DOI:
10.1039/C7QM00236J
(Research Article)
Mater. Chem. Front., 2017,
1, 2103-2110
Novel vinylene-bridged donor–acceptor copolymers: synthesis, characterization, properties and effect of cyano substitution†
Received
25th May 2017
, Accepted 17th July 2017
First published on 18th July 2017
Abstract
Two novel vinylene-bridged π-extended building blocks, (2E,2′E)-3,3′-(2,5-bis(octyloxy)-1,4-phenylene)bis(2-(5-bromothiophen-2-yl)acrylonitrile) (BOPACN) and 5,5′-((1E,1′E)-(2,5-bis(octyloxy)-1,4-phenylene)bis(ethene-1,2-diyl))bis(2-bromothiophene) (BOPA), were synthesized. They were further copolymerized with two diketopyrrolopyrrole (DPP) units possessing different alkyl side chains, and four donor–acceptor copolymers, namely PBOPACN-1, PBOPACN-2, PBOPA-1 and PBOPA-2, were synthesized. The optical, thermal, and electrochemical properties and field-effect properties of all copolymers were systematically investigated by theoretical calculations and experimental methods. Field-effect transistors based on these copolymers exhibited p-type charge transport characteristics in air. The performance implies that cyano substituents exert an important influence on semiconducting behaviours. Furthermore, 2D-GIXRD and theoretical calculations demonstrate that the presence of cyano substituents improves molecular planarity and generates highly-ordered molecular aggregates for PBOPACN-1 and PBOPACN-2.
Introduction
Organic field-effect transistors (OFETs) have received much attention in the past few years due to their potential applications in flexible and printed electronic devices.1–3 Much effort has been devoted to improving the device performance of OFETs including device fabrication and processing,4 electrode modification5 and the optimization of the device structure, as well as developing new small-molecule semiconductors,6 monomers and their derived copolymers to enrich the library of organic semiconductors.7 The third-generation donor–acceptor (D–A) copolymers, in which the donor and acceptor units are alternately distributed along the polymeric conjugated backbones, are widely synthesized and investigated.8,9 In the ground state, there are partial charge transfers between the donor and acceptor units, which result in a dipole moment in the molecular backbone. The dipole–dipole interactions among molecules are further in favour of the arrangement of the polymer molecules and facilitate charge-carrier transport. Moreover, the re-hybridization of frontier orbitals between the donor and acceptor units generally elevates the highest occupied molecular orbital (HOMO) energy level and lowers the lowest unoccupied molecular orbital (LUMO) energy level of the polymer at the same time, leading to a reduced energy gap of the polymers.10 Therefore, the semiconducting properties of polymer semiconductors could be readily tuned by polymerization of different donor and acceptor units.
In recent years, a vinylene unit has been incorporated into conjugated backbones due to its intrinsic advantages.11–13 Firstly, the vinylene unit would bring in rigid and π-extended conjugation, which is beneficial for enlarging the overlapping between π-orbitals, and enhancing the charge carrier mobility. Secondly, the vinylene unit could reduce the backbone torsion, thus leading to a planar conjugated backbone and an increased intensity of intramolecular charge transfer among polymer backbones. This effect was discussed in vinylene-flanked naphthalene diimide copolymers reported by Fei et al. and Zhang et al.14,15 Encouraged by these achievements, the structure–property relationships of the vinylene unit were further studied, indicating that the positions of alkyl side chains incorporated into the vinylene-bridged aromatic ring also have an impact on the semiconducting behaviours of the resulting polymer semiconductors. Specifically, when the alkyl side chains are incorporated close to a vinylene unit, higher charge transport properties are afforded.13 Moreover, vinylene units with substituted groups on the double bonds have also been developed, indicating that the HOMO and LUMO energy levels are less influenced than those with substituted groups on their aromatic rings.16 However, the building blocks with double vinylene units or multiple vinylene units are seldom investigated, partly because of the increased synthetic difficulty,17 which will further reduce the steric hindrance and enhance the conjugation length at the same time.
Besides, heteroatoms or heteroatom-substituted groups are one of the important constructing elements in developing high-performance organic semiconductors.18,19 They could effectively modify the energy levels, intramolecular and intermolecular interactions, conjugated backbone conformation and molecular aggregation state in the solid state, which will further elevate charge transport mobility. Fluorine atoms, for example, could induce F⋯S and F⋯H noncovalent interactions for polymer semiconductors.20 Other halogen elements like chlorine were also adopted to enhance the semiconducting properties.21 In recent years, cyano groups have been regarded as important electron-withdrawing groups similar to halogen atoms.22 In 2014, Kwon et al. developed a cyano-substituted polymer, which exhibited impressive electron mobility.23 However, when double cyano groups were incorporated onto the vinylene unit, the resulting building blocks and polymers showed twisted conjugated backbones. Generally, the studies of cyano groups in polymer semiconductors warrant further investigations.
In this work, we designed and synthesized two novel vinylene-bridged π-extended building blocks 5,5′-((1E,1′E)-(2,5-bis(octyloxy)-1,4-phenylene)bis(ethene-1,2-diyl))bis(2-bromothiophene) (BOPACN) and (2E,2′E)-3,3′-(2,5-bis(octyloxy)-1,4-phenylene)bis(2-(5-bromothiophen-2-yl)acrylonitrile) (BOPA), which not only extend the conjugation length but also reduce the steric hindrance. On the basis of the two new constructing units, we developed four D–A copolymers, namely PBOPACN-1, PBOPACN-2, PBOPA-1 and PBOPA-2, respectively. All polymers have good thermal stability and broad absorption. Their electrochemical properties were also investigated by theoretical and experimental methods, indicating that the cyano groups could effectively lower the HOMO and LUMO energy levels. Field-effect transistors based on these copolymers exhibited typical p-type charge transport characteristics in air with a hole mobility of 0.13 cm2 V−1 s−1. Devices based on PBOPACN-1 exhibited higher mobilities than those of PBOPA-1 after annealing, whereas devices of PBOPACN-2 exhibited almost identical mobility with those of PBOPA-2. Theoretical calculations and 2D-GIXRD characterization revealed that the higher mobilities of PBOPACN-1 and PBOPACN-2 are ascribed to improved molecular planarity and highly-ordered molecular aggregates.
Experimental section
Materials and methods
All the materials were purchased from Sigma Aldrich, ACROS and J&K Scientific Ltd. All the monomers were characterized with 1H NMR, 13C NMR, mass spectra (MS) and high-resolution MS. Nuclear magnetic resonance (NMR) spectra were acquired using a Bruker AVANCE 300 spectrometer with deuterated chloroform (CDCl3), dichloromethane (CD2Cl2) and dimethyl sulfoxide (DMSO) as the solvents. According to the molecular weight, mass spectra (MS) were conducted using EI-MS or MALDI-TOF. EI-MS and high-resolution EI-MS were performed using a Shimadzu QP 2010 gas chromatograph mass spectrometer. MALDI-TOF mass spectrometric measurements were performed on a Bruker Autoflex III type MALDI-TOF. HR-MALDIs were tested using a Bruker Solarix type Fourier transform ion cyclotron resonance mass spectrometer. UV-vis-NIR absorption spectra were measured using a Jasco V-570 type spectrophotometer in solution and a film. Cyclic voltammetry (CV) experiments were conducted on an electrochemistry workstation (CHI660C, Chenhua Shanghai) using a three-electrode system. Thermogravimetric analysis (TGA) was carried out using a DTG-60 instrument under nitrogen flow, heating from room temperature (RT) to 550 °C, with a heating rate of 10 °C min−1.
