
Charge mobility enhancement for diketopyrrolopyrrole-based conjugated polymers by partial replacement of branching alkyl chains with linear ones†
Jing
Ma
ab,
Zitong
Liu
 *a,
Zhijie
Wang
ab,
Yizhou
Yang
ab,
Guanxin
Zhang
a,
Xisha
Zhang
ab and
Deqing
Zhang
*ab
*a,
Zhijie
Wang
ab,
Yizhou
Yang
ab,
Guanxin
Zhang
a,
Xisha
Zhang
ab and
Deqing
Zhang
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: dqzhang@iccas.ac.cn; zitong_@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 8th September 2017
Abstract
Conjugated D–A polymers with high charge mobilities have received increasing attention in recent years. In this paper, we report a simple, but efficient approach to improve the charge mobilities of conjugated polymers by partial replacement of branching alkyl chains with linear ones. With this strategy in mind, we designed and investigated three new DPP-quaterthiophene terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3 containing both linear (n-dodecane) and branching (2-decyltetradecyl) alkyl chains with different ratios. On the basis of GIWAXS data, interchain packing order is improved for terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3, in comparison with PDPP4T with only branching alkyl chains. The incorporation of linear alkyl chains can affect the interchain packing mode, and PDPP4T-3 with more linear alkyl chains among the three terpolymers adopts the edge-on chain arrangement on the substrate. Field effect transistor measurements indicate that PDPP4T-1, PDPP4T-2 and PDPP4T-3 exhibit higher hole mobilities than PDPP4T. Moreover, the hole mobilities increase in the following order: PDPP4T-1 < PDPP4T-2 < PDPP4T-3 by increasing the content of linear alkyl chains in these terpolymers. The thin film mobility of PDPP4T-3 can reach 6.1 cm2 V−1 s−1, which can be further increased to 9.1 cm2 V−1 s−1 after incorporating NMe4I.
Introduction
Conjugated donor–acceptor (D–A) polymers exhibit promising semiconducting properties and thus they have received increasing attention in recent years.1–5 These conjugated D–A polymers have been successfully utilized to fabricate organic field-effect transistors (OFETs),6–12 organic photovoltaic cells (OPVs)13–16 and other devices with good performances.17,18 The electron donors and acceptors in the conjugated backbones are expected to influence the electronic structures, HOMO/LUMO levels and interchain interactions, thus they play a vital role in determining the optoelectronic properties of conjugated polymers in the solid state. For this reason, various electron donors and acceptors have been devised and employed for the construction of conjugated D–A polymers.2,3,19–24Meanwhile, recent studies have indicated that side alkyl chains in these conjugated polymers not only endow their good solubilities in organic solvents, but also affect the interchain packing and thin film morphology.25–27 As a result, side-chain engineering has become a useful approach to tune the interchain packing and thus improve semiconducting performances for conjugated polymers. For instance, charge mobility enhancement was observed for conjugated D–A polymers by modifying side alkyl chains.8,9,28–30 Apart from pure alkyl chains, side chains with functional groups, such as siloxane-terminated alkyl chains,31–34 urea-containing alkyl chains,7 fluoroalkyl chains35 and oligo(ethylene glycol) chains,36–38 have been linked to conjugated polymers, aiming to improve their semiconducting performances.
Because branching alkyl chains enable the conjugated polymers to possess good solubilities in organic solvents, most of the reported conjugated D–A polymers contain branching alkyl chains. However, branching alkyl chains can preclude the dense packing of polymer chains due to steric hindrance.25 One approach to reduce the steric hindrance of branching alkyl chains is to move the branching points away from the backbones. Pei and coworkers reported the charge mobility enhancement for isoindigo–bithiophene conjugated polymers by using this approach.28 Alternatively, linear alkyl chains can promote interchain interactions through interdigitations.39–41 Chen, Fréchet and co-workers reported furan-containing DPP polymers with either linear hexadecyl or branching 2-butyloctyl chains. The π–π stacking distance for the polymer with n-hexadecyl chains is shorter than that with 2-butyloctyl chains, and as a result the charge mobility was enhanced after replacing the 2-butyloctyl chains with linear ones.30 However, conjugated D–A polymers with linear chains are usually poorly soluble in organic solvents even under heating.
In this article, we report a new approach to weaken steric hindrance by partial replacement of bulky branching groups in the conjugated polymers with linear alkyl chains. With this idea in mind, we designed and investigated conjugated D–A terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3 (Scheme 1), which contain alternating DPP and quaterthiophene as electron acceptors and donors in their backbones. The selection of DPP and quaterthiophene units is based on the following considerations: (i) the monomers are either easily accessible or commercially available and (ii) the resulting conjugated D–A polymers such as PDPP4T show relatively high charge mobilities. The choice of 2-decyltetradecyl as branching side chains is intended to endow the polymer with better solubilities in organic solvents. As a result, more linear side chains can be incorporated in the polymers. As shown in Scheme 1, DPP units in PDPP4T-1, PDPP4T-2 and PDPP4T-3 are connected to either 2-decyltetradecyl chains or n-dodecane chains. The ratios of linear/branching chains are 1/20 for PDPP4T-1, 1/10 for PDPP4T-2, and 1/5 for PDPP4T-3. For comparison, PDPP4T was also prepared. The results reveal that (i) PDPP4T-1, PDPP4T-2 and PDPP4T-3 can be solution-processed as for PDPP4T; (ii) interchain packing order degree is improved for PDPP4T-1, PDPP4T-2 and PDPP4T-3 in comparison with PDPP4T, and (iii) thin film charge mobility is increased by incorporating more n-dodecane chains for these terpolymers, and the thin film of PDPP4T-3 shows the highest charge mobility of up to 6.1 cm2 V−1 s−1, which can further be improved to 9.1 cm2 V−1 s−1 after blending with tiny amounts of tetramethylammonium iodide.6
