
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-assisted azide–alkyne cycloaddition chemistry as a tool for the production of emissive boron difluoride 3-cyanoformazanates†
Stephanie M.
Barbon
ab,
Samantha
Novoa
ab,
Desiree
Bender
ab,
Hilary
Groom
c,
Leonard G.
Luyt
*adc and
Joe B.
Gilroy
*ab
aDepartment of Chemistry, The University of Western Ontario, 1151 Richmond St. N., London, Ontario, Canada N6A 5B7. E-mail: lluyt@uwo.ca; joe.gilroy@uwo.ca; Tel: +1-519-661-2111 ext. 81561 (JBG), +1-519-685-8600 ext. 53302 (LGL)
bThe Centre for Advanced Materials and Biomaterials Research (CAMBR), The University of Western Ontario, 1151 Richmond St. N., London, Ontario, Canada N6A 5B7
cDepartment of Oncology, London Regional Cancer Program, 790 Commissioners Rd. E., London, Ontario, Canada N6A 4L6
dDepartment of Medical Imaging, The University of Western Ontario, 1151 Richmond St. N., London, Ontario, Canada N6A 5B7
First published on 8th November 2016
Abstract
The synthesis and characterization of emissive boron difluoride (BF2) complexes of 3-cyanoformazanate ligands produced using copper-assisted azide–alkyne cycloaddition (CuAAC) chemistry is described. Detailed spectroscopic and electrochemical characterization of benzyl-functionalized complexes served as models and demonstrated that triazole formation at the N-aryl substituents of the formazanate ligand scaffold led to red-shifted absorption and emission maxima and more difficult electrochemical reduction compared to alkyne-substituted precursors. CuAAC chemistry was also used to append ferrocene and tetraethylene glycol substituents to the formazanate backbone. In the case of the ferrocene-substituted complexes, fluorescence was quenched and a reversible oxidation feature (in addition to the reduction features associated with formazanate complexes) was observed using cyclic voltammetry. Treatment with NOBF4 oxidized ferrocene to ferrocenium and resulted in the reestablishment of fluorescence. Tetraethylene glycol substitution produced the first water soluble BF2 formazanate, which was shown to distribute throughout the cytoplasm and nucleus of mouse fibroblast cells when studied as a fluorescence imaging agent.
Introduction
Fluorophores are a very useful class of molecules as they provide a handle that allows for the visualization of chemical phenomena at the molecular, macromolecular and bulk size regimes.1–6 For this and other reasons, the development of new classes of fluorophores has been the focus of a significant amount of research.7,8 One of the major hurdles to be overcome in the field was the facile synthetic modification of fluorophores in order to utilize them for an expanded range of applications.9–11 Significant impact was made in this area with the reports on copper-assisted azide–alkyne cycloaddition (CuAAC) chemistry.12,13 Since the original reports appeared, the number of alkyne- and azide-modified fluorophores such as those based on anthracene,14,15 pyrene,16,17 boron dipyrromethenes (BODIPYs),18–20 rhodamine21,22 and coumarin23,24 has expanded dramatically (e.g., 1–3). The onward CuAAC chemistry of these and other fluorophores has led to their use, for example, as protein labels,25 in photosensitizers,26 as molecular switches,27 as probes of DNA hybridization,28 and in radiolabeling.29 CuAAC chemistry has also been used to create a variety of fluorescence sensors for anions,30 acids and bases,31,32 nitroxide radicals,33 hydroxyl radicals,34 and various metal ions including mercury35,36 and zinc.37,38
In addition, CuAAC chemistry has been utilized to modify fluorophores for biological imaging applications. This strategy has been used both to impart water solubility (e.g., 4),39,40 and functionalities that interact with specific biological targets (e.g., 5).41–44 More recently, this strategy has been extended to include copper-free, strain-promoted azide–alkyne cycloaddition chemistry with specific utility in biological systems.45,46
Transition metal47–53 and boron54–56 complexes of formazanate ligands have received significant attention due to their rich optical and electrochemical properties. More specifically, boron difluoride (BF2) complexes of 3-cyanoformazanates have recently been shown to have many of the desirable properties associated with common fluorophores, including high fluorescence quantum yields, large Stokes shifts, facile high-yielding syntheses, and perhaps most importantly, emission in the red to near-IR region of the electromagnetic spectrum.57–59 These complexes have shown promise as fluorescence cell-imaging agents,58 as precursors to B(1)-carbenoid intermediates60 and stable radicals,61 as efficient electrochemiluminescence emitters,62 aggregation-induced emission luminogens (AIEgens),63 and building blocks for functional polymeric materials.64,65 Herein, we expand the scope of BF2 formazanate chemistry further by producing a variety of emissive materials through the use of CuAAC chemistry that cannot be readily synthesized by other methods.
Results and discussion
Benzyl-functionalized BF2 complexes of 3-cyanoformazanates
Formazans 6 and 10 were prepared via aryl diazonium coupling reactions in basic solutions of cyanoacetic acid by adapting a reported method.66 Formazan 6 was prepared from a mixture of alkyne- and methoxy-substituted aryl diazonium salts and separated from the symmetrically-substituted byproducts using column chromatography. In order to study the effect of triazole formation on the photophysical properties of BF2 complexes of 3-cyanoformazanate ligands, formazans 6 and 10 were converted to mono-alkyne-substituted complex 7 and bis-alkyne-substituted complex 11 by adaptation of established methods (Scheme 1).57 Complex 7 was first characterized by single crystal X-ray diffraction (Fig. 1), and crystallized on a two-fold rotation axis. As a result, the alkyne and methoxy substituents were disordered. However, the structure does confirm the expected connectivity and planarity throughout the molecule (dihedral angle between N4 plane and aryl substituents: 7.2°). All other structural metrics were consistent with the solid-state structures of previously reported BF2 adducts of 3-cyanoformazanate ligands.57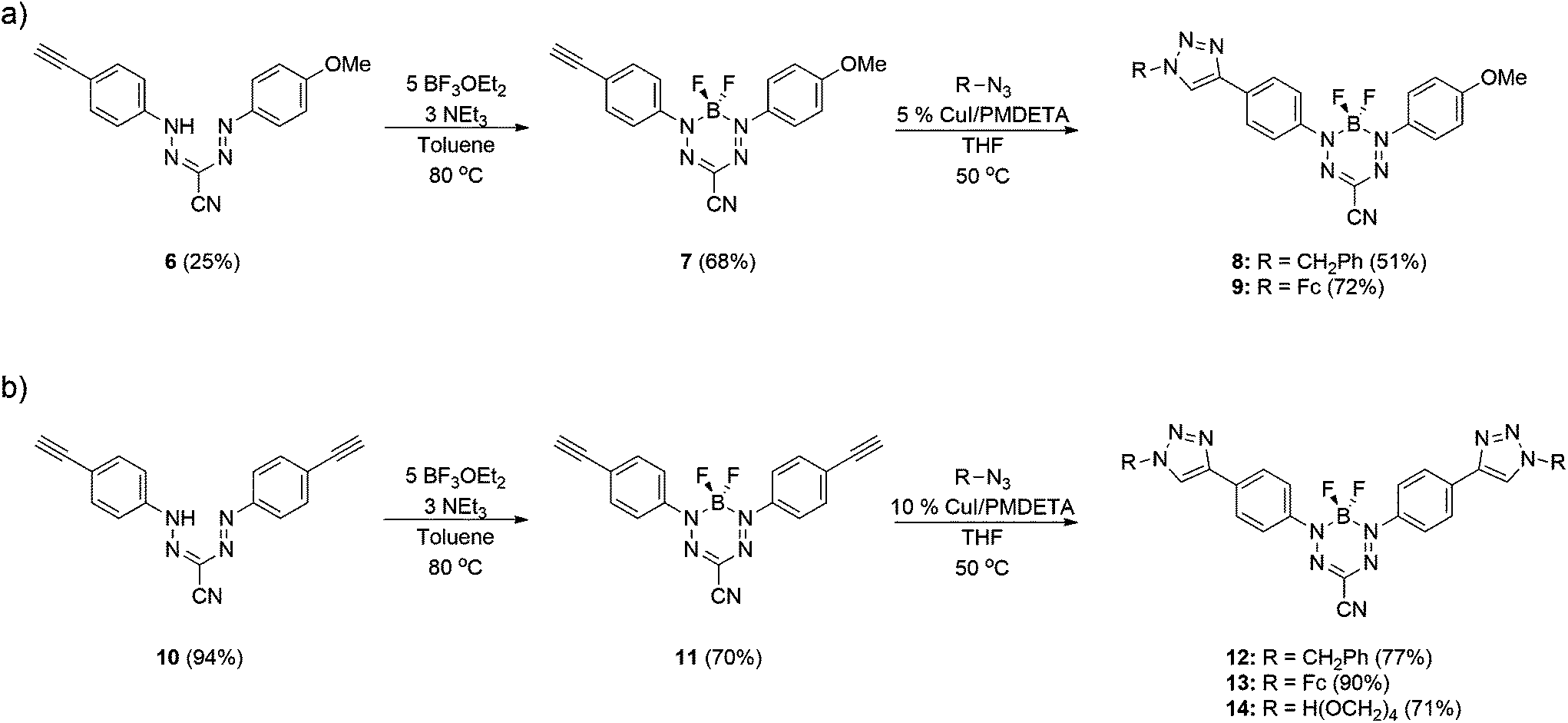 | ||
| Scheme 1 Synthesis of (a) mono- and (b) bis-functionalized BF2 complexes of 3-cyanoformazanates. | ||
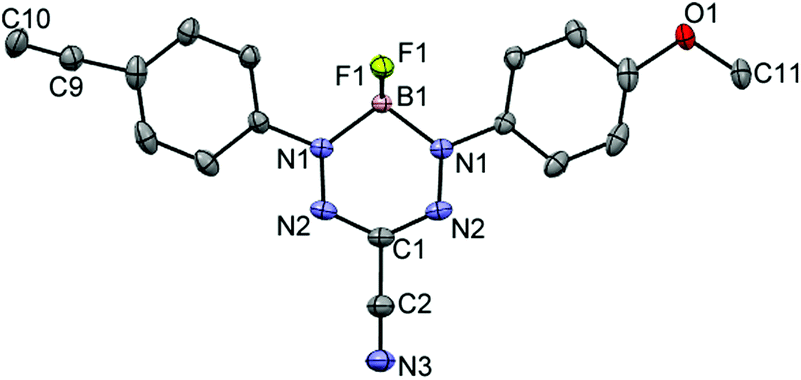 | ||
| Fig. 1 Solid-state structure of 7. Anisotropic displacement ellipsoids are shown at 50% probability and hydrogen atoms have been removed for clarity. Selected bond lengths (Å): B1–N1 1.5759(15), N1–N2 1.3023(15), N2–C1 1.3351(13), O1–C11 1.418(5), C9–C10 1.191(7). Selected bond angles (°): B1–N1–N2 124.57(10), N1–N2–C1 117.24(11). | ||
The alkyne-substituted complexes 7 and 11 underwent CuAAC chemistry with benzyl azide when combined in the presence of copper(I) iodide and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) in THF to form mono- and bis-benzyl-functionalized compounds 8 and 12 (Scheme 1). The successful conversion of alkyne-substituted complexes 7 and 11 to benzyl-functionalized complexes 8 and 12 was confirmed by 1H, 11B, 13C{1H} and 19F NMR spectroscopy (Fig. 2 and S1–S17†), UV-vis absorption/emission and IR spectroscopy, and high resolution mass spectrometry. CuAAC chemistry was also used to synthesize BF2 formazanate complexes with appended ferrocene substituents (9 and 13) and water-solubilizing tetraethylene glycol substituents (14). Initial attempts to synthesize the water-soluble complex 14 were unsuccessful, likely due to the TEG chains outcompeting PMDETA for the ligation of copper(I) when all of the reagents were combined simultaneously. We circumvented this issue by stirring CuI and PMDETA in THF for 30 min before the TEG azide was added.