Theoretical calculations were carried out using the Gaussian 09 package. The corresponding trimer was optimized by performing density functional theory (DFT) calculations at the B3LYP level with a 6-31G(d) basis set.24
Device fabrication and characterization
Devices with a bottom-gate/bottom-contact (BG/BC) configuration using n++-Si/SiO2 substrates (300 nm) were fabricated. The source–drain electrodes were prepared with Au by the photolithography technique. Then the substrates were washed successively with acetone, deionized water and ethanol. The substrates were further purified using a UV Ozone cleaner for 20 min. The octadecyltrichlorosilane (OTS) self-assembled layer was formed by using OTS under vacuum at 120 °C. Then, a layer of the polymer solution was deposited on the OTS-modified SiO2 substrates by spin-coating. The devices were annealed at different temperatures to optimize the FET performance, and they were tested using a Keithley 4200 SCS semiconductor parameter analyser. The mobility was calculated in the saturated regime using the following equation:
| IDS = (W/2L)Ciμ(VGS − VTH)2 |
in which W/L is the ratio of the channel width/length, Ci is the insulator capacitance per unit area, and VGS and VTH correspond to the gate voltage and threshold voltage, respectively. To get the best performance of all the polymers, all the FET devices were optimized at different channels. The width of all the devices is 1400 μm, while the optimal device lengths of PBOPA-1, PBOPA-2, PBOPACN-1 and PBOPACN-2 are 10, 20, 30, and 50 μm, respectively.
Synthesis of the monomers and polymers
1,4-Dibromo-2,5-bis(octyloxy)benzene (1).
2,5-Dibromobenzene-1,4-diol (2.00 g, 7.74 mmol) and K2CO3 (2.06 g, 14.93 mmol) were added to 50 mL of N,N-dimethyl formamide, and the solution was stirred for 30 min at room temperature. Then, 1-bromooctane (3.03 g, 15.69 mmol) was added. Then the mixture was heated at 80 °C and stirred overnight. The reaction mixture was allowed to cool to room temperature and extracted with dichloromethane, dried with anhydrous Na2SO4 and filtered. The solvent was removed under reduced pressure and the residues were purified by silica gel chromatography to obtain the final product as a white solid (3.00 g, 81.57%). 1H NMR (400 MHz, CDCl3, δ): 7.08 (s, 2H), 3.95–3.92 (t, 4H, J = 4.0 Hz), 1.81–1.77 (m, 4H), 1.49–1.45 (m, 4H), 1.34–1.29 (m, 16H), 0.90–0.87 (t, 6H, J = 4.0 Hz) 13C NMR (75 Hz, CDCl3, δ): 150.12, 118.54, 111.17, 70.36, 31.80, 29.27, 29.22, 29.13, 25.95, 22.66, 14.10. EI-MS: 492 [M+], HR-MALDI calcd for C22H36Br2O2: 490.1076, found 490.1081.
2,5-Bis(octyloxy)terephthalaldehyde (2).
Compound 1 (5.0 g, 10.15 mmol) was added to a two-necked flask charged with argon. Then 120 mL of dry THF was added to the flask and stirred. The mixture was cooled to −78 °C and n-butyl lithium (2.5 M, 9.14 mL, and 22.85 mmol) was added dropwise. The mixture was stirred at this temperature for 3 h. Then N-formyl piperidine (2.60 g, 22.98 mmol) was added in one portion, and the reaction mixture was kept at this temperature for 1 h. The resulting mixture was warmed up to room temperature and stirred for 2 h. The reaction was quenched with water and the mixture was extracted with dichloromethane three times, dried with anhydrous Na2SO4 and filtered. The solvent was removed under reduced pressure to afford the title product (1.11 g, 28%). 1H NMR (400 MHz, CDCl3, δ): 10.52 (s, 2H), 7.43 (s, 2H), 4.09 (t, 4H, J = 8.0 Hz), 1.87–1.80 (m, 4H), 1.49–1.45 (m, 4H), 1.33–1.29 (m, 16H), 0.89 (t, 6H, 8.0 Hz). 13C NMR (75 Hz, CDCl3, δ): 189.35, 155.23, 129.29, 111.62, 69.25, 31.78, 29.27, 29.20, 29.05, 26.02, 22.64, 14.07. EI-MS: 390 [M+], HR-EI calcd for C24H38O4: 390.2770, found 390.2776.
(2E,2′E)-3,3′-(2,5-Bis(octyloxy)-1,4-phenylene)bis(2-(5-bromothiophen-2-yl)acrylonitrile) (BOPACN).
Potassium tert-butylate (0.32 g, 2.82 mmol) was added to a two-neck flask protected by argon, then 2-(5-bromothiophen-2-yl)acetonitrile (0.57 g, 2.82 mmol) and ethanol (20 mL) were added and the solution was stirred. Compound 2 (0.5 g, 1.28 mmol) was dissolved in ethanol and the solution was added dropwise to the flask. The resulting mixture was stirred at room temperature overnight. The reaction was stopped and filtered, and the final product was purified by silica gel chromatography (0.87 g, 90%). m.p. 174 °C. 1H NMR (300 MHz, CDCl3, δ): 7.80 (s, 2H), 7.74 (s, 2H), 7.14 (d, 2H, J = 3.9 Hz), 7.06–7.04 (d, 2H, J = 3.9 Hz), 4.09 (t, 4H, J = 6.0 Hz), 1.87–1.80 (m, 4H), 1.35–1.30 (m, 20H), 0.88–0.86 (t, 6H, J = 6.6 Hz). 13C NMR (75 Hz, CHCl2CHCl2, 360 K, δ): 151.68, 141.04, 133.53, 130.98, 127.02, 125.75, 116.26, 113.66, 111.51, 105.60, 69.92, 31.52, 29.09, 28.97, 26.00, 22.35, 13.73. HR-MALDI calcd for C36H42Br2N2NaO2S2: 779.0947, found 779.0942.
(5-Bromothiophen-2-yl)methanol (5).
Compound 4 (24.86 g, 0.12 mol) was dissolved in methanol and the system was cooled to 0 °C for 10–20 min. Then NaBH4 (6.81 g, 0.18 mol) was added in batches. The reaction was monitored by thin film chromatography. Then the reaction was quenched by the addition of water. The mixture was extracted with chloroform, washed with saturated brine, dried with anhydrous Na2SO4 and filtered. The final product was purified by silica gel chromatography (20.90 g, 90%). 1H NMR (300 MHz, DMSO, δ): 7.02 (d, 1H, J = 3.6 Hz), 6.77 (dd, 1H, J1 = 4.5 Hz, J2 = 0.9 Hz), 5.55 (t, 1H, 5.7 Hz), 4.57 (d, 2H, J = 5.7 Hz). 13C NMR (75 Hz, DMSO, δ): 149.15, 130.22, 125.02, 110.10, 58.90. HR-EI calcd for C5H5BrOS: 191.9244, found: 191.9241.
2-Bromo-5-(bromomethyl)thiophene (6).