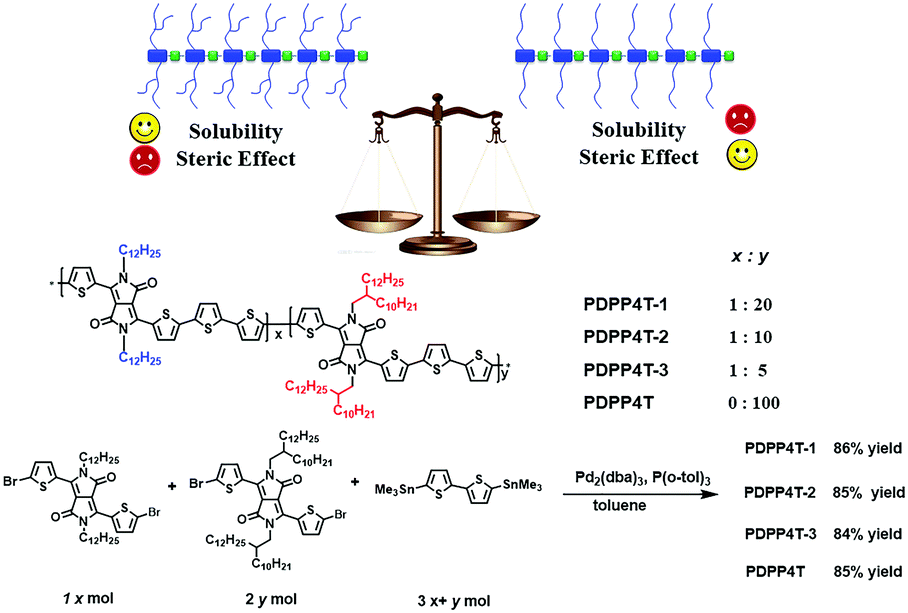 | ||
| Scheme 1 Illustration of the side-chain design rationale, and chemical structures of terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3 as well as PDPP4T and the synthetic approach. | ||
Results and discussion
Synthesis and characterization
Conjugated terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3 were prepared by co-polymerization of compounds 1, 2 and 3 under Stille coupling reaction conditions as for PDPP4T (Scheme 1). Compounds 1 and 2 were synthesized according to the reported procedures.42,43 By varying the feeding ratios between compounds 1 and 2, terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3, in which the ratios of the linear chains vs. the branching ones are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, 1:10 and 1:5, respectively, were obtained in 86%, 85% and 84% yields (see Experimental). For comparison, PDPP4T was prepared similarly in 85% yield.42,43 All polymers were purified by precipitation from methanol, followed by Soxhlet extraction sequentially with methanol, acetone and hexane to remove the remaining monomers and oligomers. The resulting polymers were extracted with chloroform and precipitated again from methanol, filtered, washed with methanol, and dried under vacuum at 45 °C for 48 h. The chemical structures of all polymers were verified by 1H NMR, solid state 13C NMR and elemental analysis (see the Experimental and ESI†).
:20, 1:10 and 1:5, respectively, were obtained in 86%, 85% and 84% yields (see Experimental). For comparison, PDPP4T was prepared similarly in 85% yield.42,43 All polymers were purified by precipitation from methanol, followed by Soxhlet extraction sequentially with methanol, acetone and hexane to remove the remaining monomers and oligomers. The resulting polymers were extracted with chloroform and precipitated again from methanol, filtered, washed with methanol, and dried under vacuum at 45 °C for 48 h. The chemical structures of all polymers were verified by 1H NMR, solid state 13C NMR and elemental analysis (see the Experimental and ESI†).
The presence of the 2-decyltetradecyl alkyl chain in these terpolymers endows PDPP4T-1, PDPP4T-2 and PDPP4T-3 with good solubility in hot CHCl3, 1,2-dichlorobenzene and other halogenated solvents. In comparison, the DPP-quaterthiophene conjugated D–A terpolymers with n-dodecane and 2-octyldodecane side chains display rather poor solubilities in 1,2-dichlorobenzene even at high temperature (>100 °C).7 The molecular weights of these terpolymers were determined with high temperature gel permeation chromatography in hot 1,3,5-trichlorobenzene at 150 °C. The Mns (number-average molecular weight) and DPIs (dispersion index) of PDPP4T-1, PDPP4T-2, PDPP4T-3 and PDPP4T were estimated to be 94 kg mol−1 (DPI = 2.3), 106 kg mol−1 (DPI = 2.1), 94 kg mol−1 (DPI = 1.8) and 86 kg mol−1 (DPI = 2.2), respectively. Thermal transitions were not detected for all polymers based on the differential scanning calorimetry (DSC) data (Fig. S1, ESI†), while thermal decompositions occurred at 366, 404 and 384 °C for PDPP4T-1, PDPP4T-2 and PDPP4T-3, respectively, based on the thermogravimetric analysis (TGA) data (Fig. S2, ESI†).
HOMO/LUMO energies and electronic absorptions
On the basis of their voltammograms (Fig. S3, ESI†), the HOMO/LUMO energy levels of PDPP4T-1, PDPP4T-2 and PDPP4T-3 were estimated to be −5.27/−3.53 eV, −5.26/−3.56 eV and −5.25/−3.57 eV, respectively. In comparison to those of PDPP4T, the HOMO energy levels of PDPP4T-1, PDPP4T-2 and PDPP4T-3 are slightly enhanced after incorporating more linear chains, whereas the LUMO levels are weakly lowered in the same order (see Table 1).| Polymer | λ max (nm) |
E
onsetredl
(V) |
E LUMO (eV) |
E
onsetoxl
(V) |
E HOMO (eV) |
E
cvg
(eV) |
E
optg
(eV) |
|
|---|---|---|---|---|---|---|---|---|
| Solution | Thin film | |||||||
| a Absorption maxima in 1,2-dichlorobenzene solution (1.0 × 10−5 M) and a spin-coated thin film. b Onset potentials (V vs. Fc/Fc+) for reduction (Eonsetredl) and oxidation (Eonsetoxl). c Estimated with the following equation: EHOMO = −(Eonsetoxl + 4.8) eV, ELUMO = −(Eonsetredl + 4.8) eV. d Based on redox potentials. e Based on the absorption spectral data. | ||||||||
| PDPP4T-1 | 782 | 720, 778 | −1.27 | −3.53 | 0.47 | −5.27 | 1.74 | 1.35 |
| PDPP4T-2 | 782 | 726, 784 | −1.24 | −3.56 | 0.46 | −5.26 | 1.70 | 1.35 |
| PDPP4T-3 | 788 | 728, 792 | −1.23 | −3.57 | 0.45 | −5.25 | 1.68 | 1.32 |
| PDPP4T | 782 | 718, 778 | −1.26 | −3.54 | 0.47 | −5.27 | 1.73 | 1.36 |
The solutions of PDPP4T-1, PDPP4T-2 and PDPP4T-3 show similar absorptions in the range of 500–1000 nm as PDPP4T as shown in Fig. 1. By increasing the content of linear chains in PDPP4T-1, PDPP4T-2 and PDPP4T-3, the shoulder absorptions at around 718–728 nm become gradually weak and the maximum absorptions are slightly red-shifted (Table 1). The absorption of PDPP4T-3 at 792 nm is red-shifted by 14 nm, in comparison to that of PDPP4T. Moreover, the optical band gaps of PDPP4T-1, PDPP4T-2 and PDPP4T-3 are lowered in comparison with that of PDPP4T, by increasing the number of linear chains in the polymers. These data imply that the conjugated backbones of PDPP4T-1, PDPP4T-2 and PDPP4T-3 become more planar by incorporating more linear chains.
