6 mmol, 1.2 equiv.) was added dropwise at 0 °C. The reaction mixture was allowed to warm-up to room temperature and stirred for 2 h. Then the reaction mixture was cooled to 0 °C in an ice bath and stirred for 5 min. To the resulting mixture, sodium borohydride (57 mg, 1.5 mmol, 3.0 equiv.) and MeOH (3 mL) were added. The reaction mixture was warmed to room temperature and stirred for 3 h. The reaction was quenched with a saturated NaHCO3 aqueous solution and extracted with dichloromethane (3 × 8 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel to afford the desired saturated N-heterocycles.General procedure B: dehydrative cyclization of olefinic amides
Into a dry 10 mL round-bottom flask equipped with a magnetic stirring bar were added successively a halo-amide (0.5 mmol, 1.0 equiv.), 2 mL of anhydrous CH2Cl2 and 2-fluoropyridine (0.6 mmol, 1.2 equiv.) under an argon atmosphere. After being cooled to 0 °C, trifluoromethanesulfonic anhydride (Tf2O) (155 mg, 93 μL, 0.55 mmol, 1.1 equiv.) was added dropwise via a syringe and the reaction was stirred for 10 min. Then the mixture was allowed to warm-up to room temperature and stirred for 2 h. The reaction mixture was concentrated under reduced pressure, and the residue was purified by flash column chromatography (FC) on silica gel (pre-neutralized with 2% Et3N in n-hexane) to afford the desired α,β-unsaturated ketimine.:10), 2-styrylpiperidine 4a (128 mg, yield: 88%). Colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.54–1.67 (m, 3H), 1.73–1.86 (m, 3H), 2.27 (s, 6H), 3.52 (d, J = 6.7 Hz, 2H), 3.67–3.75 (m, 1H), 6.03 (dd, J = 15.8, 7.8 Hz, 1H), 6.28 (d, J = 15.8 Hz, 1H), 6.78 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 2H), 7.16–7.21 (m, 1H), 7.26 (m, 4H) ppm; 13C NMR (125 MHz, CDCl3) δ 19.1 (2C), 23.6, 32.6, 35.8, 44.8, 59.6, 121.8, 126.2 (2C), 127.3, 128.5 (2C), 128.8 (2C), 129.4 (2C), 130.2, 131.6, 136.9, 144.4 ppm; IR (film) νmax: 3023, 2930, 2847, 1470, 1447, 740 cm−1; HRMS-ESI calcd for [C21H26N]+ (M + H+): 292.2060; found: 292.2058.
:31 inseparable mixture of E/Z isomers. Colorless oil; 1H NMR (400 MHz, CDCl3, data of the two geometric isomers) δ 1.40–1.65 (m, 3.5H), 1.70 (s, 2.5H), 1.75–1.87 (m, 2.6H), 1.91 (s, 0.7H), 2.00 (s, 1.6H), 2.29 (s, 4.4H), 3.45–3.59 (m, 2.3H), 3.95–4.05 (m, 0.7H), 5.23 (d, J = 9.6 Hz, 0.3H), 5.53 (d, J = 9.6 Hz, 0.7H), 6.54–6.61 (m, 0.5H), 6.76–6.84 (m, 1H), 6.89 (d, J = 7.4 Hz, 0.5H), 6.95 (d, J = 7.4 Hz, 1.5H), 7.09–7.32 (m, 4.5H) ppm; 13C NMR (100 MHz, CDCl3, data of the two geometric isomers) δ 16.1, 18.6, 18.9, 23.5, 23.6, 25.7, 32.6, 32.7, 36.1, 36.3, 44.8, 44.9, 55.2, 55.4, 121.8, 122.0, 125.7, 126.4, 126.9, 127.6, 127.7, 128.1, 128.5, 128.7, 129.5, 129.8, 130.2, 130.5, 136.7, 139.1, 141.4, 143.5, 144.3, 144.6 ppm; IR (film) νmax: 3079, 3050, 3021, 2922, 2855, 1474, 1441, 1101, 765, 699 cm−1; HRMS-ESI calcd for [C22H28N]+ (M + H+): 306.2216; found: 306.2217.