At 0 °C, PBr3 (20.30 g, 75 mmol) and three drops of pyridine were added successively to a solution of compound 5 (10.00 g, 52 mmol) in dichloromethane, and the system was kept at this temperature for 1 h. The ice bath was removed and the reaction mixture was stirred at room temperature for another 1 h. Water was added to the mixture slowly and the residue was extracted with dichloromethane, washed with sodium carbonate and saturated brine, and filtered. The title compound was acquired after the solvent was removed under reduced pressure (12.50 g, 93%). 1H NMR (300 MHz, CDCl3, δ): 6.89–6.85 (m, 2H), 4.63 (s, 2H). 13C NMR (75 Hz, CDCl3, δ): 142.07, 129.86, 128.31, 113.88, 26.21. HR-EI calcd for C5H4Br2S: 253.8400, found: 253.8397.
Diethyl((5-bromothiophen-2-yl)methyl)phosphonate (7).
Compound 6 (13.00 g, 50.78 mmol) and triethyl phosphite (8.86 g, 53.31 mmol) were added in a flask and the mixture was stirred at 120 °C overnight. After the residual triethyl phosphite was removed, the title compound was acquired as a light-yellow oil liquid (4.94 g, 31%). 1H NMR (300 MHz, CD2Cl2, δ): 6.91–6.89 (dd, 1H, J1 = 4.5 Hz, J2 = 0.6 Hz), 6.72–6.70 (m, 1H), 4.08–3.99 (m, 4H), 3.27–3.20 (dd, 2H, J1 = 3.6 Hz, J2 = 0.9 Hz), 1.26 (t, 6H, J = 7.2 Hz). 13C NMR (75 Hz, CD2Cl2, δ): 135.08, 134.94, 129.81, 129.77, 127.79, 127.67, 110.54, 110.47, 62.45, 62.36, 54.15, 53.79, 53.43, 53.07, 52.71, 29.26, 27.35, 16.24, 16.16. HR-EI calcd for C9H14BrO3PS: 311.9585, found: 311.9582.
5,5′-((1E,1′E)-(2,5-Bis(octyloxy)-1,4-phenylene)bis(ethene-2,1-diyl))bis(2-bromothiophene) (BOPA).
At 0 °C, compound 7 (353 mg, 1.13 mmol) and compound 2 (200 mg, 0.51 mmol) were added to dry tetrahydrofuran and the mixture was stirred for 10 min. Potassium tert-butoxide (1.13 mmol, 126.8 mg) was then added in batches and the system was kept stirring at this temperature for 1 h. The ice bath was removed and the mixture was kept at room temperature for another 1 h. The reaction was quenched with water, extracted with chloroform and dried with anhydrous Na2SO4 and filtered. The solvent was removed under reduced pressure. The final product was acquired as a yellow solid by silica gel chromatography (256 mg, 70.5%). m.p. 108 °C. 1H NMR (300 MHz, CD2Cl2, δ): 7.24–7.08 (m, 4H), 6.99 (s, 2H), 6.96 (d, 2H, J = 3.6 Hz), 6.80 (d, 4H, J = 3.6 Hz), 4.00 (t, 4 H, J = 6.6 Hz), 1.87–1.78 (m, 4H), 1.41–1.29 (m, 16H), 0.87 (t, 6H, J = 6.9 Hz). 13C NMR (75 Hz, CD2Cl2, δ)151.04, 145.50, 130.65, 126.09, 123.68, 121.60, 110.79, 110.42, 69.43, 31.84, 29.42, 29.40, 29.32, 26.27, 22.70, 13.89. HR-MALDI calcd for C34H44Br2O2S2: 706.1144, found: 706.1149.
General procedure for polymerization
The polymerization reaction was proceeded using the Suzuki reaction under argon protection, with monomer 3 (or 8) (0.1 mmol), the corresponding boric acid ester monomer (0.1 mmol), Pd2(dba)3 (4 mg, dba = dibenzylideneacetone), P(o-tol)3 (12 mg, o-tol = 2-methylphenyl), 2 mL of 2 M K2CO3 solution, a catalytic amount of Aliquat 336 and 5 mL of toluene. The system underwent freeze–pump–thaw cycles three times under argon and was then refluxed for 3 days. The mixture was poured into 100 mL of methanol, stirred for 2 h, and filtered. The residue was purified using a Soxhlet apparatus with methanol, acetone, and hexane to remove oligomers and the residual catalyst. The product was obtained by extraction with chloroform and chlorobenzene. The chlorobenzene fractions are used in device fabrication and characterization. Also, the molecular weights listed in the following correspond to the chlorobenzene fractions.
PBOPACN-1 (yield: 55% Mw/Mn = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 640/25094 g mol−1, polydispersity index (PDI) = 2.02)
640/25094 g mol−1, polydispersity index (PDI) = 2.02)
PBOPACN-2 (yield: 62% Mw/Mn = 41871/20004 g mol−1, PDI = 2.09)
PBOPA-1 (yield: 70% Mw/Mn = 114947/50472 g mol−1, PDI = 2.28)
PBOPA-2 (yield: 68% Mw/Mn = 124375/84488 g mol−1, PDI = 1.47)
Results and discussion
Synthesis
The synthetic routes of vinylene-bridged units, BOPA and BOPACN, and the derived donor–acceptor copolymers, PBOPACN-1, PBOPACN-2, PBOPA-1 and PBOPA-2, are shown in Schemes 1 and 2. We used 2,5-dibromobenzene-1,4-diol as the starting material to synthesize BOPACN. After an alkylation reaction under basic conditions, 2,5-dibromobenzene-1,4-diol was converted to intermediate 1 in 81% yield. Then compound 1 underwent a Br/Li exchange reaction with n-BuLi and was quenched with N-formyl piperidine to give the aldehyde in moderate yield. The Knoevenagel reaction of compound 2 and 2-(5-bromothiophen-2-yl)acetonitrile afforded the desired BOPACN in 90% yield. 5-Bromothiophene-2-carbaldehyde was adopted as the starting material to synthesize BOPA. After two reaction steps including reduction and bromination reactions, compound 6 was obtained. Intermediate 6 was then converted to phosphite, which further underwent a Horner–Wadsworth–Emmons reaction to give the desired BOPA. The four copolymers were synthesized through the Suzuki copolymerization of BOPA and BOPACN with two diketopyrrolopyrroles, respectively. All crude polymer materials were purified by the same procedures including Soxhlet extraction. The detailed purification procedures are collected in the experimental section. Infrared spectra (IR) characterization of BOPA and BOPACN indicates the introduction of a cyano group with characteristic vibronic bands at 2206 cm−1. Also, both PBOPACN-1 and PBOPACN-2 show characteristic bands of the cyano group at 2213 cm−1, while PBOPA-1 and PBOPA-2 do not show this band (see ESI,† Fig. S6), indicating that the cyano groups were introduced into the polymer backbone effectively. High-temperature gel permeation chromatography (GPC) tests were utilized to evaluate the degree of polymerization of the resulting four polymer materials. The weight-average molecular weight (Mw), the number-average molecular weight (Mn), and the polymer dispersity index (PDI) are 50.6 kDa, 25.1 kDa, and 2.02 for PBOPACN-1, 41.9 kDa, 20.0 kDa, and 2.09 for PBOPACN-2, 114.9 kDa, and 50.5 kDa, 2.28 for PBOPA-1, and 124.3 kDa, 84.5 kDa, and 1.47 for PBOPA-2, respectively. The GPC measurements show that the four copolymers have a high degree of polymerization.