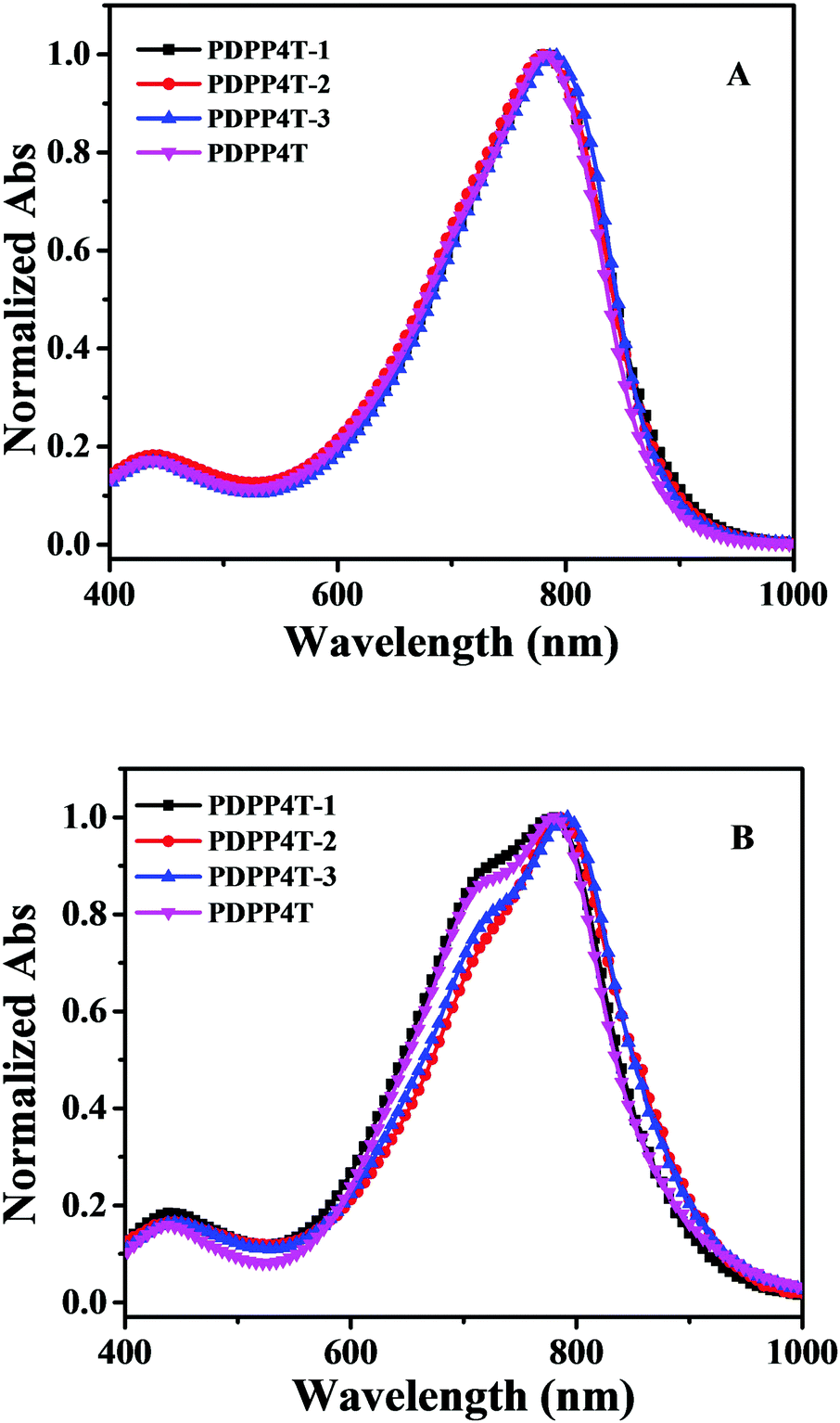 | ||
| Fig. 1 Normalized absorption spectra of PDPP4T-1, PDPP4T-2, PDPP4T-3 and PDPP4T in 1,2-dichlorobenzene solution (1.0 × 10−5 M) (A) and their thin films (B). | ||
Interchain packing and thin film morphology
Fig. 2 shows the out-of-plane and in-plane GIWAXS patterns for thin films of PDPP4T-1, PDPP4T-2 and PDPP4T-3 on OTS modified SiO2/Si substrates after thermal annealing at 160 °C. For comparison, the corresponding out-of-plane and in-plane GIWAXS patterns for a thin film of PDPP4T were also measured under the same conditions. In the out-of-plane direction, scattering signals up to 4th orders, which are owing to the lamellar packing of alkyl chains, were detected for PDPP4T-1, PDPP4T-2, PDPP4T-3 and PDPP4T. The scattering signals for PDPP4T-3 were detected at 4.2°, 8.2°, 12.3° and 17.8°, corresponding to a d-spacing of 21.01 Å. Similarly, the d-spacing owing to the lamellar stacking of alkyl chains was estimated to be 21.53 Å, 21.03 Å and 22.31 Å for PDPP4T-1, PDPP4T-2 and PDPP4T, respectively. It is interesting to note that the d-spacing was shortened in the following order: PDPP4T > PDPP4T-1 > PDPP4T-2 > PDPP4T-3. This can be attributed to the fact that the incorporation of more linear alkyl chains will enable the polymer chains to pack more closely through the interdigitations of linear alkyl chains. Furthermore, the intensities of these scattering signals are gradually enhanced by incorporating more linear alkyl chains in these terpolymers.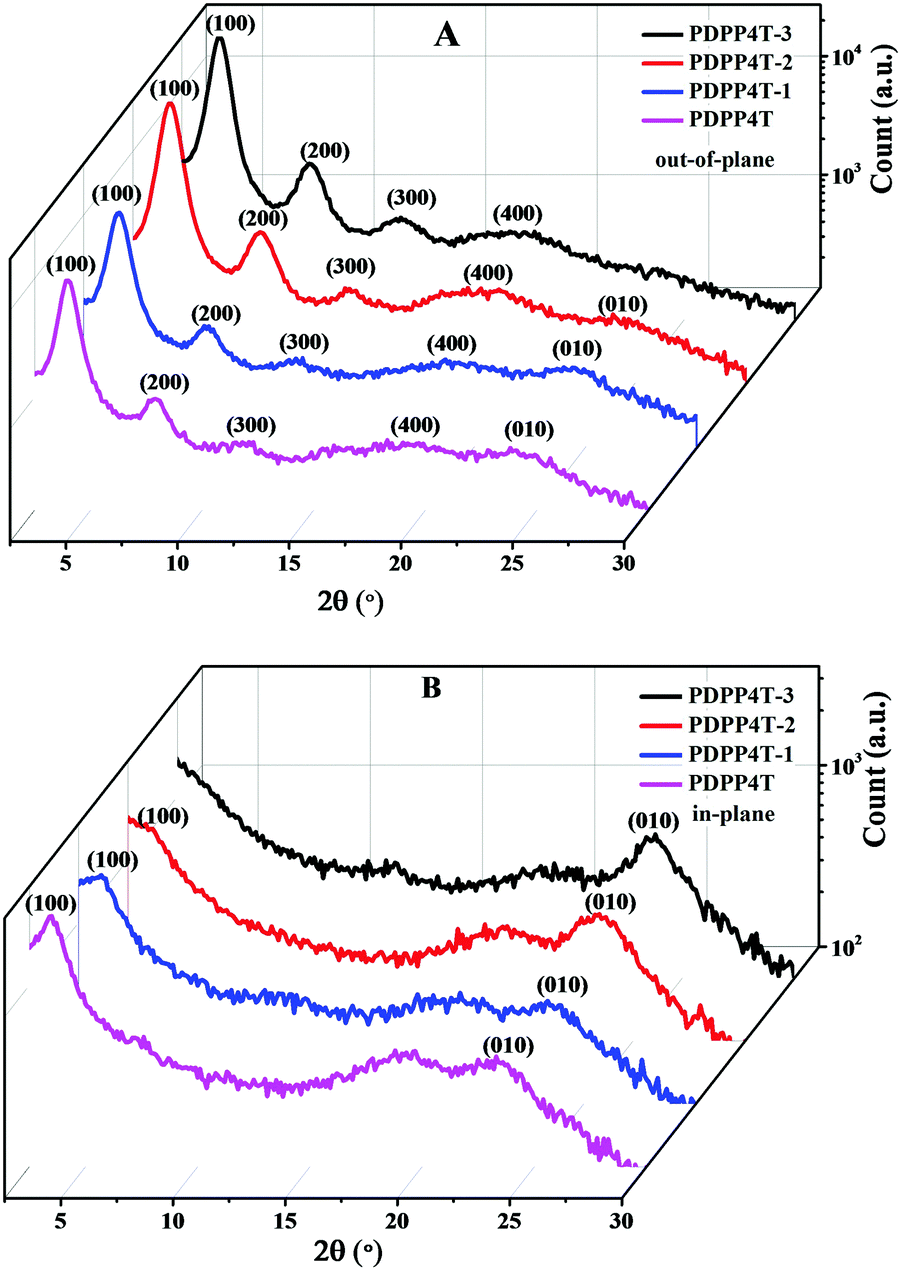 | ||