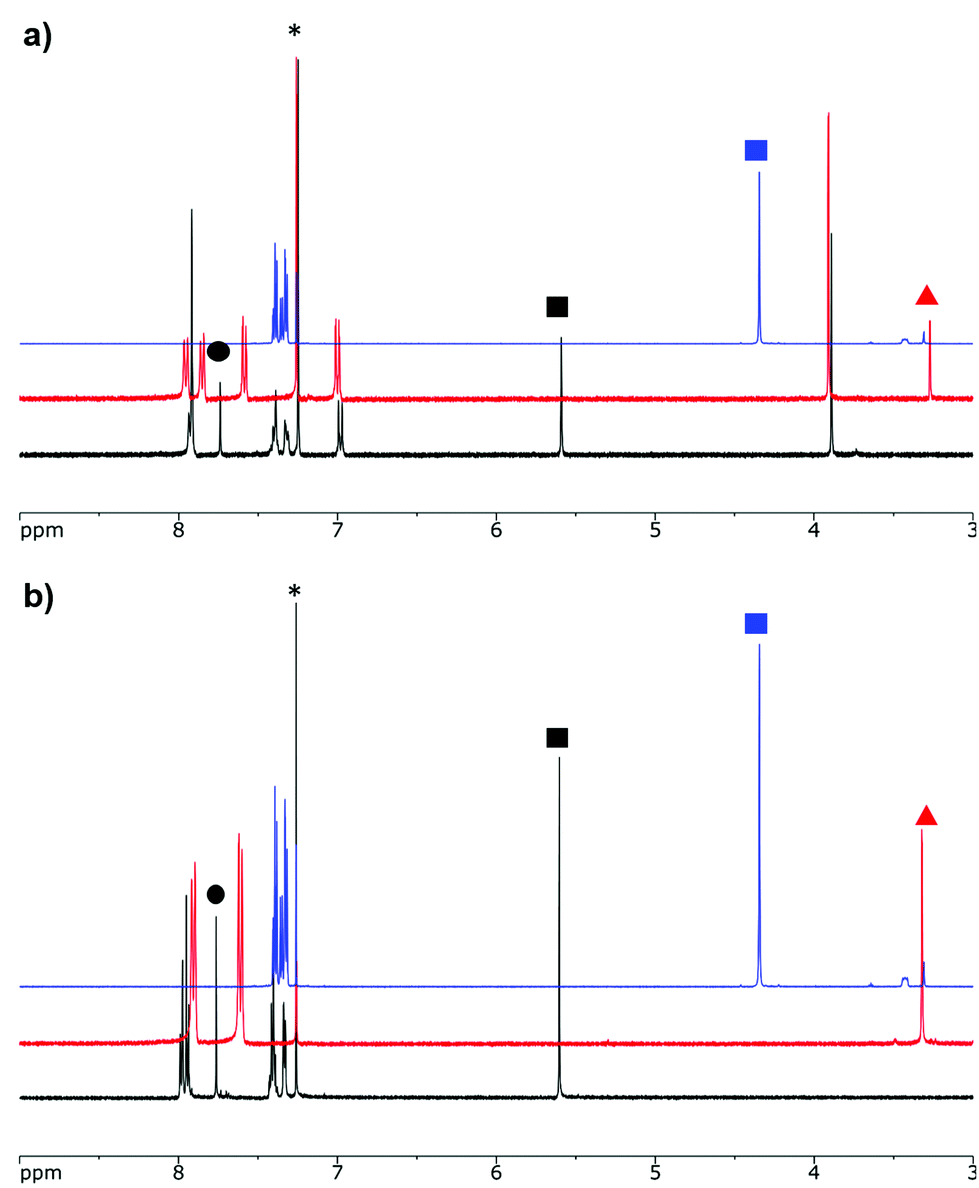 | ||
| Fig. 2 1H NMR spectra of (a) benzyl azide (blue), mono-alkyne-substituted complex 7 (red) and mono-benzyl-functionalized complex 8 (black) and (b) benzyl azide (blue), bis-alkyne-substituted complex 11 (red) and bis-benzyl-functionalized complex 12 (black) in CDCl3. The asterisks denote residual solvent signals. The blue squares denote the benzyl azide CH2 signals, the red triangles denote the alkyne CH signals, the black circles denote the triazole CH signals, and the black squares denote the CH2 signal from the benzyl azide moieties in complexes 8 and 12. | ||
The electrochemical properties of complexes 7, 8, 11 and 12 were studied using cyclic voltammetry in THF (Fig. 3 and Table 1). Each voltammogram showed two reversible one-electron reduction waves within the electrochemical window of the solvent. Considering the first reduction wave, which corresponds to the formation of a ligand-centered radical anion, bis-alkyne-substituted complex 11 is significantly easier to reduce than mono-alkyne-substituted complex 7 (−0.50 V and −0.67 V relative to the ferrocene/ferrocenium redox couple, respectively) due to the presence of the electron donating methoxy substituent in 7. The benzyl-functionalized complexes were more difficult to reduce than their alkyne-substituted precursors (8: −0.68 V; 12: −0.62 V). We have previously demonstrated that extending electronic conjugation in BF2 complexes of 3-cyanoformazanate ligands results in complexes that are easier to reduce due to a narrowing of the HOMO–LUMO gap.59 However, in the case of complexes 8 and 12, the more negative reduction potentials observed are likely due to the electron donating nature of the benzyl-substituted triazoles, which appear to be better electron donors than alkyne substituents. The second reduction waves, which correspond to the formation of ligand-centred dianions, followed the same trend.
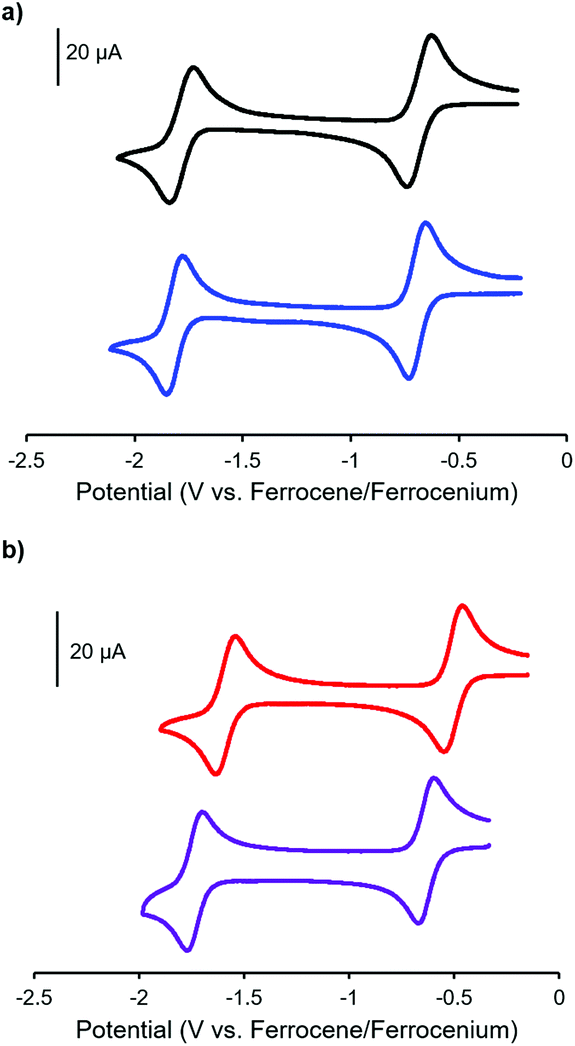 | ||
| Fig. 3 Cyclic voltammograms of (a) mono-alkyne-substituted complex 7 (black) and mono-benzyl-functionalized complex 8 (blue) and (b) bis-alkyne-substituted complex 11 (red) and bis-benzyl-functionalized complex 12 (purple) recorded at 100 mV s−1 in 1 mM THF solutions containing 0.1 M [nBu4N][PF6] as supporting electrolyte. | ||
| Solvent | λ max (nm) | ε (M−1 cm−1) | λ em (nm) |
Φ
F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
ν ST (nm) | ν ST (cm−1) |
E
ox1b (V) |
E
red1b (V) |
E
red2b (V) |
|
|---|---|---|---|---|---|---|---|---|---|---|
| a Quantum yields were measured according to published protocols using [Ru(bpy)3][PF6]2 as a relative standard and corrected for wavelength-dependent detector sensitivity (Fig. S18).68,69 b Cyclic voltammetry experiments were conducted in THF containing 1 mM analyte and 0.1 M [nBu4N][PF6] as supporting electrolyte at a scan rate of 100 mV s−1. All voltammograms were referenced internally against the ferrocene/ferrocenium redox couple. c Complexes 9 and 13 were non-emissive in the range of organic solvents employed. | ||||||||||
| 7 | CH2Cl2 | 552 | 30400 |
647 | 30 | 95 | 2660 | |||
| THF | 550 | 32500 |
650 | 27 | 100 | 2797 | — | −0.67 | −1.78 | |
| Toluene | 569 | 26800 |
648 | 36 | 79 | 2143 | ||||
| 8 | CH2Cl2 | 560 | 31000 |
660 | 30 | 100 | 2706 | |||
| THF | 560 | 33700 |
672 | 25 | 112 | 2976 | — | −0.68 | −1.81 | |
| Toluene | 579 | 37400 |
661 | 37 | 82 | 2143 | ||||
|
9c |
CH2Cl2 | 562 | 28600 |
— | — | — | — | |||
| THF | 562 | 31700 |
— | — | — | — | 0.20 | −0.67 | −1.80 | |
| Toluene | 578 | 23900 |
— | — | — | — | ||||
| 11 | CH2Cl2 | 530 | 32700 |
635 | 18 | 105 | 3120 | |||
| THF | 529 | 29600 |
638 | 19 | 109 | 3230 | — | −0.50 | −1.58 | |
| Toluene | 547 | 41700 |
637 | 36 | 90 | 2583 | ||||
| 12 | CH2Cl2 | 558 | 39600 |
679 | 46 | 121 | 3194 | |||
| THF | 563 | 34100 |
696 | 34 | 133 | 3394 | — | −0.62 | −1.72 | |
| Toluene | 577 | 33700 |
680 | 72 | 103 | 2625 | ||||
|
13c |
CH2Cl2 | 569 | 30900 |
— | — | — | — | |||
| THF | 569 | 26100 |
— | — | — | — | 0.21 | −0.56 | −1.67 | |
| Toluene | 582 | 30700 |
— | — | — | — | ||||
| 14 | CH2Cl2 | 542 | 16500 |
680 | 53 | 138 | 3744 | |||
| THF | 563 | 11900 |
697 | 20 | 134 | 3415 | — | −0.58 | −1.68 | |
| Toluene | — | — | — | — | — | — | ||||
In both the UV-vis absorption and emission spectra, the wavelength of maximum absorption (λmax) and emission (λem) red-shift by approximately 10 nm when comparing mono-benzyl-functionalized complex 8 and mono-alkyne-substituted complex 7. As may be expected, a more drastic red-shift was observed when the spectra of bis-benzyl-functionalized complex 12 and bis-alkyne-substituted complex 11 were compared. In this case, red-shifts of approximately 30 nm (absorption) and 40 nm (emission) were observed (Fig. 4 and Table 1).