:50 inseparable mixture of E/Z isomers. Colorless oil; 1H NMR (400 MHz, CDCl3, data of the two geometric isomers) δ 1.12–1.39 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H), 1.46–2.16 (m, 13H), 2.19–2.24 (m, 2H), 2.24 (s, 6H), 3.31–3.42 (m, 1H), 3.47–3.54 (m, 2H), 4.12 (q, J = 7.1 Hz, 2H), 6.35–6.43 (m, 1H), 6.76 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3, data of the two geometric isomers) δ 14.2, 19.2, 23.3, 23.3, 27.8, 28.0, 28.6, 28.7, 30.3, 30.6, 31.4, 31.5, 32.7, 32.8, 34.5, 34.7, 40.7, 40.9, 44.3, 44.6, 44.9, 54.1, 54.2, 60.1, 121.0, 122.3, 122.5, 128.4, 128.4, 128.9, 128.9, 134.9, 135.0, 172.9, 172.9 ppm; IR (film) νmax: 3042, 2918, 2853, 1731, 1478, 1445, 1155, 1097, 765 cm−1; HRMS-ESI calcd for [C24H36NO2]+ (M + H+): 370.2741; found: 370.2732.
:50 inseparable mixture of E/Z isomers. Colorless oil; 1H NMR (400 MHz, CDCl3, data of the two geometric isomers) δ 1.25 (t, J = 7.2 Hz, 3H), 1.29–1.38 (m, 1H), 1.44–1.82 (m, 5H), 1.98–2.34 (m, 4H), 2.24 (s, 3H), 2.25 (s, 3H), 2.50–2.72 (m, 2H), 2.93–2.98 (m, 1H), 3.02–3.07 (m, 1H), 3.32–3.42 (m, 1H), 3.46–3.54 (m, 2H), 4.09–4.17 (m, 2H), 5.41–5.49 (m, 0.6H), 5.50–5.58 (m, 0.6H), 5.62–5.67 (m, 0.4H), 5.69–5.74 (m, 0.4H), 6.72–6.81 (m, 1H), 6.92–6.99 (m, 2H) ppm; 13C NMR (100 MHz, CDCl3, data of the two geometric isomers) δ 14.2, 14.3, 19.2, 23.3, 23.3, 26.8, 27.2, 29.9, 30.2, 32.7, 32.8, 34.5, 42.5, 42.7, 43.5, 44.9, 54.1, 54.5, 60.5, 60.5, 120.7, 121.1, 121.1, 121.2, 122.7, 122.7, 128.3, 128.5, 128.6, 128.9, 129.4, 131.9, 135.5, 144.4, 171.4, 171.5 ppm; IR (film) νmax: 3054, 2934, 2859, 1731, 1470, 1267, 1159, 773, 736 cm−1; HRMS-ESI calcd for [C24H34NO2]+ (M + H+): 368.2584; found: 368.2581.
:10), 1,2-disubstituted piperidine 4A1 (97 mg, yield: 74%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 1.18 (t, J = 7.0 Hz, 3H), 1.41–1.53 (m, 4H), 1.60–1.67 (m, 1H), 1.68–1.77 (m, 2H), 1.82–1.91 (m, 1H), 2.25 (s, 6H), 3.37–3.46 (m, 3H), 3.48 (t, J = 6.7 Hz, 2H), 3.51–3.57 (m, 2H), 6.76 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3) δ 15.2, 19.2 (2C), 23.5, 32.7, 34.8, 35.3, 44.9, 53.6, 66.2, 67.7, 121.0, 128.4 (2C), 129.0 (2C), 144.8 ppm; IR (film) νmax: 2975, 2933, 2860, 1473, 1474, 1114, 764 cm−1; HRMS-ESI calcd for [C17H28NO]+ (M + H+): 262.2165; found: 262.2167.