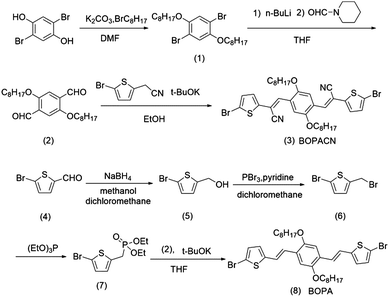 |
| | Scheme 1 Synthetic routes of BOPA and BOPACN. | |
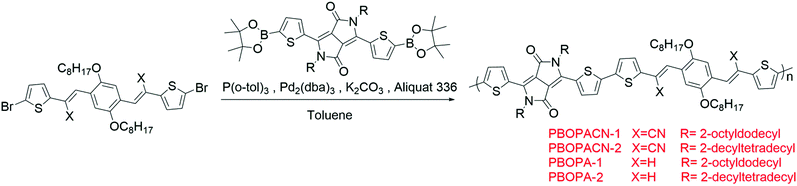 |
| | Scheme 2 Polymerization reactions of PBOPACN-1, PBOPACN-2, PBOPA-1 and PBOPA-2. | |
Optical properties
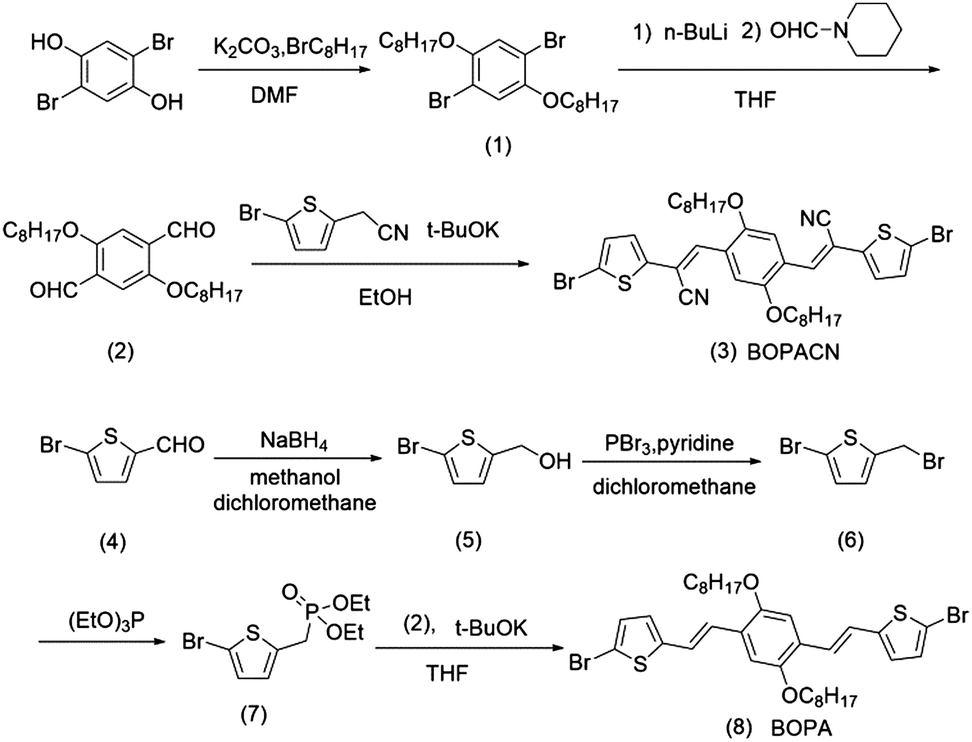
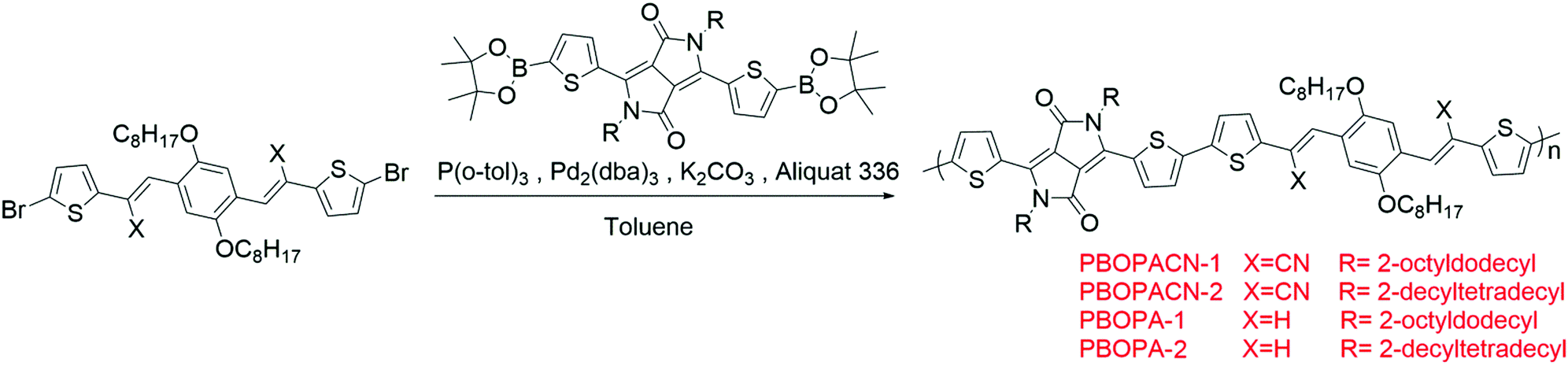
The optical properties of the four polymers were tested in chloroform solution (Fig. 1), and the corresponding data are listed in Table 1. In solution, polymers of PBOPA-1 and PBOPA-2 show different absorption spectra compared with the cyano-substituted polymers, PBOPACN-1 and PBOPACN-2. Both PBOPA-1 and PBOPA-2 solutions display identical absorption profiles due to their identical conjugated skeleton, with the maximum absorption peaks (λabsmax) at ca. 740 nm and the shoulder peaks at ca. 680 nm. The results demonstrate that the alkyl side chains do not affect the photophysical properties of the molecular backbone. Similarly, the absorption profile of PBOPACN-1 is the same as that of PBOPACN-2 in solution, with a blue-shifted λabsmax at ca. 708 nm and the shoulder peaks at around 760 nm (Fig. 1a). Compared with PBOPA-1 and PBOPA-2, the absorption spectra of PBOPACN-1 and PBOPACN-2 are broadened and show an obvious bathochromic shift, indicating that the optical energy gap is decreased after the introduction of cyano groups. Their thin film absorption spectra are depicted in Fig. 1b. Apparently, the absorption spectra of all polymer thin films become similar with broadened main absorption peaks from 500 nm to 900 nm. The changes can be attributed to the similar planarity of the conjugated backbones of all polymers in the solid state, implying that the presence of a cyano group does not destroy the conjugated backbone planarity of PBOPACN-1 and PBOPACN-2.