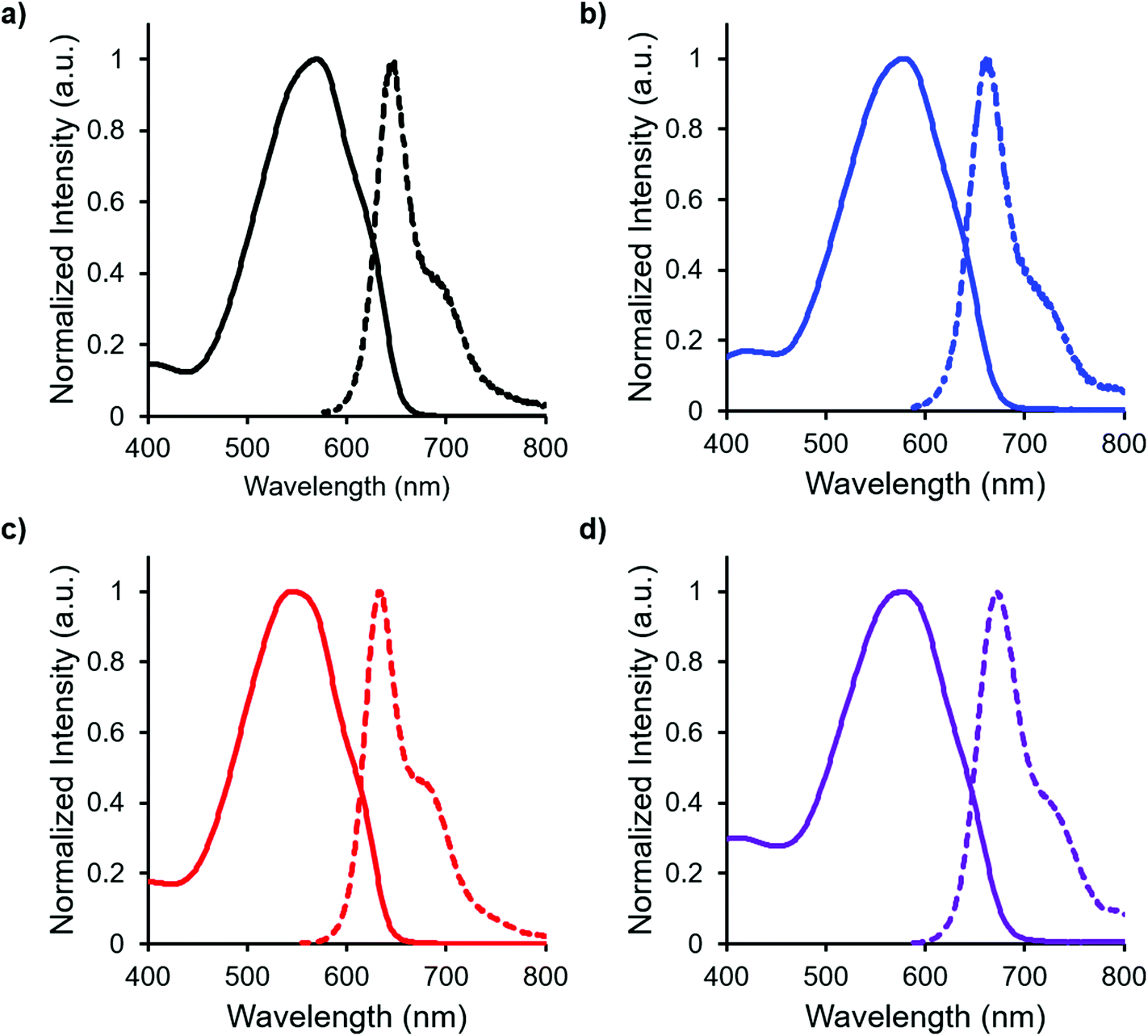 | ||
| Fig. 4 UV-vis absorption spectra (solid line) and emission spectra (dashed line) of (a) mono-alkyne-substituted complex 7, (b) mono-benzyl-functionalized complex 8, (c) bis-alkyne-substituted complex 11 and (d) bis-benzyl-functionalized complex 12, recorded for 10−5 M toluene solutions. | ||
Complexes 7, 8, 11 and 12 exhibit moderate fluorescence quantum yields in THF, CH2Cl2, and toluene (Table 1). Mono-alkyne-substituted complex 7 has a quantum yield of 30% in CH2Cl2, which is almost double that of bis-alkyne-substituted complex 11 (ΦF = 18% in CH2Cl2), likely due to its asymmetric structure.67 Mono-benzyl-functionalized complex 8 has the same quantum yield as complex 7, while bis-benzyl-functionalized complex 12 showed the most intense emission at a quantum yield of 46%. The Stokes shifts observed (79–133 nm; 2143–3394 cm−1) for these complexes were typical of other BF2 complexes of 3-cyanoformazanates.57
Ferrocene-functionalized BF2 complexes of 3-cyanoformazanates
Complexes 9 and 13, which bear one or two ferrocene substituents, are dark purple/blue (Fig. S19 and S20†) and non-emissive in solution. The lack of emission is likely due to excited-state electron transfer from the BF2 formazanate moieties to the ferrocenes.70–73 In order to further explore the apparent fluorescence quenching, we sequentially oxidized the ferrocene units in 9 and 13 and studied their absorption and emission properties in CH2Cl2 (Fig. 5). NOBF4 was chosen as an oxidant as the gaseous byproducts were assumed not to contribute to the spectra collected upon oxidation and the oxidation potential (ca. 1.0 V relative to the ferrocene/ferrocenium redox couple)74 was not in a range that would oxidize the 3-cyanoformazanate ligand backbone. In CH2Cl2, the absorption maxima associated with the π → π* transition of the BF2 formazanate backbone of complexes 9 and 13 were blue-shifted, by up to 20 nm in complex 13, upon sequential addition of NOBF4. The blue-shifts appear to arise due to the removal of electron density from the ferrocene moieties upon oxidation, rendering the complexes less electron rich overall. This effect has been observed previously for similar complexes, whereby the introduction of electron withdrawing groups blue-shifted absorption maxima.57 Perhaps more significant, was the observation that, upon conversion of ferrocene to ferrocenium, the solutions of 9 and 13 that had been treated with NOBF4 became emissive. The emission intensity gradually increased with the subsequent addition of oxidizing agent until a maximum intensity (ΦF = 7% for 9 and 14% for 13) was reached when a full equivalent of NOBF4 per ferrocene unit had been added, while λem was unchanged. These observations corroborate our hypothesis that the emission of these complexes was quenched as a result of excited-state electron transfer from the formazanate backbone to ferrocene.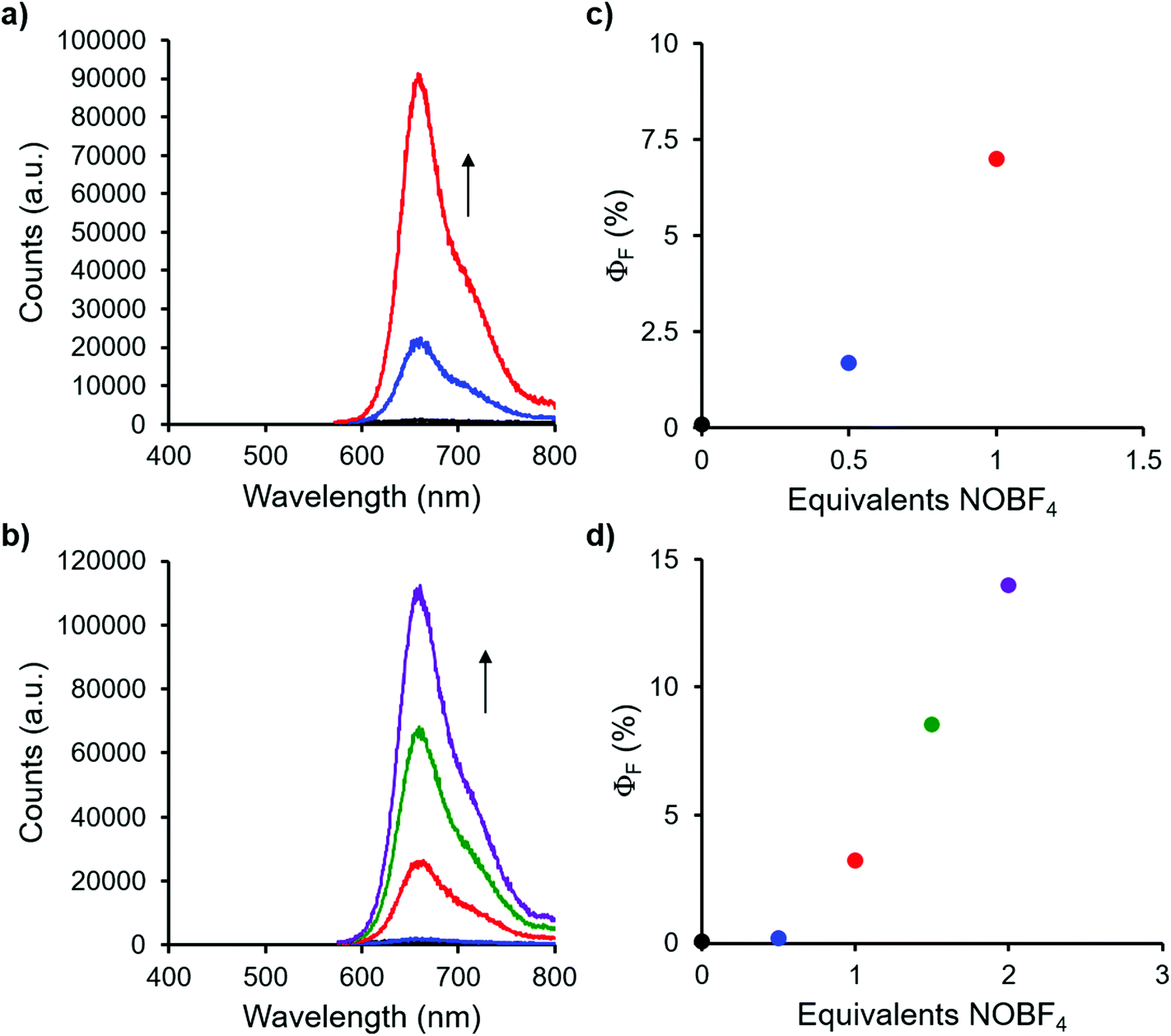 | ||
| Fig. 5 Emission spectra of solutions of ferrocene-substituted complexes (a) 9 and (c) 13 treated with 0 equiv. (black), 0.5 equiv. (blue), 1.0 equiv. (red), 1.5 equiv. (green) and 2.0 equiv. (purple) of NOBF4, recorded in degassed CH2Cl2 at a concentration of 10−5 M. The arrows indicate the trends upon addition of NOBF4. The corresponding quantum yields for each solution are plotted in panels (b) 9 and (d) 13. | ||
BF2 complexes of formazanate ligands generally exhibit very well-behaved reduction chemistry.56–61 However, their oxidation is rarely observed within the electrochemical window of most organic solvents. Ferrocene can be reversibly oxidized, and so the redox properties of compounds that contain both ferrocene and formazanate moieties may have unusual properties, including charge-transfer characteristics. Similar compounds based on different fluorophores have been previously used as viscosity probes,75 redox-active fluorescent switches,70,76 and ion pair recognition receptors.77