:10) to give 6-styryl-δ-lactam 28 in 80% yield (24 mg). Colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.66–1.72 (m, 1H), 1.76–1.85 (m, 3H), 2.27 (s, 6H), 2.36–2.44 (m, 2H), 3.69–3.74 (m, 1H), 6.02 (dd, J = 15.8, 7.9 Hz, 1H), 6.26 (d, J = 15.8 Hz, 1H), 6.77 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 7.5 Hz, 2H), 7.15–7.22 (m, 1H), 7.24–7.28 (m, 4H) ppm; 13C NMR (100 MHz, CDCl3) δ 19.0 (2C), 21.6, 33.9, 35.7, 59.7, 122.0, 126.3 (2C), 127.4, 128.5 (2C), 128.8 (2C), 129.6 (2C), 130.6, 131.2, 136.9, 144.1, 178.9 ppm; IR (film) νmax: 3033, 2923, 2847, 1707, 1473 cm−1; HRMS calcd for [C21H23NNaO]+ (M + Na+): 328.1672; found: 328.1675.
20a (82 mg, yield: 71%). Pale yellow wax. 1H NMR (500 MHz, CDCl3) δ 1.36–1.41 (m, 1H), 1.63–1.84 (m, 5H), 2.23 (t, J = 11.5 Hz, 1H), 2.35 (s, 3H), 2.70 (t, J = 9.4 Hz, 1H), 3.03–3.10 (m, 1H), 3.78 (s, 3H), 6.06 (dd, J = 15.8, 7.0 Hz, 1H), 6.46 (d, J = 15.8 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 23.5, 25.1, 32.7, 43.7, 55.4, 56.3, 68.5, 114.2 (2C), 127.4 (2C), 128.3, 129.4, 132.1, 159.4 ppm; MS (ESI) m/z 232 (M + H+, 100%).
(±)-Caulophyllumine B (5): Brown powder. IR (film) νmax: 3334, 3030, 2927, 2847, 2789, 1518, 1457, 1137, 1073 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.30–1.42 (m, 1H), 1.57–1.84 (m, 5H), 2.13–2.23 (m, 1H), 2.33 (s, 3H), 2.58–2.67 (m, 1H), 3.01–3.09 (m, 1H), 5.98 (dd, J = 15.8, 9.0 Hz, 1H), 6.43 (d, J = 15.8 Hz, 1H), 6.78–6.83 (m, 2H), 7.18–7.23 (m, 2H), 7.39 (br s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 23.5, 25.1, 32.6, 43.9, 56.3, 68.5, 116.2 (2C), 127.6 (2C), 127.9, 128.1, 131.9, 157.1 ppm; MS (ESI) m/z 218 (M + H+, 100%).
:10), enimine 22a (129 mg, yield: 89%) as a 55:45 inseparable mixture of E/Z isomers. Brown oil. 1H NMR (500 MHz, CDCl3, data of the two geometric isomers) δ 1.80 (s, 1.3H), 1.96 (s, 3.0H), 2.00 (s, 1.7H), 2.01–2.05 (m, 3.5H), 2.33–2.47 (m, 2.6H), 2.81–2.88 (m, 0.45H), 2.93–2.98 (m, 0.45H), 3.00–3.08 (m, 0.55H), 3.22–3.30 (m, 0.45H), 5.62 (s, 0.45H), 6.33 (s, 0.55H), 6.79–6.84 (m, 0.55H), 6.86–6.90 (m, 0.45H), 6.93–7.03 (m, 2H), 7.11–7.38 (m, 5H) ppm; 13C NMR (125 MHz, CDCl3, data of the two geometric isomers) δ 17.8, 18.0, 18.1, 18.2, 24.0, 24.1, 35.0, 38.8, 39.3, 40.5, 40.5, 40.6, 117.9, 122.4, 122.5, 122.5, 126.1, 126.3, 126.5, 126.5, 126.7, 126.8, 126.8, 126.9, 127.6, 127.6, 127.7, 128.6, 128.6, 144.1, 144.1, 148.2, 148.4, 150.4, 152.3, 165.2, 166.6 ppm; IR (film) νmax: 3071, 3033, 2917, 2847, 1639, 1620, 1595, 771, 704 cm−1. HRMS-ESI calcd for [C21H24N]+ (M + H+): 290.1903; found: 290.1904.