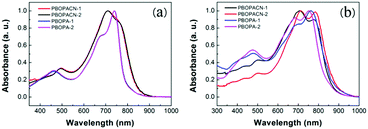 |
| | Fig. 1 UV-vis absorption spectra of PBOPACN-1, PBOPACN-2, PBOPA-1 and PBOPA-2 in chloroform solution (a) and in thin films (b). | |
Table 1 The data of UV-vis absorption spectra and cyclic voltammetry tests
| Polymer |
λ
max (nm) |
E
cvHOMO (eV) |
E
cvLUMO (eV) |
| Solution |
Film |
|
PBOPACN-1
|
708 |
708 |
−5.42 |
−3.38 |
|
PBOPACN-2
|
708 |
713 |
−5.41 |
−3.39 |
|
PBOPA-1
|
740 |
762 |
−5.24 |
−3.32 |
|
PBOPA-2
|
740 |
751 |
−5.27 |
−3.28 |
Thermal properties
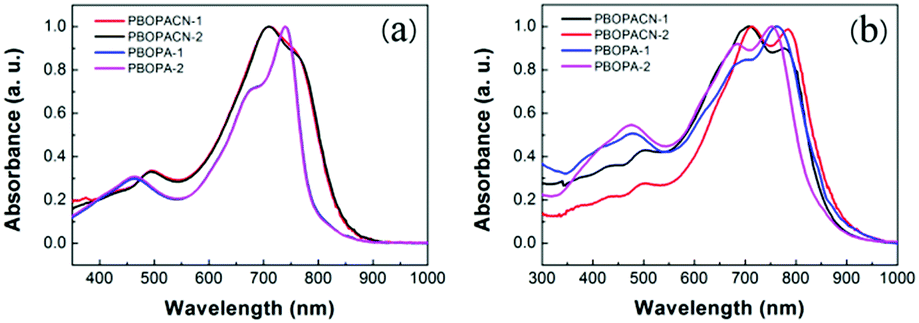
We performed thermogravimetric analyses (TGA) and differential scanning calorimetric (DSC) tests to investigate the thermal properties of the four polymers. All materials have high decomposition temperatures (Tdec, with 5% weight loss), suggesting all polymers have excellent thermal stability (Fig. 2a). PBOPA-1 and PBOPA-2 show high Tdec values of 336 and 339 °C, respectively. Compared with PBOPA-1 and PBOPA-2, the cyano substituted copolymers, PBOPACN-1 and PBOPACN-2, exhibited a higher Tdec of 387 °C for PBOPACN-1 and 392 °C for PBOPACN-2. DSC curves are shown in Fig. 2b. PBOPA-1 and PBOPA-2 have obvious peaks in both the endothermic and exothermic processes (288 and 258 °C for PBOPA-1, and 267 and 239 °C for PBOPA-2), which could be attributed to the corresponding melting and crystallization processes,7 whereas PBOPACN-1 and PBOPACN-2 do not show any obvious peaks in their DSC curves. The absence of any peaks in PBOPACN-1 and PBOPACN-2 indicates that the introduction of cyano groups in the polymer backbone enhances the intermolecular interactions.
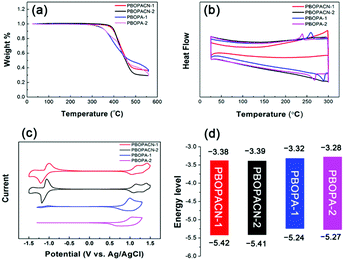 |
| | Fig. 2 Thermogravimetric analysis (a) and differential scanning calorimetry analysis results (b) of all four polymers. Cyclic voltammetry curves (c) and graphical representation (d) of the energy levels of PBOPACN-1, PBOPACN-2, PBOPA-1 and PBOPA-2. | |
Electrochemical properties
The electrochemical properties of all the materials were examined by performing cyclic voltammetry experiments with a Pt electrode as the counter electrode and an Ag/AgCl electrode as the reference electrode, and a solution of Bu4NPF6 (0.1 M) in acetonitrile was used as the electrolyte. The results and the graphical representation are shown in Fig. 2c and d. PBOPA-1 and PBOPA-2 show obvious oxidation peaks and negligible reduction peaks. Depending on their respective onsets of oxidation potential (Eoxonset), the HOMO energy levels were estimated to be −5.24 eV for PBOPA-1 and −5.27 eV for PBOPA-2 calculated according to the equation of EHOMO = −(4.40 + Eoxonset) eV. On the basis of their respective onset of reduction potential (Eredonset, see the enlarged curves in Fig. S1, ESI†), the LUMO energy levels of PBOPA-1 and PBOPA-2 are −3.32 and −3.28 eV, respectively, calculated using the equation of ELUMO = −(4.40 + Eredonset) eV. It is worth noting that, however, PBOPACN-1 and PBOPACN-2 not only show obvious oxidation peaks but also display clear reduction peaks. The enhanced reduction peaks suggest that the electron-withdrawing cyano groups make the two copolymers more readily reduced than the two cyano-unsubstituted copolymers. Based on the same calculation equations, The HOMO/LUMO energy levels are −5.42/−3.38 eV for PBOPACN-1 and −5.41/−3.39 eV for PBOPACN-2. Obviously, the introduction of the cyano group in the donor part has an important effect on the energy levels: it effectively lowers the HOMO and LUMO energy levels of the resulting polymers.
Theoretical calculations
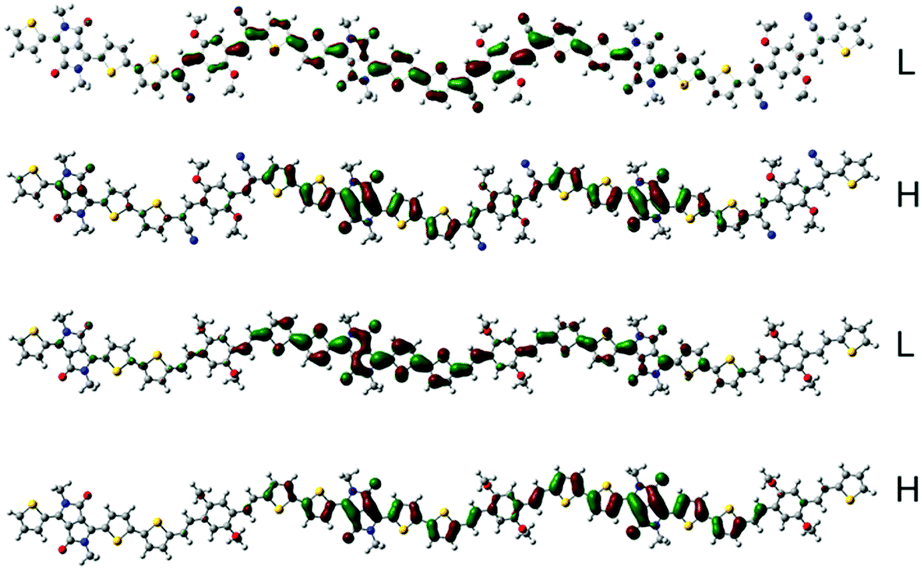
To estimate the polymer conformations and intrinsic electrical properties, density functional theory (DFT) calculations for trimers of the copolymers were conducted using the Gaussian 09 package at the B3LYP/6-31G(d) level. The optimized structures and frontier molecular orbitals of the polymers are shown in Fig. 3. To simplify the calculation, the alkyl side chains in the trimers were replaced by methyl groups. Moreover, the optimized structures and frontier molecular orbitals of the monomers, BOPACN and BOPA, are shown in Fig. S2 (ESI†). The calculated results of BOPACN and BOPA show that they both possess planar conjugated backbones, implying that the introduction of the cyano group does not distort the backbone planarity of the BOPACN unit. The calculated results of PBOPA (PBOPA-1 and PBOPA-2) and PBOPACN (PBOPACN-1 and PBOPACN-2) also indicate that all the polymers have good conjugated backbone planarity (Fig. S3, ESI†). As shown in Fig. 3, all the HOMOs and LUMOs of the four polymers delocalize along the backbone well. The calculated HOMO/LUMO energy levels are −4.83/−3.27 eV for PBOPACN, and −4.61/−2.89 eV for PBOPA. It is well known that the LUMO energy level of an acceptor unit and the HOMO energy level of a donor unit generally affect the LUMO and HOMO energy levels of the polymers, respectively. PBOPACN has low-lying HOMO energy levels due to the cyano substituents having strong electron-withdrawing ability. Also, the introduction of cyano groups mainly affects the HOMO levels of polymers but has less influence on the LUMO levels. The results are consistent with the CV measurements.