Cyclic voltammetry studies of complexes 9 and 13 revealed two characteristic BF2 formazanate reduction waves, and indicated that each complex was more difficult to reduce than the alkyne-substituted precursors (7 and 11), due to the electron-rich nature of ferrocene (Table 1). Oxidation waves corresponding to one electron for 9 and two electrons for 13 were observed at a potentials of 0.20 V and 0.21 V, which corresponds to the oxidation of the ferrocene groups (Fig. 6). The coincident appearance of the ferrocene waves in the cyclic voltammogram of 13 confirmed that there was little to no electronic communication between the ferrocene units via the BF2 formazanate spacer. This observation was consistent with those noted for polymers and model compounds derived from BF2 complexes of triarylformazanate ligands, whereby triazole groups were shown to limit the overall degree of electronic conjugation along the polymer backbone.65
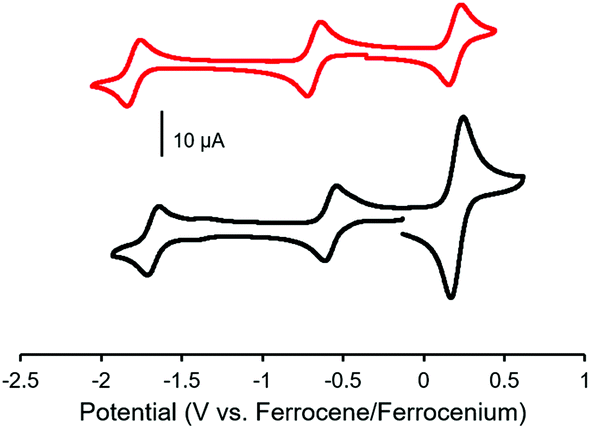 | ||
| Fig. 6 Cyclic voltammograms of mono-ferrocene substituted BF2 complex 9 (red) and bis-ferrocene substituted complex 13 (black) recorded at 100 mV s−1 in 1 mM THF solutions containing 0.1 M [nBu4N][PF6] as supporting electrolyte. | ||
Tetraetylene glycol-functionalized BF2 complexes of 3-cyanoformazanates
Complex 14, which was targeted in an effort to maximize hydrophilicity, is soluble in both polar organic solvents and water. Interestingly, the optical properties were highly dependent on the polarity of the solvent used. In relatively non-polar solvents such as CH2Cl2 and THF, 14 was moderately fluorescent (ΦF: 53% and 20%, respectively). However, the fluorescence intensity dropped off significantly in more polar solvents (ΦF: 5% in H2O), and there was also a blue-shift in the wavelength of maximum emission (Table 2, Fig. 7), as is often observed for fluorescent dyes in highly polar solvents.78–82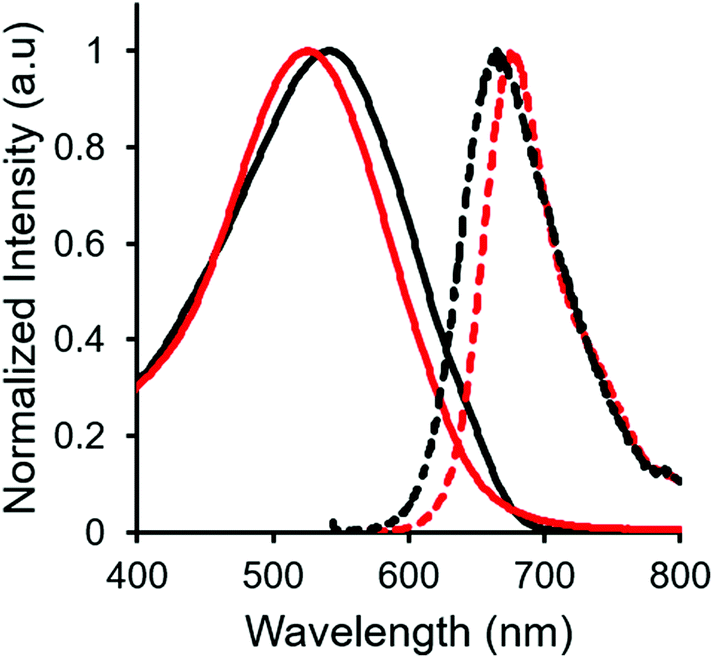 | ||
| Fig. 7 UV-vis absorption (solid line) and emission (dashed line) spectra of TEG-functionalized complex 14 in CH2Cl2 (black) and H2O (red), recorded for degassed solutions at a concentration of 10−5 M. | ||
| Solvent | λ max (nm) | ε (M−1 cm−1) | λ em (nm) |
Φ
Fa (%) |
ν ST (nm) | ν ST (cm−1) |
|---|---|---|---|---|---|---|
| a Quantum yields were measured according to published protocols using [Ru(bpy)3][PF6]2 as a relative standard and corrected for wavelength-dependent detector sensitivity (Fig. S18).68,69 | ||||||
| CH2Cl2 | 542 | 16500 |
680 | 53 | 138 | 3744 |
| THF | 563 | 11900 |
697 | 20 | 134 | 3415 |
| MeOH | 521 | 19900 |
681 | 6 | 160 | 4510 |
| MeCN | 524 | 18200 |
697 | 10 | 173 | 4737 |
| DMSO | 526 | 17600 |
716 | <1 | 190 | 5045 |
| H2O | 527 | 15100 |
698 | 5 | 171 | 4649 |
While the emission intensity of 14 was relatively low in water, many other fluorophores with relatively low quantum yields have shown promise as cell-imaging agents, especially dyes which emit in the red to near-IR region.83–87 With these factors in mind, complex 14 was introduced into mouse fibroblast cells and its utility as an imaging agent studied using fluorescence confocal microscopy.
Complex 14 was successfully incorporated into the cells, as evidenced by the fluorescence micrographs shown in Fig. 8. Specifically, complex 14 was distributed throughout the cell structure and clearly penetrated the cell nucleus. The dark spots visualized in Fig. 8c are believed to be DNA-free nucleoli, and appear to be the only features of the cells that were not stained by 14. Despite the widespread incorporation of complex 14 throughout the cell, Fig. 8b and d demonstrate our ability to differentiate between cytoplasm and nucleus when 4′,6-diamidino-2-phenylindole (DAPI), a selective nuclear stain, was employed concurrently. These results differed significantly when compared to previous studies involving BF2 complexes of 3-cyanoformazanates bearing p-anisole substituents.58 The hydrophobic nature of the anisole-based dye resulted in selective staining of the cell cytoplasm and required the use of DMSO to achieve cellular uptake.58 While we have not produced a site-specific imaging agent as part of this study, we have demonstrated our ability to create hydrophilic analogs of the parent complexes and shown that, in doing so, we can drastically alter their cellular uptake. This work sets the stage for future iterations whereby site-specific staining will be achieved through the introduction of hydrophilic, tailor-made peptide chains.