:20), enimine 22b (173 mg, yield: 87%) as a 55:45 inseparable mixture of E/Z isomers. Brown oil. 1H NMR (400 MHz, CDCl3, data of the two geometric isomers) δ 1.91–1.94 (m, 1H), 2.00–2.12 (m, 9H), 2.42–2.49 (m, 1H), 2.51–2.58 (m, 1H), 2.79–2.85 (m, 1H), 5.51–5.56 (m, 0.45H), 6.17–6.25 (m, 0.55H), 6.86 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 7.5 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3, data of the two geometric isomers) δ 17.8, 18.0, 18.2, 18.4, 29.5, 32.4, 34.0, 34.5, 122.3, 122.4, 123.5, 126.0, 127.2, 127.5, 127.8, 129.9, 149.9, 165.0, 166.6, 177.9, 179.4 ppm; IR (film) νmax: 3062, 3014, 2953, 2930, 2866, 1739, 1704, 1678, 1476 cm−1; HRMS-ESI calcd for [C14H18N]+ (M + H+): 200.1434; found: 200.1438.
:10), allylic amine 33 (92 mg, yield: 80%). Colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.93 (t, J = 7.3 Hz, 3H), 1.33–1.41 (m, 2H), 1.45–1.56 (m, 3H), 1.65–1.74 (m, 1H), 1.88–2.00 (m, 2H), 2.34–2.48 (m, 2H), 2.73 (t, J = 7.3 Hz, 2H), 3.34–3.40 (m, 1H), 6.11 (br s, 1H), 7.21–7.26 (m, 1H), 7.28–7.33 (m, 2H), 7.38–7.42 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 14.0, 20.6, 20.8, 27.7, 29.0, 32.4, 46.6, 53.9, 125.3 (2C), 126.8, 127.0, 128.2 (2C), 138.6, 141.9 ppm; IR (film) νmax: 3305, 3055, 3033, 2924, 2853, 1447, 752, 691 cm−1. HRMS-ESI calcd for [C16H24N]+ (M + H+): 230.1903; found: 230.1906.
:20), enimine 23 (110 mg, yield: 76%). Brown oil. 1H NMR (400 MHz, CDCl3) δ 1.70–1.76 (m, 4H), 2.09 (s, 6H), 2.10–2.16 (m, 2H), 2.79–2.86 (m, 2H), 6.89 (t, J = 7.5 Hz, 1H), 7.03 (d, J = 7.5 Hz, 2H), 7.26–7.31 (m, 1H), 7.32–7.42 (m, 5H) ppm; 13C NMR (100 MHz, CDCl3) δ 18.0, 24.7, 25.2, 29.5, 31.9, 122.6, 126.0 (2C), 127.2, 127.7 (2C), 128.1 (2C), 129.0, 129.7 (2C), 136.8, 139.2, 148.1, 171.4 ppm; IR (film) νmax: 3063, 3025, 2922, 2851, 1686, 1590, 1449, 1254, 1134, 765, 699 cm−1; HRMS-ESI calcd for [C21H24N]+ (M + H+): 290.1903; found: 290.1910.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (21332007) and to the Program for Changjiang Scholars and the Innovative Research Team in University of Ministry of Education, China, for financial support.Notes and references
- For selected reviews, see: (a) B. M. Trost, Science, 1991, 254, 1471–1477 CAS; (b) D. Seebach, Angew. Chem., Int. Ed. Engl., 1990, 29, 1320–1367 CrossRef.
- (a) C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed; (b) H. Pellissier, Tetrahedron, 2013, 69, 7171–7210 CrossRef CAS.