| Fig. 2 The out-of-plane (A) and in-plane (B) GIWAX patterns of thin films of PDPP4T-1, PDPP4T-2 and PDPP4T-3 as well as PDPP4T deposited on OTS-modified SiO2/Si substrates after thermal annealing at 160 °C. | ||
Apart from the scattering signals due to the lamellar stacking of alkyl chains, weak scattering signals at ∼24° (owing to interchain π–π stacking) were detected for thin films of PDPP4T-1, PDPP4T-2 and PDPP4T, but not for the thin film of PDPP4T-3, in the out-of-plane direction. PDPP4T-1, PDPP4T-2, PDPP4T-3 and PDPP4T exhibited scattering signals at 23.8°, 23.6°, 23.4° and 23.3°, respectively, in the in-plane direction. The corresponding π–π packing distances for PDPP4T-1, PDPP4T-2, PDPP4T-3 and PDPP4T were estimated to be 3.79 Å, 3.76 Å, 3.73 Å and 3.81 Å, respectively. Moreover, scattering signals at ∼3.5° (due to the lamellar stacking of alkyl chains) were also observed for PDPP4T and PDPP4T-1, and a shoulder signal at ∼3.5° was also observed for PDPP4T-2, but not for PDPP4T-3. These results can lead to the following conclusions: (i) the interchain π–π stacking distances become short after replacing more branching alkyl chains with linear alkyl chains, and (ii) the incorporation of linear alkyl chains can alter the arrangement of polymer chains on the substrate. For PDPP4T, PDPP4T-1 and PDPP4T-2 both edge-on and face-on packing modes coexist, whereas polymer chains of PDPP4T-3 are packed only in the edge-on mode. The variation of interchain packing order and packing mode is in good agreement with the observation that charge mobility is largely enhanced by replacing the branching alkyl chains with linear alkyl chains.