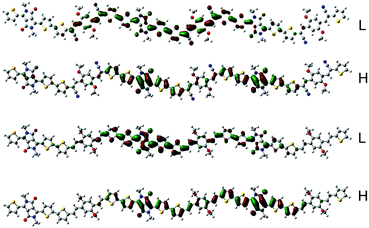 |
| | Fig. 3 The HOMO (H) and LUMO (L) representations of the two trimers (the above is PBOPACN and the bottom is PBOPA). | |
Charge transport properties
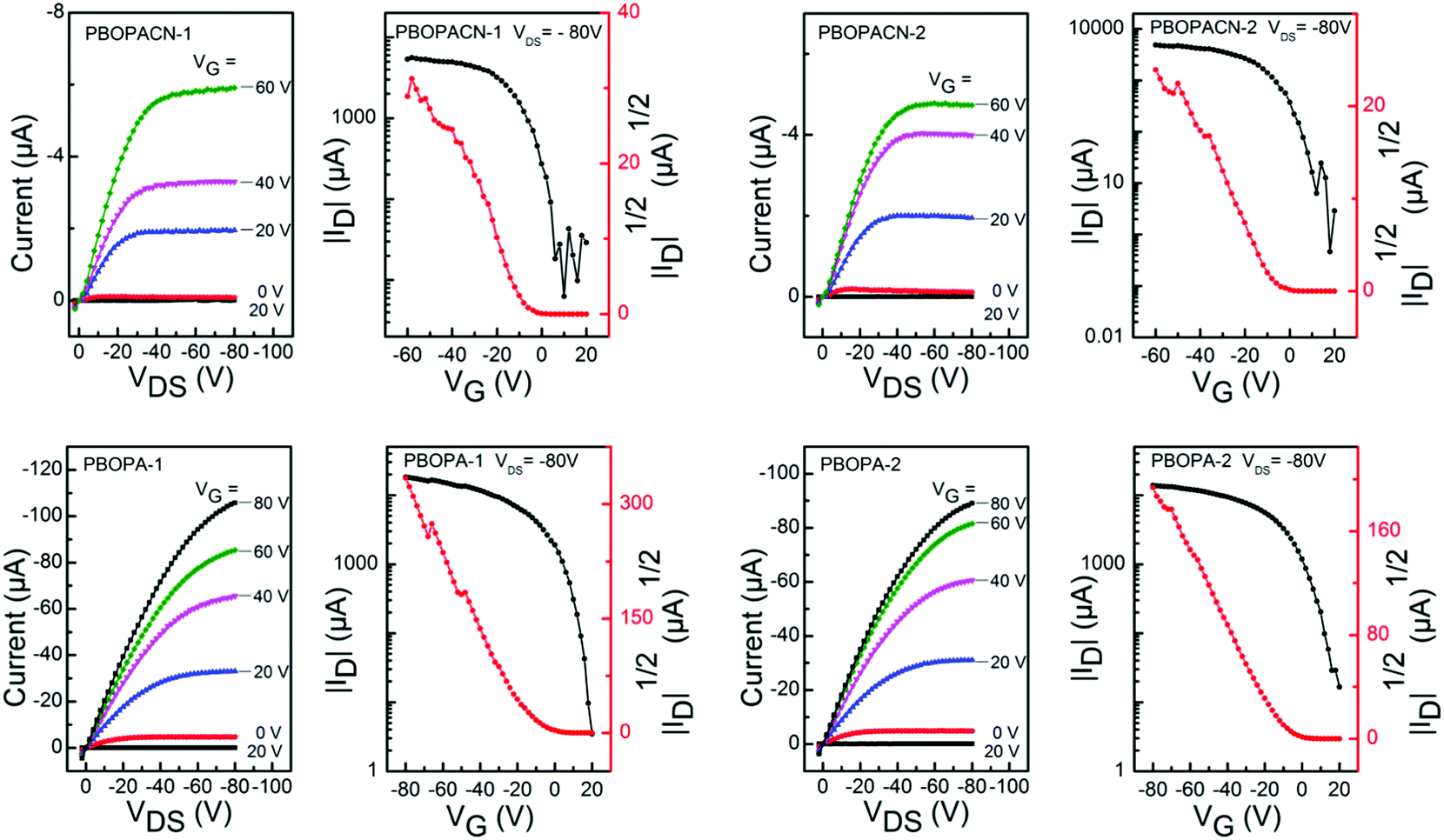
We fabricated OFET devices with the bottom-gate/bottom-contact (BG/BC) configuration to investigate the charge transport properties of the four polymer materials. The typical transfer and output curves are shown in Fig. 4. Procedures of fabricating OFETs are described in the experimental section. The corresponding parameters are listed in Table 2. Under ambient conditions, PBOPACN-1 and PBOPACN-2 showed hole mobilities of 2.74 × 10−2 and 4.90 × 10−2 cm2 V−1 s−1, and after annealing at 180 °C their mobilities increased to 1.01 × 10−1 and 1.20 × 10−1 cm2 V−1 s−1, respectively. By contrast, PBOPA-1 and PBOPA-2 showed higher hole mobilities of 4.80 × 10−2 and 7.10 × 10−2 cm2 V−1 s−1 at room temperature, respectively. Even after annealing at the optimized temperature of 100 °C, the highest mobilities are 7.57 × 10−2 cm2 V−1 s−1 for PBOPA-1 and 1.29 × 10−1 cm2 V−1 s−1 for PBOPA-2. It is worthwhile to note that PBOBACN-1 and PBOPACN-2-based FET devices exhibited ambipolar charge transport characteristics under a nitrogen atmosphere. The hole mobility (μh) and electron mobility (μe) tested in a glove box are 1.62 × 10−2 cm2 V−1 s−1 and 2.29 × 10−2 cm2 V−1 s−1 for PBOBACN-1, respectively, and 1.36 × 10−2 cm2 V−1 s−1 and 2.01 × 10−3 cm2 V−1 s−1 for PBOBACN-2, respectively (see Fig. S4, ESI†). However, for the PBOPA-1 and PBOPA-2-based devices, no ambipolar charge transport characteristics were obtained. Such results show that the introduction of cyano groups could effectively enhance the electron transport, thus causing the charge-transport polarity of the polymer semiconductors to change from p-type to ambipolar characteristics. In addition, the enhanced charge transport properties of PBOPACN-1 and PBOPACN-2 compared with PBOPA-1 and PBOPA-2 could be attributed to the improved molecular aggregation induced by the introduction of cyano groups.