- (a) P. W. Tan, J. Seayad and D. J. Dixon, Angew. Chem., Int. Ed., 2016, 55, 13436–13440 CrossRef CAS PubMed; (b) J. Boudreault, F. Lévesque and G. Bélanger, J. Org. Chem., 2016, 81, 9247–9268 CrossRef CAS PubMed; (c) S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato and N. Chida, J. Am. Chem. Soc., 2016, 138, 5246–5249 CrossRef CAS PubMed; (d) A. W. Gregory, A. Chambers, A. Hawkins, P. Jakubec and D. J. Dixon, Chem. – Eur. J., 2015, 21, 111–114 CrossRef CAS PubMed; (e) M. Mewald, J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2014, 53, 11634–11639 CrossRef CAS PubMed; (f) S. Bonazzi, B. Cheng, J. S. Wzorek and D. A. Evans, J. Am. Chem. Soc., 2013, 135, 9338–9341 CrossRef CAS PubMed; (g) P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc., 2012, 134, 17482–17485 CrossRef CAS PubMed; (h) J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572–4576 CrossRef CAS PubMed; (i) G. Bélanger, M. Dupuis and R. Larouche-Gauthier, J. Org. Chem., 2012, 77, 3215–3221 CrossRef PubMed; (j) B. Peng, D. H. O'Donovan, I. D. Jurberg and N. Maulide, Chem. – Eur. J., 2012, 18, 16292–16296 CrossRef CAS PubMed; (k) G. Vincent, R. Guillot and C. Kouklovsky, Angew. Chem., Int. Ed., 2011, 50, 1350–1353 CrossRef CAS PubMed; (l) K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2010, 49, 6369–6372 CrossRef CAS PubMed; (m) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037–3040 CrossRef CAS PubMed; (n) C. Madelaine, V. Valerio and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 1583–1586 CrossRef CAS PubMed; (o) G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, J. Org. Chem., 2006, 71, 704–712 CrossRef PubMed.
- (a) N. J. Sisti, E. Zeller, D. S. Grierson and F. W. Fowler, J. Org. Chem., 1997, 62, 2093–2097 CrossRef CAS PubMed; (b) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254–14255 CrossRef CAS PubMed; (c) M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096–10097 CrossRef CAS PubMed; (d) Q.-L. Dong, G.-S. Liu, H.-B. Zhou, L. Chen and Z.-J. Yao, Tetrahedron Lett., 2008, 49, 1636–1640 CrossRef CAS; (e) S.-L. Cui, J. Wang and Y.-G. Wang, J. Am. Chem. Soc., 2008, 130, 13526–13527 CrossRef CAS PubMed.
- For recent reviews; (a) D. Seebach, Angew. Chem., Int. Ed., 2011, 50, 96–101 CrossRef CAS PubMed; (b) T. Sato and N. Chida, J. Synth. Org. Chem., Jpn., 2016, 74, 599–610 CrossRef CAS; (c) D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421–4428 CrossRef CAS PubMed; (d) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697–3736 CrossRef CAS; (e) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147–3150 RSC; (f) V. Pace and W. Holzer, Aust. J. Chem., 2013, 66, 507–510 CAS.
- For direct transformation of tertiary amides, see: (a) M. Nakajima, T. Sato and N. Chida, Org. Lett., 2015, 17, 1696–1699 CrossRef CAS PubMed; (b) M. Nakajima, Y. Oda, T. Wada, R. Minamikawa, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 17565–17571 CrossRef CAS PubMed; (c) M. Yoritate, T. Meguro, N. Matsuo, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 8210–8216 CrossRef CAS PubMed; (d) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462–5466 CrossRef CAS PubMed; (e) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512–516 CrossRef CAS PubMed; (f) B. Peng, D. Geerdink and N. Maulide, J. Am. Chem. Soc., 2013, 135, 14968–14971 CrossRef CAS PubMed; (g) V. Valerio, D. Petkova, C. Madelaine and N. Maulide, Chem. – Eur. J., 2013, 19, 2606–2610 CrossRef CAS PubMed; (h) Y. Yanagita, H. Nakamura, K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Chem. – Eur. J., 2013, 19, 678–684 CrossRef CAS PubMed; secondary amides: (i) V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750–2753 CrossRef CAS PubMed; (j) V. Pace, L. Castoldi, A. D. Mamuye and W. Holzer, Synthesis, 2014, 2897–2909 CrossRef; (k) V. Pace, L. Castoldi and W. Holzer, Chem. Commun., 2013, 49, 8383–8385 RSC; (l) Y. Oda, T. Sato and N. Chida, Org. Lett., 2012, 14, 950–953 CrossRef CAS PubMed; (m) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228–234 CrossRef CAS PubMed.