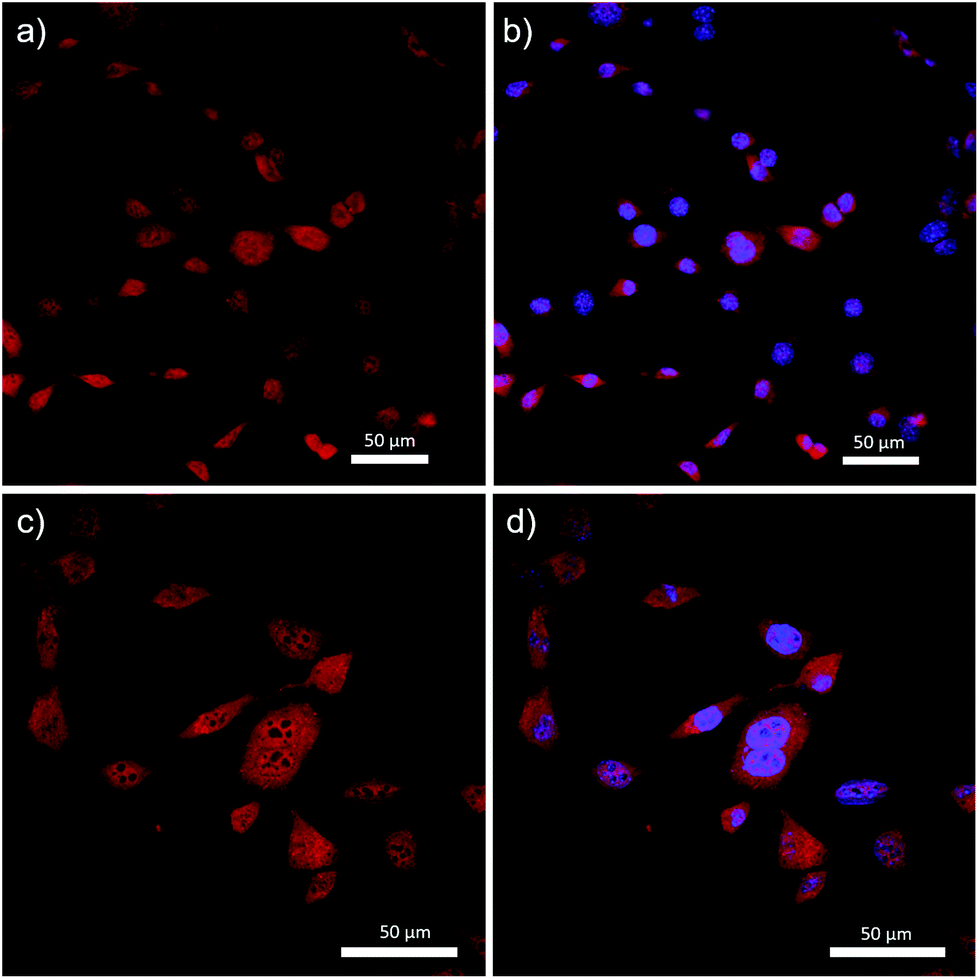 | ||
| Fig. 8 Confocal fluorescence micrographs of mouse fibroblast cells stained with TEG-functionalized complex 14 and DAPI. Images (a) and (c) were visualized with excitation at 579 nm and emission collected between 620–720 nm. Images (b) and (d) are an overlay of images (a) and (c) with those obtained from excitation at 405 nm and emission collected between 425–475 nm. | ||
Conclusions
As a result of this work, we have demonstrated the effect of triazole formation on the spectroscopic and electrochemical properties of BF2 complexes of 3-cyanoformazanates. Complexes functionalized with benzyl groups showed that the formation of triazole rings resulted in red-shifted wavelengths of maximum absorption and emission, as well as increased fluorescence quantum yields. Triazole formation also resulted in BF2 complexes that were more difficult to reduce electrochemically. We further demonstrated how CuAAC chemistry could be used to expand the scope of BF2 formazanate chemistry. The introduction of ferrocene into the BF2 formazanate scaffold via CuAAC resulted in the formation of a non-emissive complex with rich electrochemistry. Specifically, oxidation waves corresponding to one electron per ferrocene and two reduction waves (one electron each) originating from the formazanate backbones were observed by cyclic voltammetry. We also demonstrated that the stepwise conversion of ferrocene to ferrocenium was accompanied by an increase in emission intensity, confirming our hypothesis that quenching arose due to excited-state electron transfer from the formazanate backbone to ferrocene. Finally, CuAAC chemistry was used to append water-solubilizing TEG chains to a BF2 formazanate complex. The resulting complex was used to image mouse fibroblast cells, and demonstrated our ability to control cellular uptake via the modification of hydrophobicity/hydrophilicity.Experimental section
General considerations
Reactions and manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques unless otherwise stated. Solvents were obtained from Caledon Laboratories, dried using an Innovative Technologies Inc. solvent purification system, collected under vacuum and stored under a nitrogen atmosphere over 4 Å molecular sieves. Reagents were purchased from Sigma-Aldrich or Alfa Aesar and used as received. TEG-N388 and Fc-N389 were prepared according to literature procedures.
NMR spectra were recorded on 400 MHz (1H: 399.8 MHz, 11B: 128.3 MHz, 19F: 376.1 MHz) or 600 MHz (1H: 599.5 MHz, 13C{1H}: 150.8 MHz) Varian INOVA instruments. 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) or DMSO-d6 (2.50 ppm) and 13C{1H} NMR spectra were referenced to CDCl3 (77.2 ppm) or DMSO-d6 (39.5 ppm). 11B spectra were referenced to BF3·OEt2 at 0 ppm and 19F spectra were referenced to CFCl3 at 0 ppm. Mass spectrometry data were recorded in positive-ion mode using a high-resolution Finnigan MAT 8200 spectrometer using electron impact ionization or a Micromass LCT electrospray time-of-flight mass spectrometer. UV-vis absorption spectra were recorded using a Cary 5000 instrument. Four separate concentrations were run for each sample and molar extinction coefficients were determined from the slope of a plot of absorbance against concentration. FT-IR spectra were recorded on a KBr disk or using an attenuated total reflectance (ATR) attachment using a Bruker Vector 33 FT-IR spectrometer. Emission spectra were obtained using a Photon Technology International QM-4 SE spectrofluorometer. Excitation wavelengths were chosen based on λmax from the respective UV-vis absorption spectrum in the same solvent. Emission quantum yields were estimated relative to [Ru(bpy)3][PF6]2 and corrected for wavelength dependent detector sensitivity (Fig. S18†).68,69
Electrochemical methods
Cyclic voltammetry experiments were performed with a Bioanalytical Systems Inc. (BASi) Epsilon potentiostat and analyzed using BASi Epsilon software. Electrochemical cells consisted of a three-electrode setup including a glassy carbon working electrode, platinum wire counter electrode and silver wire pseudo reference electrode. Experiments were run at scan rates of 100 mV s−1 in degassed THF solutions of the analyte (∼1 mM) and supporting electrolyte (0.1 M nBu4PF6). Cyclic voltammograms were referenced against an internal standard (∼1 mM ferrocene) and corrected for internal cell resistance using the BASi Epsilon software.X-ray crystallography details
Single crystals of mono-alkyne-substituted complex 7 suitable for X-ray diffraction studies were grown by slow evaporation of a concentrated CH2Cl2 solution. The sample was mounted on a MiTeGen polyimide micromount with a small amount of Paratone N oil. X-ray measurements were made on a Bruker Kappa Axis Apex2 diffractometer at a temperature of 110 K. The data collection strategy included a number of ω and φ scans which collected data over a range of angles, 2θ. The frame integration was performed using SAINT.90 The resulting raw data was scaled and absorption corrected using a multi-scan averaging of symmetry equivalent data using SADABS.91 The structure was solved by using a dual space methodology using the SHELXT program.92 All non-hydrogen atoms were obtained from the initial solution. The hydrogen atoms were introduced at idealized positions and were allowed to refine isotropically. The structural model was fit to the data using full matrix least-squares based on F2. The calculated structure factors included corrections for anomalous dispersion from the usual tabulation. The structure was refined using the SHELXL-2014 program from the SHELXT suite of crystallographic software.93 See Table 3 and CCDC 1500180 for additional crystallographic data.| 7 | |
|---|---|
| a R 1 = ∑(|Fo| − |Fc|)/∑Fo. b wR2 = [∑(w(Fo2 − Fc2)2)/∑(wFo4)]1/2. c GOF = [∑(w(Fo2 − Fc2)2)/(no. of reflns. − no. of params.)]1/2. | |
| Chemical formula | C17H12BF2N5O |
| FW (g mol−1) | 351.13 |
| Crystal habit | Purple prism |
| Crystal system | Monoclinic |
| Space group | C2/c |
| T (K) | 110 |
| λ (Å) | 0.71073 |
| a (Å) | 10.248(6) |
| b (Å) | 14.634(7) |
| c (Å) | 11.146(5) |
| α (°) | 90 |
| β (°) | 107.352(18) |
| γ (°) | 90 |
| V (Å3) | 1595.5(14) |
| Z | 4 |
| ρ (g cm−3) | 1.462 |
| μ (cm−1) | 0.111 |
|
R
1,a wR2b [I > 2σ] |
0.0428, 0.0963 |
| R 1, wR2 (all data) | 0.0770, 0.1107 |
| GOFc | 1.038 |
Cell imaging studies
Mouse fibroblast C3H/10T1/2 cells (ATCC) were released from the tissue culture flask and seeded onto cover slips in a 12-well tissue culture plate at approximately 50000 cells per well. The cells were incubated overnight in Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich) containing 10% fetal bovine serum (FBS, Sigma-Aldrich) with penicillin streptomycin at 37 °C in a 5% CO2 atmosphere. Cell media was aspirated and the cells were washed once with serum free media and twice with phosphate buffer saline (PBS, Sigma-Aldrich). Water-soluble complex 14 (8 mg) was dissolved into 5 mL DMEM (serum free), filter sterilized with a 0.2 μm filter and diluted to 100 μM with serum free DMEM. The dye stock was incubated at 37 °C for 1 h with the cells. The cells were washed twice with PBS, fixed with 4% paraformaldehyde and mounted onto slides containing Pro-Long Antifade mounting medium with DAPI (ThermoFisher Scientific). Fluorescence microscopy images were obtained using an Olympus FluoView FV 1000 confocal microscope. Images based on the fluorescence of DAPI were obtained using 2% laser strength, with excitation at 405 nm and emission collected between 425 and 475 nm. Images based on the fluorescence of complex 14 were obtained using 10% laser strength, with excitation at 579 nm and emission collected between 620 and 720 nm.
Mono-alkyne-substituted formazan 6
In air, cyanoacetic acid (1.00 g, 11.7 mmol) and NaOH (4.70 g, 117 mmol) were mixed with deionized H2O (60 mL) and the solution was stirred in an ice bath for 20 min. Meanwhile, in a separate flask, 4-ethynylaniline (1.10 g, 9.36 mmol) was mixed with 12 M HCl (2.3 mL, 28 mmol) in deionized H2O (2.3 mL). The solution was cooled in an ice bath for 15 min before a cooled solution of sodium nitrite (0.75 g, 10 mmol) in deionized H2O (7 mL) was added dropwise. The resulting reaction mixture, which contained the corresponding diazonium salt, was stirred in an ice bath for an additional 20 min. In a separate flask, p-anisidine (1.4 g, 12 mmol) was mixed with 12 M HCl (2.9 mL, 35 mmol) in deionized H2O (2.9 mL). The solution was cooled in an ice bath for 15 min before a cooled solution of sodium nitrite (0.93 g, 13 mmol) in deionized H2O (8 mL) was added dropwise. The resulting reaction mixture, which contained diazonium salt, was stirred in an ice bath for an additional 20 min. The diazonium salt solutions were then mixed together and stirred in an ice bath for 10 min. This solution was then added dropwise to the cyanoacetic acid solution described above. The solution turned dark red after approximately 2 min. After complete addition, the mixture was stirred in an ice bath for an additional 60 min before it was neutralized with 1 M HCl. The resulting red-brown solid was isolated by vacuum filtration and purified by flash chromatography using a gradient strategy (starting at 1:1 n-hexanes:CH2Cl2 and ending with 2:8 n-hexanes:CH2Cl2) where the second coloured fraction contained the desired product. Removal of the solvent in vacuo afforded mono-alkyne-substituted formazan 6 as a dark red microcrystalline solid. Yield = 0.89 g, 25%. M.p. 212–213 °C. 1H NMR (400.1 MHz, DMSO-d6): δ 12.49 (br s, 1H, NH), 8.08 (d, 3JHH = 9 Hz, 2H, aryl CH), 7.54 (d, 4H, aryl CH), 7.18 (d, 3JHH = 9 Hz, 2H, aryl CH), 4.19 (s, 1H, alkyne CH), 3.91 (s, 3H, OCH3).13C{1H} NMR (100.6 MHz, DMSO-d6): δ 163.2, 145.9, 142.5, 132.8, 126.9, 126.2, 116.6, 115.5, 114.8, 112.6, 83.3, 80.5, 55.7. FT-IR(ATR): 3308 (m), 3230 (m), 2942 (s), 2837 (s), 2224 (m), 2099 (m), 1605 (m), 1579 (m), 1514 (s), 1249 (s), 1183 (s), 1164 (s), 1140 (s), 1110 (m), 1028 (m) cm−1. UV-Vis (CH2Cl2): λmax 444 nm (ε = 26400 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C17H13N5O]+: 303.1120; exact mass found: 303.1111; difference: −3.0 ppm.