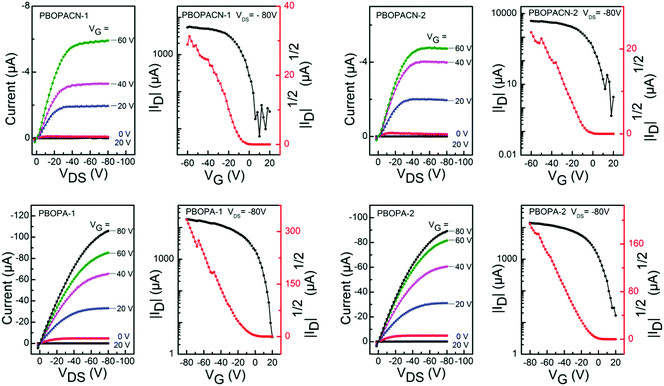 |
| | Fig. 4 Typical p-type transfer and output curves of OFETs based on PBOPACN-1 (180 °C), PBOPACN-2 (180 °C), PBOPA-1 (100 °C) and PBOPA-2 (100 °C) measured in air. | |
Table 2 The summary of the OFET devices with the BG/BC configuration measured in air
| Polymer |
T
an (°C) |
Mobility (max) (cm2 V−1 s−1) |
I
on/Ioff |
V
th (V) |
|
PBOPACN-1
|
RT |
2.74 × 10−2 |
104–105 |
−10–0 |
| 180 |
1.01 × 10−1 |
104–105 |
−10–0 |
|
|
|
PBOPACN-2
|
RT |
4.90 × 10−2 |
105–106 |
−10–0 |
| 180 |
1.20 × 10−1 |
105–106 |
−10–0 |
|
|
|
PBOPA-1
|
RT |
4.80 × 10−2 |
105–106 |
−5–0 |
| 100 |
7.57 × 10−2 |
105–106 |
−5–0 |
|
|
|
PBOPA-2
|
RT |
7.10 × 10−2 |
104–105 |
−5–0 |
| 100 |
1.29 × 10−1 |
104–105 |
−5–0 |
Thin film microstructural characterization
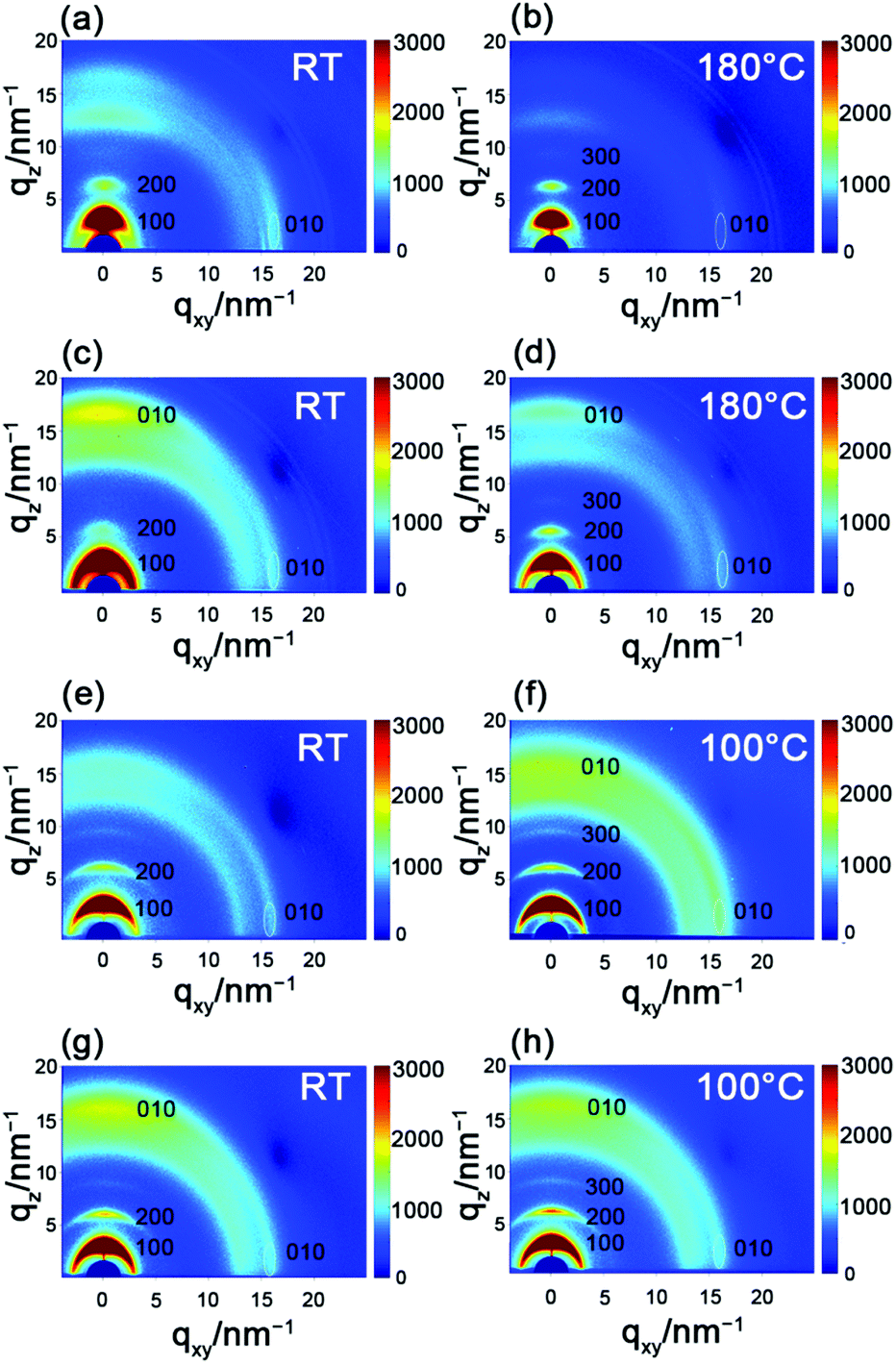
To explore the correlation between the performance and the microstructure of the corresponding films, 2D-GIXRD was used to gain insight into the molecular arrangement (Fig. 5). The as-spun PBOPACN-1 thin film exhibited ordered molecular arrangement with (h00) diffraction peaks up to the third order in the out-of-plane direction, along with a (010) diffraction peak appearing in the in-plane direction. Meanwhile, a (010) diffraction peak also appeared in the out-of-plane direction, which implies that PBOPACN-1 in a thin film adopts a bimodal arrangement (i.e. mixed face-on and edge-on). After annealing at 180 °C, the (010) diffraction peak in the out-of-plane direction disappeared, implying that the polymer adopts the edge-on arrangement in the annealed thin film. However, the as-spun PBOPACN-2 thin film possesses a bimodal arrangement, in which the face-on packing mode is dominant. After annealing at 180 °C, the polymer thin film still maintained the bimodal arrangement, in which the face-on arrangement becomes weaker than that of the as-spun thin film. As the edge-on packing mode is considered to be in favor of charge transport, such observations could explain the increased mobility of the annealed thin film compared to the as-spun ones. As shown in Fig. 5e and g, both the as-spun PBOPA-1 and PBOPA-2 thin films adopt a random arrangement. They keep the same packing even when annealed at 100 °C. All the polymer films show π–π spacing distances of around 3.90 Å, while the d–d spacing distances are obviously different. The d–d spacing distances of PBOPACN-1 and PBOPA-1 are shorter than those of PBOPACN-2 and PBOPA-2, which is consistent with the alkyl chain length (Table 3). The above characterizations are responsible for the different charge transport mobilities between the four copolymers. AFM characterization was further conducted to obtain the surface morphology of all the polymers (see Fig. S5, ESI†). PBOPACN-1 and PBOPACN-2 show rough surface morphologies with apparent crystalline domains in both as-spun and annealed thin films, while PBOPA-1 and PBOPA-2 give more uniform surface morphologies with smaller domains. Because the greater crystallites in network thin films are well-recognized to be beneficial for charge transport, the AFM images are reasonable for their charge transport mobilities.