- For direct transformations of secondary amides, see: (a) P.-Q. Huang, Y.-H. Huang, H. Geng and J.-L. Ye, Sci. Rep., 2016, 6, 28801 CrossRef PubMed; (b) P.-Q. Huang and Y.-H. Huang, Chin. J. Chem., 2017, 34 DOI:10.1002/cjoc.201600700; (c) P.-Q. Huang, Y.-H. Huang and K.-J. Xiao, J. Org. Chem., 2016, 81, 9020–9027 CrossRef CAS PubMed; (d) A.-E. Wang, Z. Chang, Y.-P. Liu and P.-Q. Huang, Chin. Chem. Lett., 2015, 26, 1055–1058 CrossRef CAS; (e) P.-Q. Huang, W. Ou and J.-L. Ye, Org. Chem. Front., 2015, 2, 1094–1106 RSC; (f) J.-F. Zheng, X.-Y. Qian and P.-Q. Huang, Org. Chem. Front., 2015, 2, 927–935 RSC; (g) J.-F. Zheng, Z.-Q. Xie, X.-J. Chen and P.-Q. Huang, Acta Chim. Sin., 2015, 73, 705–715 CrossRef CAS; (h) K.-J. Xiao, A.-E. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314–8317 CrossRef CAS PubMed. For direct transformations of tertiary amides, see: (i) P.-Q. Huang, W. Ou and F. Han, Chem. Commun., 2016, 52, 11967–11970 RSC; (j) P.-Q. Huang, Y. Wang, K.-J. Xiao and Y.-H. Huang, Tetrahedron, 2015, 71, 4248–4254 CrossRef CAS; (k) P.-Q. Huang, H. Geng, Y.-S. Tian, Q.-R. Peng and K.-J. Xiao, Sci. China: Chem., 2015, 58, 478–482 CrossRef CAS.
- (a) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC; (b) J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 CrossRef CAS PubMed.
- (a) C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405–5452 RSC; (b) K. T. H. Wanner and G. Höfner, Arch. Pharm., 1990, 323, 977–986 CrossRef CAS; (c) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed; (d) S. H. Lim, S. Ma and P. Beak, J. Org. Chem., 2001, 66, 9056–9062 CrossRef CAS PubMed; (e) C. Serino, N. Stehle, Y. S. Park, S. Florio and P. Beak, J. Org. Chem., 1999, 64, 1160–1165 CrossRef CAS.
- (a) Z. Ali and I. A. Khan, Phytochemistry, 2008, 69, 1037–1042 CrossRef CAS PubMed; (b) V. L. M. Madgula, Z. Ali, T. Smillie, I. A. Khan, L. A. Walker and S. I. Khan, Planta Med., 2009, 75, 329–332 CrossRef CAS PubMed; (c) S. Kankala, R. K. Kankala, R. Balaboina, N. S. Thirukovela, R. Vadde and C. S. Vasam, Bioorg. Med. Chem. Lett., 2014, 24, 1180–1183 CrossRef CAS PubMed; (d) M. Takadoi, K. Yamaguchi and S. Terashima, Bioorg. Med. Chem., 2003, 11, 1169–1186 CrossRef CAS PubMed.