Mono-alkyne-substituted BF2 complex 7
Formazan 6 (0.40 g, 1.3 mmol) was dissolved in dry toluene (75 mL). NEt3 (0.40 g, 0.55 mL, 3.9 mmol) was then added slowly, and the solution was stirred for 10 min before BF3·OEt2 (0.94 g, 0.81 mL, 6.6 mmol) was added and the solution was heated to 80 °C with stirring for 16 h. The solution became dark purple during this time and after cooling to 22 °C, deionized H2O (10 mL) was added to quench any excess boron-containing compounds. The toluene solution was then washed with deionized H2O (3 × 20 mL), dried over MgSO4, gravity filtered, and concentrated in vacuo. The resulting compound was purified by flash chromatography (THF, neutral alumina) to afford mono-alkyne-substituted complex 7 as a dark-purple solid. Yield = 0.31 g, 68%. M.p. 199–201 °C. 1H NMR (399.8 MHz, CDCl3): δ 7.96 (d, 3JHH = 9 Hz, 2H, aryl CH), 7.85 (d, 3JHH = 9 Hz, 2H, aryl CH), 7.59 (d, 3JHH = 9 Hz, 2H, aryl CH), 7.00 (d, 3JHH = 9 Hz, 2H, aryl CH), 3.91 (s, 3H, OCH3), 3.27 (s, 1H, alkyne CH). 13C{1H} NMR (150.7 MHz, CDCl3): δ 163.0, 142.9, 136.8, 133.0, 125.3, 124.5, 122.5, 115.0, 114.0, 82.5, 80.7, 55.8. 11B NMR (128.3 MHz, CDCl3): δ −0.7 (t, 1JBF = 31 Hz). 19F NMR (376.1 MHz, CDCl3): δ −133.7 (q, 1JFB = 31 Hz). FT-IR (ATR): 3282 (m), 2928 (s), 2840 (s), 2240 (m), 1593 (s), 1505 (m), 1407 (s), 1343 (s), 1328 (s), 1307 (s), 1262 (s), 1166 (s), 1138 (s) cm−1. UV-Vis (CH2Cl2): λmax 552 nm (ε = 30400 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C17H12BF2N5O]+: 351.1103; exact mass found: 351.1108; difference: +1.4 ppm.
Mono-benzyl-functionalized complex 8
A solution of PMDETA (0.002 g, 0.003 mL, 0.01 mmol) in dry THF (2 mL) was degassed via 3 freeze–pump–thaw cycles before CuI (0.003 g, 0.014 mmol) was added and the resulting mixture was stirred for 15 min at 22 °C. Benzyl azide (0.04 g, 0.04 mL, 0.3 mmol) and mono-alkyne-substituted complex 7 (0.1 g, 0.3 mmol) were then added and the reaction mixture was stirred at 22 °C for 16 h. The THF solution was then purified by flash chromatography (THF, neutral alumina) and recrystallized from MeOH to afford mono-benzyl-functionalized complex 8 as a dark-purple microcrystalline solid. Yield = 0.14 g, 51%. M.p. 188–189 °C. 1H NMR (400.1 MHz, CDCl3): δ 7.95–7.93 (m, 6H, aryl CH), 7.75 (s, 1H,![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.42–7.40 (m, 3H, aryl CH), 7.34–7.32 (m, 2H, aryl CH), 7.00 (d, 3JHH = 9 Hz, 2H, aryl CH) 5.60 (s, 2H, CH2), 3.90 (s, 3H, CH2). 13C{1H} NMR (150.7 MHz, CDCl3): δ 162.7, 146.6, 142.6, 136.8, 134.4, 132.9, 129.2, 128.9, 128.1, 126.4, 125.2, 123.3, 120.5, 114.9, 114.3, 55.8, 54.3. 11B NMR (128.3 MHz, CDCl3): δ −0.7 (t, 1JBF = 29 Hz). 19F NMR (376.1 MHz, CDCl3): δ −134.2 (q, 1JFB = 29 Hz). FT-IR(ATR): 3123 (m), 2849 (m), 2250 (m), 1603 (s), 1509 (m), 1460 (m), 1375 (s), 1344 (s), 1326 (s), 1308 (s) cm−1. UV-Vis (CH2Cl2): λmax 560 nm (ε = 31000 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C24H19BF2N8O]+: 484.1743; exact mass found: 484.1759; difference: +3.3 ppm.
CH), 7.42–7.40 (m, 3H, aryl CH), 7.34–7.32 (m, 2H, aryl CH), 7.00 (d, 3JHH = 9 Hz, 2H, aryl CH) 5.60 (s, 2H, CH2), 3.90 (s, 3H, CH2). 13C{1H} NMR (150.7 MHz, CDCl3): δ 162.7, 146.6, 142.6, 136.8, 134.4, 132.9, 129.2, 128.9, 128.1, 126.4, 125.2, 123.3, 120.5, 114.9, 114.3, 55.8, 54.3. 11B NMR (128.3 MHz, CDCl3): δ −0.7 (t, 1JBF = 29 Hz). 19F NMR (376.1 MHz, CDCl3): δ −134.2 (q, 1JFB = 29 Hz). FT-IR(ATR): 3123 (m), 2849 (m), 2250 (m), 1603 (s), 1509 (m), 1460 (m), 1375 (s), 1344 (s), 1326 (s), 1308 (s) cm−1. UV-Vis (CH2Cl2): λmax 560 nm (ε = 31000 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C24H19BF2N8O]+: 484.1743; exact mass found: 484.1759; difference: +3.3 ppm.
Mono-ferrocene-functionalized BF2 complex 9
A solution of PMDETA (0.002 g, 0.002 mL, 0.01 mmol) in THF (3 mL) was degassed via 3 freeze–pump–thaw cycles before CuI (0.002 g, 0.01 mmol) was added and the resulting mixture was stirred for 30 min. Ferrocenyl azide (0.024 g, 0.11 mmol) and mono-alkyne-substituted complex 7 (0.037 g, 0.11 mmol) were then added and the reaction mixture was stirred at 50 °C for 18 h. Upon cooling to 22 °C, the resulting dark purple solution was filtered through a plug of celite and purified by flash chromatography (CH2Cl2, silica gel) to afford ferrocene-functionalized complex 9 as a dark purple powder. Yield = 0.04 g, 72%. Melting point not observed (m.p. >250 °C). 1H NMR (400.1 MHz, CDCl3): δ 8.06 (s, 1H, triazole CH), 8.02–7.95 (m, 6H, aryl CH), 7.01–7.00 (m, 2H, aryl CH), 4.90 (s, 2H, ferrocene CH), 4.32 (s, 2H, ferrocene CH), 4.26 (s, 6H, ferrocene CH), 3.91 (s, 3H, OCH3). 13C{1H} NMR (150.7 MHz, CDCl3): δ 162.9, 146.3, 142.9, 137.0, 132.9, 126.7, 125.4, 123.6, 120.0, 115.1, 114.4, 93.6, 70.4, 67.1, 62.4, 62.3, 56.0. 11B NMR (128.3 MHz, CDCl3): δ −0.7 (t, 1JBF = 30 Hz). 19F NMR (376.1 MHz, CDCl3): δ −134.1 (q, 1JFB = 30 Hz). FT-IR(ATR): 3092 (w), 2926 (m), 2842 (m), 2241 (m), 1597 (s), 1505 (m), 1342 (m), 1261 (s), 1167 (s), 1029 (m), 965 (m), 832 (m), 733 (s) cm−1. UV-Vis (CH2Cl2): λmax 562 nm (ε = 28600 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C27H21BF2FeN8O]+: 578.1249; exact mass found: 578.1261; difference: +2.1 ppm.
Bis-alkyne-substituted formazan 10
In air, cyanoacetic acid (0.47 g, 5.5 mmol) was dissolved in deionized H2O (75 mL) containing NaOH (2.20 g, 55 mmol). This colourless solution was stirred for 45 min in an ice bath. Meanwhile, 4-ethynylaniline (1.30 g, 12 mmol) was mixed with concentrated HCl (2.85 mL) in deionized H2O (30 mL). This solution was cooled in an ice bath for 10 min before a solution of sodium nitrite (1.01 g, 15 mmol) in deionized H2O (15 mL) was cooled in an ice bath, and then added slowly to the 4-ethynylaniline solution over a 10 min period. This mixture was stirred in an ice bath for 30 min, and then added slowly to the basic cyanoacetic acid solution. A dark red/orange colour persisted almost immediately, and a dark red/orange precipitate formed after a few min. The mixture was stirred in an ice bath for an additional 16 h before ethyl acetate (250 mL) was added and the organic layer was isolated, washed with deionized H2O (3 × 100 mL), dried over MgSO4, gravity filtered and concentrated in vacuo. The resulting residue was purified by flash chromatography (CH2Cl2, neutral alumina) to afford bis-alkyne-substituted formazan 7 as a dark red solid. Yield = 1.55 g, 94%. Melting point not observed (>250 °C). 1H NMR (599.5 MHz, CDCl3) δ 12.60 (s, 1H, NH), 7.60 (s, 8H, aryl CH), 3.24 (s, 2H, alkyne CH). 13C{1H} NMR (100.6 MHz, DMSO-d6): δ 146.8, 133.0, 126.9, 121.7, 120.2, 112.5, 83.2, 82.8. FT-IR (KBr): 3397 (br, s), 3261 (m), 2915 (m), 2850 (m), 2221 (s), 1653 (s), 1540 (m), 1268 (m), 1193 (m), 1142 (m) cm−1. UV-vis (CH2Cl2): λmax = 434 nm (ε = 22800 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C18H11N5]+: 297.1014; exact mass found: 297.1018; difference: +1.3 ppm.