Fig. 3 shows AFM images of all polymer thin films after thermal annealing at 160 °C. Thin nanofibers were observed for the thin film of PDPP4T, which assembled into aggregates of different sizes. PDPP4T-1, PDPP4T-2 and PDPP4T-3 show a similar thin film morphology. Interestingly, the RRMSs (root-mean-square roughness) were found to be smaller with increasing linear chain content in the terpolymers. The RRMSs were 0.657 nm for PDPP4T-1, 0.500 nm for PDPP4T-2, 0.383 nm for PDPP4T-3, and 0.836 nm for PDPP4T. According to previous reports,44,45 the relatively large and continuous nanofiber-aggregates with uniform thin film morphology are in favor of charge carrier transport.
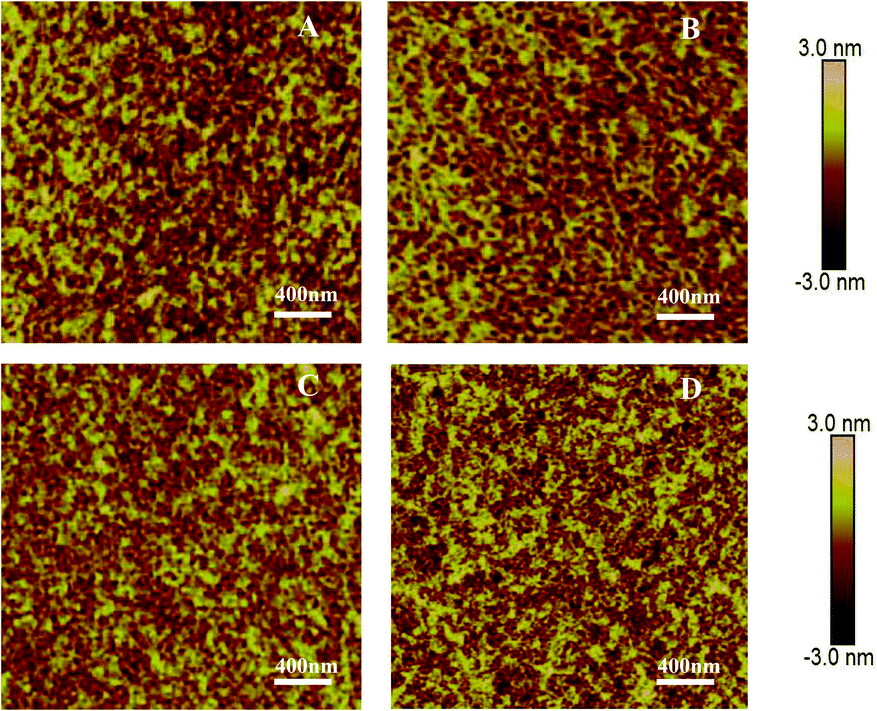 | ||
| Fig. 3 AFM height images of thin films of PDPP4T-1 (A), PDPP4T-2 (B) PDPP4T-3 (C) and PDPP4T (D) deposited on OTS-modified SiO2/Si substrates after thermal annealing at 160 °C. | ||
Thin film charge mobilities
The charge carrier mobilities of thin films of PDPP4T-1, PDPP4T-2 and PDPP4T-3 as well as PDPP4T were estimated with the respective bottom-gate/bottom-contact (BGBC) field-effect transistors (FETs) which were fabricated with conventional procedures (see the ESI†). Both as-prepared thin films and those after thermal annealing at different temperatures were utilized for fabrication of the FETs. As an example, Fig. 4 shows the transfer and output curves of the FETs with thin films of these polymers after thermal annealing at 160 °C. As expected, these polymer thin films exhibit typical p-type semiconducting properties. As listed in Table 2, the hole mobilities of all these polymers increased after thermal annealing, but their threshold voltages (VTh) and current on/off ratios (Ion/Ioff) remained almost unchanged. Moreover, maximum hole mobilities were achieved for all these polymers after thermal annealing at 160 °C, but their hole mobilities started to decrease after boosting the annealing temperature to 200 °C. For instance, μmaxh increased from 1.1 cm2 V−1 s−1 for the as-prepared FET of PDPP4T-1 to 3.5 cm2 V−1 s−1 after thermal annealing at 160 °C, while it was reduced to 3.0 cm2 V−1 s−1 after further thermal annealing at 200 °C.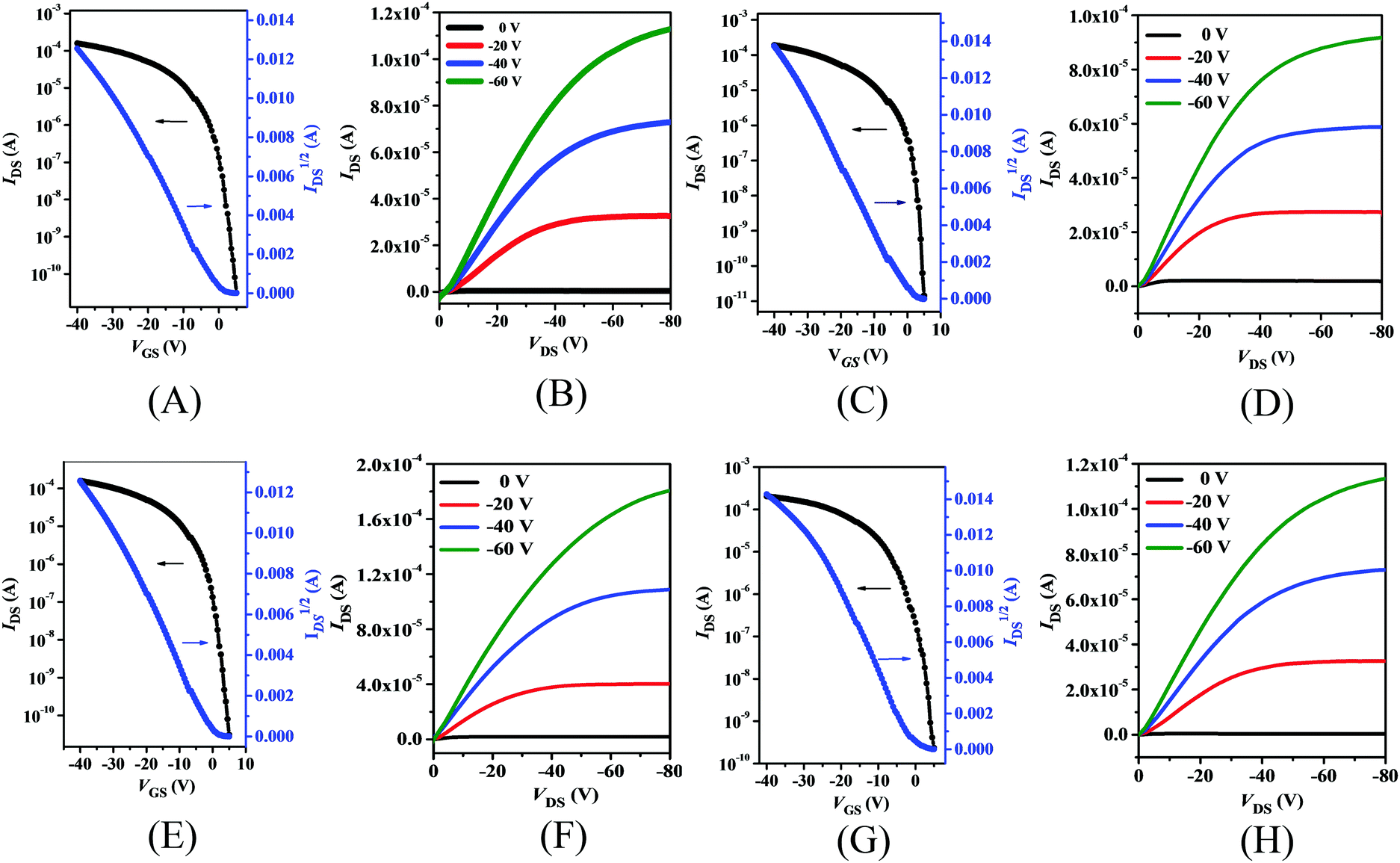 | ||
| Fig. 4 The transfer and output curves of BGBC FETs with thin films of PDPP4T-1 (A and B), PDPP4T-2 (C and D), PDPP4T-3 (E and F) and PDPP4T (G and H) after thermal annealing at 160 °C; the channel length (L) and width (W) were 50 μm and 1440 μm, respectively. | ||
| Polymer | Temp. (°C) | μ h /cm2 V−1 s−1 | V Th/V | I on/Ioff |