- J. Robertson and K. Stevens, Nat. Prod. Rep., 2014, 31, 1721–1788 RSC.
- J. P. Michael, Nat. Prod. Rep., 2007, 24, 191–222 RSC.
- (a) N. Zill, A. Schoenfelder, N. Girard, M. Taddei and A. Mann, J. Org. Chem., 2012, 77, 2246–2253 CrossRef CAS PubMed; (b) R. W. Fitch, H. M. Garraffo, T. F. Spande, H. J. C. Yeh and J. W. Daly, J. Nat. Prod., 2003, 66, 1345–1350 CrossRef CAS PubMed; (c) C.-M. Si, Z.-Y. Mao, H.-Q. Dong, Z.-T. Du, B.-G. Wei and G.-Q. Lin, J. Org. Chem., 2015, 80, 5824–5833 CrossRef CAS PubMed.
- (a) A. Bischler and B. Napieralski, Ber., 1893, 26, 1903–1908 CrossRef; (b) G. Fodor and S. Nagubandi, Tetrahedron, 1980, 36, 1279–1300 CrossRef CAS. For Movassaghi's modern version, see: (c) M. Movassaghi and M. D. Hill, Org. Lett., 2008, 10, 3485–3488 CrossRef CAS PubMed.
- (a) O. Schnider and J. Hellerbach, Helv. Chim. Acta, 1950, 33, 1437–1448 CrossRef CAS; (b) S. Ushioda, M. Fujihara and S. Sugasawa, Yakugaku Zasshi, 1965, 85, 231–234 CAS , and references cited therein.
- (a) R. E. Gawley and S. Chemburkar, Tetrahedron Lett., 1986, 27, 2071–2074 CrossRef CAS; (b) R. E. Gawley and S. Chemburker, Heterocycles, 1989, 29, 1283–1292 CrossRef CAS; (c) A. L. Marquart, B. L. Podlogar, E. W. Huber, D. A. Demeter, N. P. Peet, H. J. R. Weintraub and M. R. Angelastro, J. Org. Chem., 1994, 59, 2092–2100 CrossRef CAS.
- (a) R. Zhu, A. H. Snyder, Y. Kharel, L. Schaffter, Q. Sun, P. C. Kennedy, K. R. Lynch and T. L. Macdonald, J. Med. Chem., 2007, 50, 6428–6435 CrossRef CAS PubMed; (b) N. Ribeiro, F. Thuaud, Y. Bernard, C. Gaiddon, T. Cresteil, A. Hild, E. C. Hirsch, P. P. Michel, C. G. Nebigil and L. Désaubry, J. Med. Chem., 2012, 55, 10064–10073 CrossRef CAS PubMed; (c) I. U. Khan, S. Kattela, A. Hassan and C. R. D. Correia, Org. Biomol. Chem., 2016, 14, 9476–9480 RSC.
- 2-Fluoropyridine was introduced by Movassaghi in conjunction with Tf2O for the activation of secondary amides: J. W. Medley and M. Movassaghi, J. Org. Chem., 2009, 74, 1341–1344 CrossRef CAS PubMed . The Tf2O-2-F-pyridine system has been extensively used by Charette's,6l Pace's,6i–k and our groups7a–h for the activation of secondary amides towards nucleophilic additions.
- X. Ji and H. Huang, Org. Biomol. Chem., 2016, 14, 10557–10566 CAS.
- (a) A. Khanna, C. Maung, K. R. Johnson, T. T. Luong and D. L. Van Vranken, Org. Lett., 2012, 14, 3233–3235 CrossRef CAS PubMed; (b) P. R. Krishna and B. K. Reddy, Tetrahedron Lett., 2010, 51, 6262–6264 CrossRef.
- P.-Q. Huang, Y. Wang, S.-P. Luo, H. Geng, Y.-P. Ruan and A.-E. Wang, Tetrahedron Lett., 2015, 56, 1255–1258 CrossRef CAS.