Bis-alkyne-substituted BF2 complex 11
Formazan 10 (1.00 g, 3.40 mmol) was dissolved in dry toluene (100 mL). NEt3 (1.03 g, 1.42 mL, 10.2 mmol) was then added slowly and the solution was stirred for 10 min before BF3·OEt2 (2.39 g, 2.08 mL, 16.8 mmol) was added and the solution was heated with stirring at 80 °C for 18 h. The solution gradually turned from dark red to dark purple during this time. After cooling to 22 °C, deionized H2O (10 mL) was added to quench any excess reactive boron-containing compounds. The purple toluene solution was then washed with deionized H2O (3 × 50 mL), dried over MgSO4, gravity filtered and concentrated in vacuo. The resulting residue was purified by flash chromatography (CH2Cl2, neutral alumina) to afford bis-alkyne-substituted complex 11 as a dark purple microcrystalline solid. Yield = 0.82 g, 70%. Melting point = 218 °C (decomp.). 1H NMR (599.5 MHz, CDCl3) δ 7.91 (d, 3JHH = 9 Hz, 4H, aryl CH), 7.61 (d, 3JHH = 9 Hz, 4H, aryl CH), 3.32 (s, 2H, alkyne CH). 13C{1H} NMR (100.6 MHz, CDCl3): δ 143.0, 133.4, 126.0, 123.2, 113.8, 82.6, 81.8. 11B NMR (128.3 MHz, CDCl3): δ −0.8 (t, 1JBF = 31 Hz). 19F NMR (376.1 Hz, CDCl3): δ −132.0 (q, 1JFB = 31 Hz). FT-IR (KBr): 3290 (s), 3189 (m), 3108 (m), 3092 (m), 2244 (s), 1589 (s), 1391 (m), 1331 (s), 1187 (m), 1147 (s), 1033 (m), 958 (m), 838 (s) cm−1. UV-vis (CH2Cl2): λmax = 530 nm (ε = 32700 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C18H10N5BF2]+: 345.0997; exact mass found: 345.0999; difference: +0.6 ppm.
Bis-benzyl-functionalized BF2 complex 12
A solution PMDETA (0.0075 g, 0.0091 mL, 0.044 mmol) in THF (3 mL) was degassed via 3 freeze–pump–thaw cycles before CuI (0.0083 g, 0.044 mmol) was added and the resulting mixture was stirred for 30 min. Benzyl azide (0.174 g, 1.30 mmol) and bis-alkyne-substituted complex 11 (0.150 g, 0.44 mmol) were then added and the reaction mixture was stirred at 50 °C for 18 h. Upon cooling to 22 °C, the resulting dark purple solution was filtered through a plug of celite and purified by flash chromatography (CH2Cl2, silica gel) to afford bis-benzyl-functionalized complex 12 as a dark-purple solid. Yield = 0.21 g, 77%. Melting point not observed (>250 °C). 1H NMR (599.5 MHz, CDCl3) δ 7.99–7.94 (m, 8H, aryl CH), 7.76 (s, 2H, triazole CH), 7.43–7.38 (m, 6H, aryl CH), 7.34–7.33 (m, 4H, aryl CH), 5.60 (s, 4H, benzyl CH2). 13C{1H} NMR (150.7 MHz, CDCl3): δ 146.7, 142.8, 134.4, 133.8, 129.4, 129.2, 128.3, 126.7, 123.8, 120.7, 114.2, 54.6. 11B NMR (128.3 MHz, CDCl3): δ −0.7 (t, 1JBF = 29 Hz). 19F NMR (376.1 Hz, CDCl3): δ −133.1 (q, 1JFB = 29 Hz). FT-IR (KBr): 3109 (m), 3100 (s), 3035 (m), 2915 (m), 2246 (s), 1608 (s), 1497 (m), 1456 (s), 1326 (s), 1226 (m), 1074 (s), 1027 (s), 976 (m), 826 (m) cm−1. UV-vis (CH2Cl2): λmax = 558 nm (ε = 39600 M−1 cm−1). Mass Spec. (EI, +ve mode): exact mass calculated for [C32H25N11BF2]+: 612.2356; exact mass found: 612.2362; difference: +1.0 ppm.
Bis-ferrocene-functionalized BF2 complex 13
A solution of PMDETA (0.0019 g, 0.0023 mL, 0.011 mmol) in THF (4 mL) was degassed via 3 freeze–pump–thaw cycles before CuI (0.0021 g, 0.011 mmol) was added and the resulting mixture was stirred for 30 min. Ferrocenyl azide (0.050 g, 0.22 mmol) and bis-alkyne-substituted complex 11 (0.038 g, 0.11 mmol) were then added and the reaction mixture was stirred at 50 °C for 18 h. The resulting dark purple solution was filtered through a plug of celite and purified by flash chromatography (CH2Cl2, silica gel) to afford ferrocene-functionalized complex 13 as a dark purple powder. Yield = 0.080 g, 90%. Melting point not observed (>250 °C). 1H NMR (599.5 MHz, CDCl3) δ 8.08 (s, 2H, triazole CH), 8.05 (s, 8H, aryl CH), 4.91 (m, 4H, ferrocene CH), 4.34–4.33 (m, 4H, ferrocene CH), 4.28–4.26 (m, 10H, ferrocene CH). Due to the poor solubility of 13, and despite a saturated solution in THF-d8 being subjected to 10000 scans (8 h) on a 600 MHz spectrometer, a publication quality 13C{1H} NMR spectrum was not obtained. 11B NMR (128.3 MHz, CDCl3): δ −0.6 (t, 1JBF = 28 Hz). 19F NMR (376.1 Hz, CDCl3): δ −132.8 (q, 1JFB = 28 Hz). FT-IR (KBr): 3131 (w), 3092 (m), 2929 (m), 2246 (m), 1605 (s), 1381 (m), 1341 (s), 1226 (m), 1181 (s), 1027 (m), 966 (s), 816 (s) cm−1. UV-vis (CH2Cl2): λmax = 569 nm (ε = 30900 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C38H29N11BF2Fe2]+: 800.1367; exact mass found: 800.1380; difference: +1.6 ppm.
Bis-TEG-functionalized BF2 complex 14
A solution of PMDETA (0.008 g, 0.009 mL, 0.04 mmol) in THF (3 mL) was degassed via 3 freeze–pump–thaw cycles before CuI (0.008 g, 0.04 mmol) was added and the resulting solution was stirred for 30 min. TEG-azide (0.29 g, 1.3 mmol) and bis-alkyne-substituted complex 11 (0.15 g, 0.44 mmol) were then added and the reaction mixture was stirred at 50 °C for 18 h. The resulting dark purple solution was filtered through a plug of celite and purified by precipitation of a concentrated THF solution into n-hexanes thrice to yield a dark purple solid. Yield = 0.24 g, 71%. 1H NMR (599.5 MHz, CDCl3) δ 8.20 (s, 2H, triazole CH), 8.00 (s, 8H, aryl CH), 4.63–4.61 (m, 4H, ethylene glycol CH2), 3.93–3.90 (m, 4H, ethylene glycol CH2), 3.71–3.57 (m, 24H, ethylene glycol CH2). 13C{1H} NMR (150.7 MHz, CDCl3): 145.9, 142.4, 134.0, 126.5, 123.6, 122.6, 114.1, 72.4, 70.5, 70.4, 70.3, 70.1, 69.4, 61.5, 50.5 ppm. 11B NMR (128.3 MHz, CDCl3): δ −0.7 (t, 1JBF = 29 Hz). 19F NMR (376.1 Hz, CDCl3): δ −133.1 (q, 1JFB = 29 Hz). FT-IR (KBr): 3132 (br), 3317 (w), 2915 (m), 2868 (m), 2242 (w), 1604 (s), 1527 (w), 1461 (m), 1360 (m), 1333 (s), 1227 (m), 1179 (s), 1121 (s), 1069 (s), 1029 (s), 967 (s), 845 (m), 765 (w) cm−1. UV-vis (CH2Cl2): λmax = 542 nm (ε = 16500 M−1 cm−1). UV-vis (H2O): λmax = 527 nm (ε = 15100 M−1 cm−1). Mass Spec. (ESI, +ve mode): exact mass calculated for [C34H45N11O8BF2]+: 784.3514; exact mass found: 784.3540; difference: +3.3 ppm.
Acknowledgements
We would like to thank the University of Western Ontario, the Natural Science and Engineering Research Council (NSERC) of Canada (J. B. G.: DG, 435675, L. G. L.: DG, 326972 and S. M. B.: CGS D scholarship), the Ontario Ministry of Research and Innovation (J. B. G.: ERA, ER14-10-147) and the Canada Foundation for Innovation (J. B. G.: JELF, 33977) for funding this work. Finally we thank Prof. Elizabeth R. Gillies and Prof. Mark S. Workentin for access to instrumentation in their labs.Notes and references
- S. Weiss, Science, 1999, 283, 1676–1683 CrossRef CAS PubMed.
- J. Eid, A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. deWinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach and S. Turner, Science, 2009, 323, 133–138 CrossRef CAS PubMed.
- B. Huang, W. Wang, M. Bates and X. Zhuang, Science, 2008, 319, 810–813 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- E. Kim, Y. Lee, S. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- Z. Yang, A. Sharma, J. Qi, X. Peng, D. Y. Lee, R. Hu, D. Lin, J. Qu and J. S. Kim, Chem. Soc. Rev., 2016, 45, 4651–4667 RSC.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2014, 9, 855–866 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef.
- Q. Liu and Y. Tor, Org. Lett., 2003, 5, 2571–2572 CrossRef CAS PubMed.
- B. Khanna, S. R. Parkin and K. D. Revell, Tetrahedron Lett., 2012, 53, 6383–6387 CrossRef CAS.
- O. Karagollu, M. Gorur, F. Gode, B. Sennik and F. Yilmaz, Macromol. Chem. Phys., 2015, 216, 939–949 CrossRef CAS.
- R. Chopra, P. Kaur and K. Singh, Anal. Chim. Acta, 2015, 864, 55–63 CrossRef CAS PubMed.
- B. Brizet, V. Goncalves, C. Bernhard, P. D. Harvey, F. Denat and C. Goze, Chem. – Eur. J., 2014, 20, 12933–12944 CrossRef CAS PubMed.
- B. Dhokale, T. Jadhav, S. M. Mobin and R. Misra, Dalton Trans., 2015, 44, 15803–15812 RSC.
- H. R. A. Golf, H.-U. Reissig and A. Wiehe, Org. Lett., 2015, 17, 982–985 CrossRef CAS PubMed.
- X. Chen, Q. Wu, L. Henschke, G. Weber and T. Weil, Dyes Pigm., 2012, 94, 296–303 CrossRef CAS.