|---|---|---|---|---|
| a The mobilities were provided in “highest/average’’ form, and the performance data were obtained based on more than 10 different FETs. | ||||
| PDPP4T-1 | RT | 1.1/1.0 | −7–5 | 105–106 |
| 160 | 3.5/3.3 | −6–10 | 106–107 | |
| 200 | 3.0/2.7 | −5–5 | 105–106 | |
| RT | 1.4/1.1 | −2–5 | 106–107 | |
| PDPP4T-2 | 160 | 5.7/5.5 | −5–8 | 106–107 |
| 200 | 3.5/3.2 | −2–8 | 106–107 | |
| RT | 1.7/1.4 | −3–10 | 106–107 | |
| PDPP4T-3 | 160 | 6.1/5.9 | −6–4 | 106–107 |
| 200 | 4.2/3.9 | −5–5 | 106–107 | |
| RT | 1.1/0.9 | −3–10 | 105–106 | |
| PDPP4T | 160 | 3.0/2.3 | −5–4 | 106–107 |
| 200 | 2.2/2.0 | 0–10 | 105–106 | |
As listed in Table 2, the respective hole mobilities of PDPP4T-1, PDPP4T-2 and PDPP4T-3 are higher than that of PDPP4T either before or after thermal annealing. Moreover, the hole mobilities are increased in the following order: PDPP4T-1 < PDPP4T-2 < PDPP4T-3. The maximum hole mobilities of the as-prepared FETs increased from 1.1 cm2 V−1 s−1 for PDPP4T-1 to 1.4 cm2 V−1 s−1 for PDPP4T-2 and 1.7 cm2 V−1 s−1 for PDPP4T-3, while the maximum hole mobilities after thermal annealing at 160 °C were estimated to be 3.5 cm2 V−1 s−1, 5.7 cm2 V−1 s−1 and 6.1 cm2 V−1 s−1 for PDPP4T-1, PDPP4T-2 and PDPP4T-3, respectively. These results clearly indicate that the partial replacement of branching alkyl chains with linear ones can effectively improve the hole mobilities of these DPP-based conjugated D–A polymers.
This agrees well with the fact that interchain packing order degree is enhanced and polymer chains intend to pack in an edge-on mode after incorporation of more linear alkyl chains in the polymers as discussed above.
Based on our previous report that hole mobilities of several conjugated D–A polymers with branching alkyl chains can be enhanced upon incorporation of tetramethylammonium iodide (NMe4I) in the polymer thin films,6 we further investigated the semiconducting performances of PDPP4T-1, PDPP4T-2 and PDPP4T-3 after addition of NMe4I. Thin films of these polymers containing NMe4I at a molar ratio of 30/1 (between the repeating unit of each polymer vs.NMe4I) were prepared according to the reported procedures.6 On the basis of the respective transfer curves (Fig. S4, ESI†), maximum hole mobilities for PDPP4T-1/NMe4I, PDPP4T-2/NMe4I and PDPP4T-3/NMe4I were estimated to be 5.5 cm2 V−1 s−1, 8.7 cm2 V−1 s−1 and 9.1 cm2 V−1 s−1, respectively, which are obviously higher than the respective mobilities for PDPP4T-1, PDPP4T-2 and PDPP4T-3 without addition of NMe4I.