- (a) S. Wang, Y. Shi and W.-S. Tian, Chin. J. Chem., 2015, 33, 637–642 CrossRef CAS; (b) X. Zhang, W. Wei and R. Tan, Sci. China: Chem., 2015, 58, 1097–1109 CrossRef CAS.
- S. M. Colegate, P. R. Dorling and C. R. Huxtable, Aust. J. Chem., 1984, 37, 1503–1509 CrossRef CAS.
- M. Kikuchi, S. Niikura, N. Chiba, N. Terauchi and M. Asaoka, Chem. Lett., 2007, 36, 736–737 CrossRef CAS.
- (a) C. Wang, Q. Song and Z. Xi, Tetrahedron, 2004, 60, 5207–5214 CrossRef CAS; (b) K. Tamao, K. Kobayashi and Y. Ito, J. Am. Chem. Soc., 1988, 110, 1286–1288 CrossRef CAS.
- (a) X.-N. Wang, G. N. Winston-McPherson, M. C. Walton, Y. Zhang, R. P. Hsung and K. A. DeKorver, J. Org. Chem., 2013, 78, 6233–6244 CrossRef CAS PubMed; (b) K. A. DeKorver, R. P. Hsung, A. G. Lohse and Y. Zhang, Org. Lett., 2010, 12, 1840–1843 CrossRef CAS PubMed; (c) H. Zhang, B. Wang, H. Yi, T. Sun, Y. Zhang and J. Wang, Chem. Commun., 2016, 52, 13285–13287 RSC.
- (a) B. Groenendaal, E. Ruijter and R. V. A. Orru, Chem. Commun., 2008, 5474–5489 RSC; (b) M. Shimizu, I. Hachiya and I. Mizota, Chem. Commun., 2009, 874–889 RSC.
- (a) S. Bouquillon, F. Hénin and J. Muzart, Synth. Commun., 2001, 31, 39–45 CrossRef CAS; (b) F. Maywald and P. Eilbracht, Synlett, 1996, 380–382 CrossRef CAS.
- (a) L. Wang, S. Li, M. Blmel, A. R. Philipps, A. Wang, R. Puttreddy, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 11110–11114 CrossRef CAS PubMed; (b) J.-Y. Liu, H. Lu, C.-G. Li, Y.-M. Liang and P.-F. Xu, Synlett, 2016, 1287–1291 Search PubMed; (c) Y. Zhi, K. Zhao, T. Shu and D. Enders, Synthesis, 2016, 238–244 CAS; (d) A. Lee, A. Younai, C. K. Price, J. Izquierdo, R. K. Mishra and K. A. Scheidt, J. Am. Chem. Soc., 2014, 136, 10589–10592 CrossRef CAS PubMed.
- (a) S. Agasti, A. Dey and D. Maiti, Chem. Commun., 2016, 52, 12191–12194 RSC; (b) E. Stridfeldt, A. Seemann, M. J. Bouma, C. Dey, A. Ertan and B. Olofsson, Chem. – Eur. J., 2016, 22, 16066–16070 CrossRef CAS PubMed.
- A. G. Martinez, R. M. Alvarez, J. O. Barcina, S. M. Cerero, E. T. Vilar, A. G. Fraile, M. Hanack and L. R. Subramanian, J. Chem. Soc., Chem. Commun., 1990, 1571–1572 RSC.
- For a recent report on enantioselective reduction of ketimines, see: (a) S. Li, G. Li, W. Meng and H. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed. For a related review, see: (b) P.-G. Echeverria, T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2016, 48, 2523–2539 CrossRef CAS.
- G. B. Bajracharya, Z. Huo and Y. Yamamoto, J. Org. Chem., 2005, 70, 4883–4886 CrossRef CAS PubMed.
Footnotes |
| † In memory of the late Professor Zhi-Tang Huang. |
| ‡ Electronic supplementary information (ESI) available: 1H and 13C NMR Spectra of all products. See DOI: 10.1039/c6qo00720a |
| This journal is © the Partner Organisations 2017 |