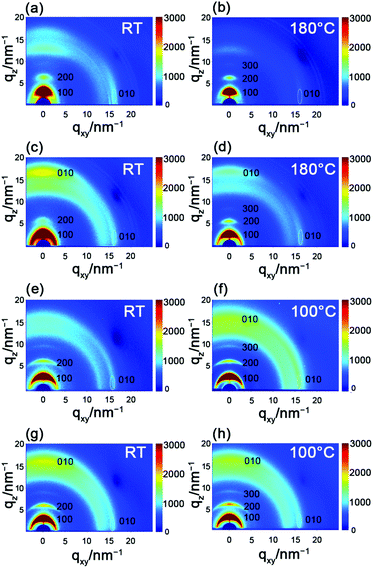 |
| | Fig. 5 2D-GIXRD characterization of PBOPACN-1 (a and b), PBOPACN-2 (c and d), PBOPA-1 (e and f) and PBOPA-2 (g and h). | |
Table 3 Summary of the data from the 2D-GIXRD characterization
| Polymer |
Temp (°C) |
Out-of-plane |
In-plane |
|
d–d spacing (Å) |
π–π spacing (Å) |
|
PBOPACN-1
|
RT |
19.02 |
3.92 |
| 180 |
19.37 |
3.92 |
|
|
|
PBOPACN-2
|
RT |
20.53 |
3.94 |
| 180 |
21.23 |
3.91 |
|
|
|
PBOPA-1
|
RT |
18.67 |
3.92 |
| 100 |
18.69 |
3.92 |
|
|
|
PBOPA-2
|
RT |
20.39 |
3.95 |
| 100 |
20.67 |
3.95 |
Conclusions
Two novel vinylene-bridged π-extended building blocks, BOPA and BOPACN, were designed and synthesized, and copolymerized with two diketopyrrolopyrroles with different alkyl side chains affording four donor–acceptor copolymers. The optical, thermal, and electrochemical properties and field-effect properties of all copolymers were systematically investigated in theoretical calculations and experimental methods, indicating that the cyano group could enhance the thermal stability, broaden the absorption profiles, and lower the HOMO energy level when the cyano group was introduced to the donor units of the polymer semiconductors. Moreover, cyano groups do not destroy the planarity of the polymer backbone; conversely, they enhance the interactions between molecules and the aggregation states were changed effectively. Furthermore, the cyano group could induce an inversion of charge polarity for organic semiconductors. The above results highlight the importance of the cyano group in constructing high-performance polymer semiconductors.
Acknowledgements
The authors are highly thankful for financial support by the National Natural Science Foundation of China (21673258, 51473021, 21474116 and 51233006), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB12030100), and the National Key Research and Development Program of China (2016YFB0401100). The GIXRD results were tested at the BL14B1 Station of Shanghai Synchrotron Radiation Facility (SSRF), 23A1 Station of National Synchrotron Radiation Research Center (NSRRC, Taiwan), and 1W1A Station of Beijing Synchrotron Radiation Facility. The authors gratefully acknowledge the assistance of scientists from the three stations during the experiments.
References
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed.
- F. Zhang, C.-a. Di, N. Berdunov, Y. Hu, Y. Hu, X. Gao, Q. Meng, H. Sirringhaus and D. Zhu, Adv. Mater., 2013, 25, 1401–1407 CrossRef CAS PubMed.
- B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef PubMed.
- H. Xin, C. Ge, X. Yang, H. Gao, X. Yang and X. Gao, Chem. Sci., 2016, 7, 6701–6705 RSC.
- Z. Zhao, Z. Wang, X. Zhang, S. Gao, X. Yang, Z. Duan and X. Gao, ChemPlusChem, 2015, 80, 57–61 CrossRef CAS.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- Q. Wu, M. Wang, X. Qiao, Y. Xiong, Y. Huang, X. Gao and H. Li, Macromolecules, 2013, 46, 3887–3894 CrossRef CAS.
- I. McCulloch, R. S. Ashraf, L. Biniek, H. Bronstein, C. Combe, J. E. Donaghey, D. I. James, C. B. Nielsen, B. C. Schroeder and W. Zhang, Acc. Chem. Res., 2012, 45, 714–722 CrossRef CAS PubMed.
- C. Wei, J. Zou, R. Zhu, J. Huang, D. Gao, L. Wang, W. Zhang, Y. Liao and G. Yu, Dyes Pigm., 2017, 136, 434–440 CrossRef CAS.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- Z. Fei, P. Pattanasattayavong, Y. Han, B. C. Schroeder, F. Yan, R. J. Kline, T. D. Anthopoulos and M. Heeney, J. Am. Chem. Soc., 2014, 136, 15154–15157 CrossRef CAS PubMed.
- Z. Fei, Y. Han, J. Martin, F. H. Scholes, M. Al-Hashimi, S. Y. AlQaradawi, N. Stingelin, T. D. Anthopoulos and M. Heeney, Macromolecules, 2016, 49, 6384–6393 CrossRef CAS.
- L. Zhang, B. D. Rose, Y. Liu, M. M. Nahid, E. Gann, J. Ly, W. Zhao, S. J. Rosa, T. P. Russell, A. Facchetti, C. R. McNeill, J.-L. Brédas and A. L. Briseno, Chem. Mater., 2016, 28, 8580–8590 CrossRef CAS.
- H. Huang, Z. Chen, R. P. Ortiz, C. Newman, H. Usta, S. Lou, J. Youn, Y.-Y. Noh, K.-J. Baeg, L. X. Chen, A. Facchetti and T. Marks, J. Am. Chem. Soc., 2012, 134, 10966–10973 CrossRef CAS PubMed.
- S. H. Yu, K. H. Park, Y.-H. Kim, D. S. Chung and S.-K. Kwon, Macromolecules, 2017, 50, 4227–4234 CrossRef CAS.
- J. Huang, Z. Mao, Z. Chen, D. Gao, C. Wei, W. Zhang and G. Yu, Chem. Mater., 2016, 28, 2209–2218 CrossRef CAS.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 29, 1606217 CrossRef PubMed.
- W. Zhang, K. Shi, J. Huang, D. Gao, Z. Mao, D. Li and G. Yu, Macromolecules, 2016, 49, 2582–2591 CrossRef CAS.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- Q. Wu, R. Li, W. Hong, H. Li, X. Gao and D. Zhu, Chem. Mater., 2011, 23, 3138–3140 CrossRef CAS.
- H. J. Yun, S. J. Kang, Y. Xu, S. O. Kim, Y. H. Kim, Y. Y. Noh and S. K. Kwon, Adv. Mater., 2014, 26, 7300–7307 CrossRef CAS PubMed.
- X. Gao, C.-a. Di, Y. Hu, X. Yang, H. Fan, F. Zhang, Y. Liu, H. Li and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3697–3699 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00236j |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Dong
Gao
a,
Liping
Wang
*c,
Yi
Liao
*d and
Gui
Yu
a,
Dong
Gao
a,
Liping
Wang
*c,
Yi
Liao
*d and
Gui
Yu