Conclusions
In this paper, we report three DPP-quaterthiophene terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3 containing both linear (n-dodecane) and branching (2-decyltetradecyl) alkyl chains with different ratios. Characterization of these polymer thin films with GIWAXS reveals that PDPP4T-1, PDPP4T-2 and PDPP4T-3 display a higher interchain packing order degree than PDPP4T which contains only branching alkyl chains. Furthermore, the partial replacement of bulky branching alkyl chains with linear ones results in the enhancement of interchain interactions. Interchain π–π stacking distances are shortened by increasing the content of linear chains in these terpolymers. Polymer chains of PDPP4T-3 which contains more linear alkyl chains among the terpolymers are packed in the edge-on mode, whereas both edge-on and face-on packing modes co-exist for PDPP4T-1 and PDPP4T-2 as well as PDPP4T. Accordingly, terpolymers PDPP4T-1, PDPP4T-2 and PDPP4T-3 show higher hole mobilities than PDPP4T. The thin film hole mobilities increase in the following order: PDPP4T-1 < PDPP4T-2 < PDPP4T-3. A hole mobility of up to 6.1 cm2 V−1 s−1 is achieved for PDPP4T-3, which can be further improved to 9.1 cm2 V−1 s−1 after incorporating NMe4I. These results clearly demonstrate that partial replacement of branching alkyl chains with linear ones is an efficient approach to improve interchain packing order and thin film crystallinity, thus boosting thin film charge mobilities. The application of this simple but efficient approach to other conjugated polymers, in particular n-type and ambipolar conjugated D–A polymers is under way.Experimental
Materials
The reagents and starting materials were commercially available and used without any further purification, if not specified. Compounds 1 and 2 were synthesized according to the previous report.42,43General synthetic method for PDPP4T-1, PDPP4T-2 and PDPP4T-3
Monomer 1 (x mol), monomer 2 (y mol), monomer 3 [1 equiv. based on (x + y) mol], P(o-tol)3 (0.08 equiv.) and Pd2(dba)3 (0.01 equiv.) were dissolved in dry toluene (5 mL). The solution was purged with N2 for 1 h. The reaction mixture was stirred at 110 °C for 72 h. The resulting mixture was poured into methanol (50 mL) and stirred for 1 h. The precipitate was filtered off and subjected to Soxhlet extraction for 3 days successively with methanol, acetone, and hexane. The resulting polymer was extracted with chloroform and precipitated again from methanol, filtered, and dried under vacuum at 45 °C for 48 h.Synthesis of PDPP4T-1
Monomer 1 (10.0 mg, 0.012 mmol), monomer 2 (291.7 mg, 0.24 mmol), monomer 3 (129.9 mg, 0.252 mmol), P(o-tol)3 (6.12 mg, 0.02 mmol), and Pd2(dba)3 (2.41 mg, 0.0025 mmol) were used. The purified polymer was collected to give a deep green solid (252 mg, 86% yield). 1H NMR (500 MHz; 1,1,2,2-tetrachloroethane-d2; 100 °C): δ = 8.79 (s, br, 2H), 7.01–6.86 (m, br, 6H), 4.03 (s, br, 4H), 2.06 (s, br, 1.9H), 1.47–1.26 (d, br, 78H), 0.88 (s, br, 11.7H).13C NMR (100 MHz; solid): δ = 160.63, 140.75, 137.00, 128.65, 124.20, 108.41, 45.33, 38.95, 32.78, 30.64, 23.53, 14.85. Mw/Mn (GPC) = 223/94 kg mol−1. Anal. calcd for (C1446H2178N42O42S84)n: C, 73.85; H, 9.34; N, 2.50; S, 11.45. Found: C, 73.75; H, 9.12; N, 2.44; S, 11.26.Synthesis of PDPP4T-2
Monomer 1 (10.0 mg, 0.012 mmol), monomer 2 (145.8 mg, 0.12 mmol), monomer 3 (68.0 mg, 0.132 mmol), P(o-tol)3 (3.36 mg, 0.01 mmol), and Pd2(dba)3 (1.26 mg, 0.0013 mmol) were used. The purified polymer was collected to give a deep green solid (128 mg, 85% yield). 1H NMR (500 MHz; 1,1,2,2-tetrachloroethane-d2; 100 °C): δ = 8.89 (s, br, 2H), 7.05–6.91 (m, br, 6H), 4.03 (s, br, 4H) 2.00 (s, br, 1.8H), 1.43–1.26 (d, br, 76.4H), 0.90 (s, br, 11.5H). 13C NMR (100 MHz; solid): δ = 160.62, 140.93, 136.66, 128.32, 123.91, 108.34, 45.17, 39.32, 32.73, 30.58, 23.49, 14.82. Mw/Mn (GPC) = 224/106 kg mol−1. Anal. calcd for (C746H1118N22O22S44)n: C, 73.70; H, 9.27; N, 2.53; S, 11.60. Found: C, 73.62; H, 9.02; N, 2.50; S, 11.36.Synthesis of PDPP4T-3
Monomer 1 (13.7 mg, 0.017 mmol), monomer 2 (100 mg, 0.086 mmol), monomer 3 (50.8 mg, 0.1 mmol), P(o-tol)3 (2.5 mg, 0.008 mmol), and Pd2(dba)3 (0.9 mg, 0.001 mmol) were used. The purified polymer was collected to give a deep green solid (93 mg, 84% yield). 1H NMR (500 MHz; 1,1,2,2-tetrachloroethane-d2; 100 °C): δ = 8.93 (s, br, 2H), 7.32–6.96 (m, br, 6H), 4.08 (s, br, 4H), 2.04 (s, br, 1.6H), 1.47–1.31 (d, br, 73.3H), 0.93 (s, br, 11H).13C NMR (100 MHz; solid): δ = 160.52, 140.70, 136.72 128.68, 124.04, 108.49, 45.45, 39.53, 32.69, 30.53, 23.45, 14.77. Mw/Mn (GPC) = 177/94 kg mol−1. Anal. calcd for (C396H588N12O12S24)n: C, 73.42; H, 9.15; N, 2.59; S, 11.88. Found: C, 73.27; H, 8.95; N, 2.58; S, 11.33.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support of the Strategic Priority Research Program of the CAS (XDB12010300), NSFC (21372226 and 21661132006) and 973 grant of MOST (No. 2013CB933501). The GIXRD data were measured at 1W1A, Beijing Synchrotron Radiation Facility. The authors gratefully acknowledge the assistance of scientists of the Diffuse X-ray Scattering Station during the experiments.Notes and references
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Z. Liu, G. Zhang, Z. Cai, X. Chen, H. Luo, Y. Li, J. Wang and D. Zhang, Adv. Mater., 2014, 26, 6965–6977 CrossRef CAS PubMed.