- P. Gobbo, P. Gunawardene, W. Luo and M. S. Workentin, Synlett, 2015, 1169–1174 CrossRef CAS.
- Y. Su, L. Li, H. Wang, X. Wang and Z. Zhang, Chem. Commun., 2016, 52, 2185–2188 RSC.
- V. Evans, P. Duncanson, M. Motevalli and M. Watkinson, Dyes Pigm., 2016, 135, 36–40 CrossRef CAS.
- M. Albrecht, A. Lippach, M. P. Exner, J. Jerbi, M. Springborg, N. Budisa and G. Wenz, Org. Biomol. Chem., 2015, 13, 6728–6736 CAS.
- S. Guo, L. Ma, J. Zhao, B. Küçüköz, A. Karatay, M. Hayvali, H. G. Yaglioglu and A. Elmali, Chem. Sci., 2014, 5, 489–500 RSC.
- C. Beyer and H.-A. Wagenknecht, J. Org. Chem., 2010, 75, 2752–2755 CrossRef CAS PubMed.
- S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2012, 77, 5–16 CrossRef CAS PubMed.
- Z. Liu, Y. Li, J. Lozada, P. Schaffer, M. J. Adam, T. J. Ruth and D. M. Perrin, Angew. Chem., Int. Ed., 2013, 52, 2303–2307 CrossRef CAS PubMed.
- V. Martí-Centelles and P. D. Beer, Chem. – Eur. J., 2015, 21, 9397–9404 CrossRef PubMed.
- R. Arumugaperumal, V. Srinivasadesikan, M. V. Ramakrishnam Raju, M.-C. Lin, T. Shukla, R. Singh and H.-C. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 26491–26503 CAS.
- X. Ma, J. Zhang, J. Cao, X. Yao, T. Cao, Y. Gong, C. Zhao and H. Tian, Chem. Sci., 2016, 7, 4582–4588 RSC.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
- N. B. Yapici, S. Jockusch, A. Moscatelli, S. R. Mandalapu, Y. Itagaki, D. K. Bates, S. Wiseman, K. M. Gibson, N. J. Turro and L. Bi, Org. Lett., 2012, 14, 50–53 CrossRef CAS PubMed.
- K.-B. Li, H. Wang, Y. Zang, X.-P. He, J. Li, G.-R. Chen and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 19600–19605 CAS.
- R. S. Singh, R. K. Gupta, R. P. Paitandi, A. Misra and D. S. Pandey, New J. Chem., 2015, 39, 2233–2239 RSC.
- E. Manandhar, J. H. Broome, J. Myrick, W. Lagrone, P. J. Cragg and K. J. Wallace, Chem. Commun., 2011, 47, 8796–8798 RSC.
- S. A. Ingale and F. Seela, J. Org. Chem., 2012, 77, 9352–9356 CrossRef CAS PubMed.
- T. Uppal, N. V. S. D. K. Bhupathiraju and M. G. H. Vicente, Tetrahedron, 2013, 69, 4687–4693 CrossRef CAS.
- M. Sun, K. Müllen and M. Yin, Chem. Soc. Rev., 2016, 45, 1513–1528 RSC.
- P. Kele, X. Li, M. Link, K. Nagy, A. Herner, K. Lőrincz, S. Béni and O. S. Wolfbeis, Org. Biomol. Chem., 2009, 7, 3486–3490 CAS.
- M. Zhou, X. Zhang, M. Bai, D. Shen, B. Xu, J. Kao, X. Ge and S. Achilefu, RSC Adv., 2013, 3, 6756–6758 RSC.
- J. H. Kwak, Y. He, B. Yoon, S. Koo, Z. Yang, E. J. Kang, B. H. Lee, S.-Y. Han, Y. C. Yoo, K. B. Lee and J. S. Kim, Chem. Commun., 2014, 50, 13045–13048 RSC.
- X.-t. Zhang, Z.-y. Gu, L. Liu, S. Wang and G.-w. Xing, Chem. Commun., 2015, 51, 8606–8609 RSC.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- R. Travieso-Puente, M.-C. Chang and E. Otten, Dalton Trans., 2014, 43, 18035–18041 RSC.
- M.-C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118–4122 CrossRef CAS PubMed.
- E. Kabir, C.-H. Wu, J. I.-C. Wu and T. S. Teets, Inorg. Chem., 2016, 55, 956–963 CrossRef CAS PubMed.
- W. Schorn, D. Grosse-Hagenbrock, B. Oelkers and J. Sundermeyer, Dalton Trans., 2016, 45, 1201–1207 RSC.
- A. Mandal, B. Schwederski, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 8126–8135 CrossRef CAS PubMed.
- R. Travieso-Puente, J. O. P. Broekman, M.-C. Chang, S. Demeshko, F. Meyer and E. Otten, J. Am. Chem. Soc., 2016, 138, 5503–5506 CrossRef CAS PubMed.
- N. A. Protasenko, A. I. Poddel'sky, A. S. Bogomyakov, G. K. Fukin and V. K. Cherkasov, Inorg. Chem., 2015, 54, 6078–6080 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Organometallics, 2016, 35, 534–542 CrossRef CAS.
- J. B. Gilroy, M. J. Ferguson, R. McDonald, B. O. Patrick and R. G. Hicks, Chem. Commun., 2007, 126–128 RSC.
- M.-C. Chang and E. Otten, Chem. Commun., 2014, 50, 7431–7433 RSC.
- S. M. Barbon, P. A. Reinkeluers, J. T. Price, V. N. Staroverov and J. B. Gilroy, Chem. – Eur. J., 2014, 20, 11340–11344 CrossRef CAS PubMed.
- R. R. Maar, S. M. Barbon, N. Sharma, H. Groom, L. G. Luyt and J. B. Gilroy, Chem. – Eur. J., 2015, 21, 15589–15599 CrossRef CAS PubMed.
- S. M. Barbon, V. N. Staroverov and J. B. Gilroy, J. Org. Chem., 2015, 80, 5226–5235 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Inorg. Chem., 2015, 54, 8656–8664 CrossRef CAS PubMed.
- M.-C. Chang, A. Chantzis, D. Jacquemin and E. Otten, Dalton Trans., 2016, 45, 9477–9484 RSC.
- M. Hesari, S. M. Barbon, V. N. Staroverov, Z. Ding and J. B. Gilroy, Chem. Commun., 2015, 51, 3766–3769 RSC.
- R. R. Maar and J. B. Gilroy, J. Mater. Chem. C, 2016, 4, 6478–6482 RSC.
- S. Novoa, J. A. Paquette, S. M. Barbon, R. R. Maar and J. B. Gilroy, J. Mater. Chem. C, 2016, 4, 3987–3994 RSC.
- S. M. Barbon and J. B. Gilroy, Polym. Chem., 2016, 7, 3589–3598 RSC.
- J. B. Gilroy, P. O. Otieno, M. J. Ferguson, R. McDonald and R. G. Hicks, Inorg. Chem., 2008, 47, 1279–1286 CrossRef CAS PubMed.
- S. M. Barbon, J. T. Price, P. A. Reinkeluers and J. B. Gilroy, Inorg. Chem., 2014, 53, 10585–10593 CrossRef CAS PubMed.
- S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260–1264 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- M. Li, Z. Guo, W. Zhu, F. Marken and T. D. James, Chem. Commun., 2015, 51, 1293–1296 RSC.
- M. R. Rao, K. V. P. Kumar and M. Ravikanth, J. Organomet. Chem., 2010, 695, 863–869 CrossRef CAS.
- B. Dhokale, P. Gautam, S. M. Mobin and R. Misra, Dalton Trans., 2013, 42, 1512–1518 RSC.
- R. Misra, B. Dhokale, T. Jadhav and S. M. Mobin, Dalton Trans., 2013, 42, 13658–13666 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- X. Yin, Y. Li, Y. Zhu, X. Jing, Y. Li and D. Zhu, Dalton Trans., 2010, 39, 9929–9935 RSC.
- J. Rochford, A. D. Rooney and M. T. Pryce, Inorg. Chem., 2007, 46, 7247–7249 CrossRef CAS PubMed.
- F. Otón, M. d. C. González, A. Espinosa, C. Ramírez de Arellano, A. Tárraga and P. Molina, J. Org. Chem., 2012, 77, 10083–10092 CrossRef PubMed.
- S. Tobita, K. Ida and S. Shiobara, Res. Chem. Intermed., 2001, 27, 205–218 CrossRef CAS.
- P. J. Sadkowski and G. R. Fleming, Chem. Phys., 1980, 54, 79–89 CrossRef CAS.
- H. Nakamura and J. Tanaka, Chem. Phys. Lett., 1981, 78, 57–60 CrossRef CAS.
- L. Quan, Y. Chen, X.-J. Lv and W.-F. Fu, Chem. – Eur. J., 2012, 18, 14599–14604 CrossRef CAS PubMed.
- J. Oshima, T. Yoshihara and S. Tobita, Chem. Phys. Lett., 2006, 423, 306–311 CrossRef CAS.
- C. Patra, A. K. Bhanja, A. Mahapatra, S. Mishra, K. D. Saha and C. Sinha, RSC Adv., 2016, 6, 76505–76513 RSC.
- B. Zhou, W. Liu, H. Zhang, J. Wu, S. Liu, H. Xu and P. Wang, Biosens. Bioelectron., 2015, 68, 189–196 CrossRef CAS PubMed.
- W. Liu, B. Zhou, G. Niu, J. Ge, J. Wu, H. Zhang, H. Xu and P. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 7421–7427 CAS.
- M. V. Werrett, P. J. Wright, P. V. Simpson, P. Raiteri, B. W. Skelton, S. Stagni, A. G. Buckley, P. J. Rigby and M. Massi, Dalton Trans., 2015, 44, 20636–20647 RSC.
- Z. Yang, C. Yan, Y. Chen, C. Zhu, C. Zhang, X. Dong, W. Yang, Z. Guo, Y. Lu and W. He, Dalton Trans., 2011, 40, 2173–2176 RSC.
- P. Gobbo, W. Luo, S. J. Cho, X. Wang, M. C. Biesinger, R. H. E. Hudson and M. S. Workentin, Org. Biomol. Chem., 2015, 13, 4605–4612 CAS.
- A. C. Pauly, C. D. Varnado, C. W. Bielawski and P. Theato, Macromol. Rapid Commun., 2014, 35, 210–213 CrossRef CAS PubMed.
- Bruker-AXS, SAINT version 2013.8, Bruker-AXS, Madison, WI 53711, USA, 2013 Search PubMed.
- Bruker-AXS, SADABS version 2012.1, Bruker-Nonius, Madison, WI 53711, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1500180. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00640j |
| This journal is © the Partner Organisations 2017 |