- J. Z. Sun, A. Qin and B. Z. Tang, Polym. Chem., 2013, 4, 211–223 RSC.
- W. Zhang, Y. Han, X. Zhu, Z. Fei, Y. Feng, N. D. Treat, H. Faber, N. Stingelin, I. McCulloch, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2016, 28, 3922–3927 CrossRef CAS PubMed.
- H. Luo, C. Yu, Z. Liu, G. Zhang, H. Geng, Y. Yi, K. Broch, Y. Hu, A. Sadhanala, L. Jiang, P. Qi, Z. Cai, H. Sirringhaus and D. Zhang, Sci. Adv., 2016, 2, e1600076 Search PubMed.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- A. R. Han, G. K. Dutta, J. Lee, H. R. Lee, S. M. Lee, H. Ahn, T. J. Shin, J. H. Oh and C. Yang, Adv. Funct. Mater., 2015, 25, 247–254 CrossRef CAS.
- I. Kang, H. J. Yun, D. S. Chung, S. K. Kwon and Y. H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- X. Zhang, H. Bronstein, A. J. Kronemeijer, J. Smith, Y. Kim, R. J. Kline, L. J. Richter, T. D. Anthopoulos, H. Sirringhaus, K. Song, M. Heeney, W. Zhang, I. McCulloch and D. M. DeLongchamp, Nat. Commun., 2013, 4, 2238 Search PubMed.
- H. R. Tseng, L. Ying, B. B. Hsu, L. A. Perez, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nano Lett., 2012, 12, 6353–6357 CrossRef CAS PubMed.
- Y. Olivier, D. Niedzialek, V. Lemaur, W. Pisula, K. Müllen, U. Koldemir, J. R. Reynolds, R. Lazzaroni, J. Cornil and D. Beljonne, Adv. Mater., 2014, 26, 2119–2136 CrossRef CAS PubMed.
- Y. F. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- C. C. Chen, W. H. Chang, K. Yoshimura, K. Ohya, J. You, J. Gao, Z. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670–5677 CrossRef CAS PubMed.
- A. Giovannitti, C. B. Nielsen, D. T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Nat. Commun., 2016, 7, 13066 CrossRef CAS PubMed.
- Y. Guo, G. Yu and Y. Liu, Adv. Mater., 2010, 22, 4427–4447 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- M. Nakano, I. Osaka and K. Takimiya, Macromolecules, 2015, 48, 576–584 CrossRef CAS.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- Z. Cai, H. Luo, P. Qi, J. Wang, G. Zhang, Z. Liu and D. Zhang, Macromolecules, 2014, 47, 2899–2906 CrossRef CAS.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS.
- Z.-G. Zhang and Y. Li, Sci. China: Chem., 2014, 58, 192–209 CrossRef.
- T. Lei, J. H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- J.-H. Dou, Y.-Q. Zheng, T. Lei, S.-D. Zhang, Z. Wang, W.-B. Zhang, J.-Y. Wang and J. Pei, Adv. Funct. Mater., 2014, 24, 6270–6278 CrossRef CAS.
- M. S. Chen, O. P. Lee, J. R. Niskala, A. T. Yiu, C. J. Tassone, K. Schmidt, P. M. Beaujuge, S. S. Onishi, M. F. Toney, A. Zettl and J. M. J. Fréchet, J. Am. Chem. Soc., 2013, 135, 19229–19236 CrossRef CAS PubMed.
- J. Mei, H.-C. Wu, Y. Diao, A. Appleton, H. Wang, Y. Zhou, W.-Y. Lee, T. Kurosawa, W.-C. Chen and Z. Bao, Adv. Funct. Mater., 2015, 25, 3455–3462 CrossRef CAS.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS PubMed.
- S.-F. Yang, Z.-T. Liu, Z.-X. Cai, H.-W. Luo, P.-L. Qi, G.-X. Zhang and D.-Q. Zhang, Macromolecules, 2016, 49, 5857–5865 CrossRef CAS.
- B. Kang, R. Kim, S. B. Lee, S. K. Kwon, Y. H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS PubMed.
- R. Kim, B. Kang, D. H. Sin, H. H. Choi, S. K. Kwon, Y. H. Kim and K. Cho, Chem. Commun., 2015, 51, 1524–1527 RSC.
- S.-F. Yang, Z.-T. Liu, Z.-X. Cai, M. J. Dyson, N. Stingelin, W. Chen, H.-J. Ju, G.-X. Zhang and D.-Q. Zhang, Adv. Sci., 2017, 1700048 CrossRef PubMed.
- X. Chen, Z. Zhang, Z. Ding, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 10376–10380 CrossRef CAS PubMed.
- W.-P. Hsu, K. Levon, K.-S. Ho, A. S. Myerson and T. K. Kwei, Macromolecules, 1993, 26, 1318–1323 CrossRef CAS.
- P. Keg, A. Lohani, D. Fichou, Y. M. Lam, Y. Wu, B. S. Ong and S. G. Mhaisalkar, Macromol. Rapid Commun., 2008, 29, 1197–1202 CrossRef CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. Macdonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, W. S. Roelofs, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2013, 135, 18942–18948 CrossRef CAS PubMed.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS PubMed.
- F. Liu, Y. Gu, X. Shen, S. Ferdous, H.-W. Wang and T. P. Russell, Prog. Polym. Sci., 2013, 38, 1990–2052 CrossRef CAS.
- Z. Cai, Y. Guo, S. Yang, Q. Peng, H. Luo, Z. Liu, G. Zhang, Y. Liu and D. Zhang, Chem. Mater., 2013, 25, 471–478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization techniques, DSC curves, TGA analysis, cyclic voltammograms, device fabrication and measurements, transfer and output curves and NMR spectra. See DOI: 10.1039/c7qm00307b |
| This journal is © the Partner Organisations 2017 |