DOI:
10.1039/C7QM00229G
(Research Article)
Mater. Chem. Front., 2017,
1, 2085-2093
Synthesis and properties of a series of quinoxaline-based copolymers: an example to understand the effect of the structure of the mainchain and sidechain on the charge transport ability of the polymers†
Received
22nd May 2017
, Accepted 28th June 2017
First published on 29th June 2017
Abstract
Five D–A (donor–acceptor) copolymers P1–P5 were designed and synthesized, with the mainchain constructed with thiophene derivatives as D and a quinoxaline unit as A, and the sidechain constructed with alkyl groups or alkylthienyl groups. All the polymers showed good thermal stability for device fabrication. The absorption spectra revealed that the five polymers covered a wide absorption range from 300 to 727 nm (with P1), and to 755 nm (with P5). Characterization results showed that both the mainchain and sidechain structures played important roles in the optical, electronic and charge transport properties. OTFT devices were fabricated to evaluate the mobilities of copolymers, and P3 with benzodithiophene and quinoxaline moieties as the mainchain and pure linear alkyl groups as the sidechain manifested the best performance.
Introduction
Organic semiconductors based on π-conjugated polymers are widely studied for electronic applications.1–6 These polymers showed great advantages compared to their inorganic counterparts. Polymer based semiconductors can be solution processed, resulting in low-cost, large-area and flexible active matrix display backplanes.7–10 Also, the polymers can be molecularly engineered to have tunable optical and electronic properties through synthesis and assembly, which can make them suitable for meeting various requirements in different electro-optical devices such as organic light emitting diodes (OLEDs), polymer solar cells (PSCs), and organic thin-film transistors (OTFTs).11–13
D–A copolymers with an electron-rich donor and an electron-deficient acceptor in the mainchain were widely studied recently.14–17 These copolymers can present a wide absorption range owing to an additional intramolecular charge transfer (ICT) from D to A and improve the carrier mobility ascribed to the optimized intermolecular arrangement. The opto-electronic properties of the polymers can be easily manipulated by altering the donor or acceptor moieties. Benzothiadiazole (BTZ), quinoxaline (QX), thienopyrazine (TP), thiazolothiazole, ester substituted thieno[3,4-b]thiophene, 3,6-diary-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (DPP), and their derivatives have been commonly used as electron acceptors, while electron donors usually contain units such as thiophene, fluorene, carbazole, silafluorene, cyclopentabithiophene, benzodithiophene and so on.18–26 A wide range of copolymers with the D–A structure exhibit excellent opto-electronic properties and are extensively applied as semiconducting layers in PSCs and OTFTs to obtain high power conversion efficiency and charge mobility.27–36 However, it's still necessary to develop new copolymers to provide more options for organic semiconductors. And for different D–A copolymer systems, carefully selecting and adjusting the mainchain and sidechain units are also very significant to gain satisfactory opto-electronic performance by changing the energy level and intermolecular packing.
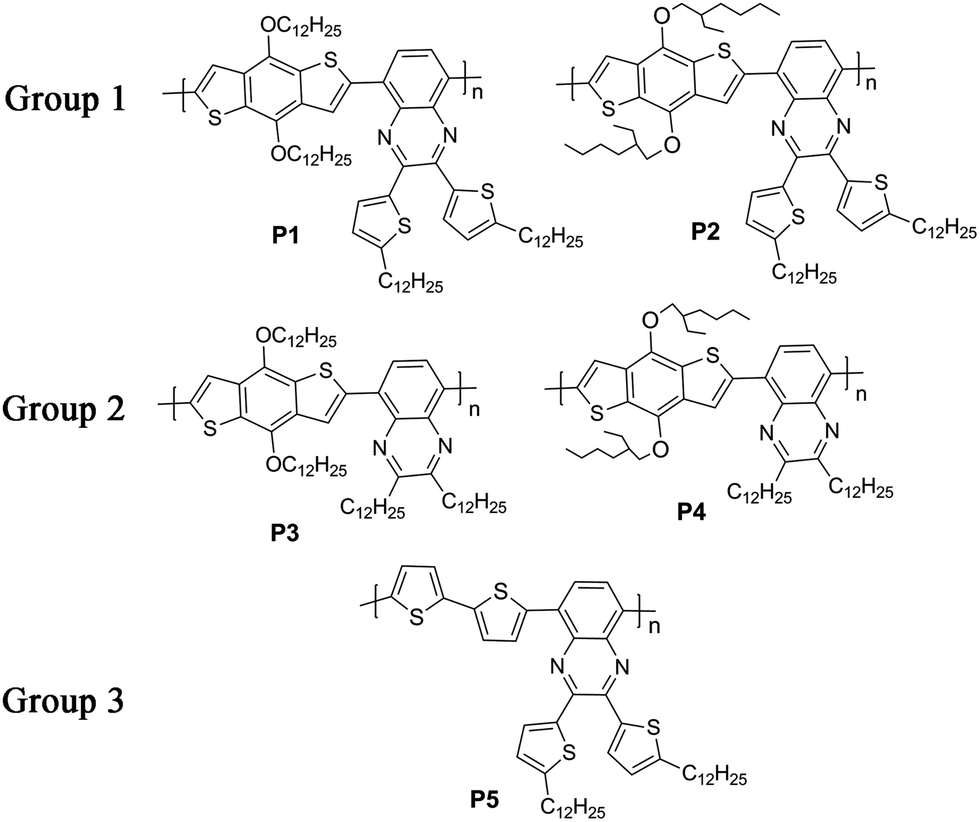
Herein, we designed and synthesized five D–A copolymers based on a quinoxaline unit as A and a benzodithiophene or a bithiophene unit as D with different kinds of alkyl groups as sidechains. Quinoxaline is a typical electron-deficient unit due to its strong electronegativity and can be easily modified by chemical methods to introduce various sidechains into the copolymers.19,37–44 As shown in Fig. 1, various sidechains were introduced into either the donor or the acceptor unit including linear alkyl groups, branched alkyl groups and alkylthienyl groups. For P1 and P2 (Group 1), they had the same acceptor moiety with a dodecylthienyl group as the sidechain on a quinoxaline unit, and different donors with a linear dodecyl or a branched ethylhexyl group as the sidechain on a benzodithiophene unit. For P3 and P4 (Group 2), they showed the same mainchain structure as P1 and P2, except that the sidechain on quinoxaline was a linear dodecyl group instead of a dodecylthienyl group. P5 (Group 3) gave a unique mainchain structure with a bithiophene unit as the donor instead of the benzodithiophene unit in the first four polymers. This article reported the synthesis and thermal, optical, and charge transport properties of these copolymers, and systematically studied the influence of polymer sidechain and mainchain variations on opto-electronic properties by comparing the five copolymers.
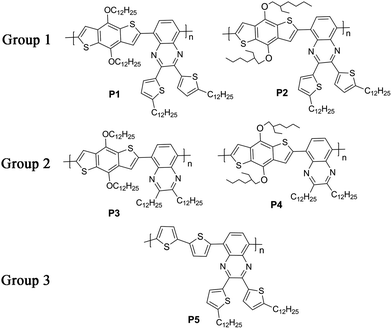 |
| | Fig. 1 The structure of five copolymers P1–P5. | |
Experimental
Materials
Toluene was dried over and distilled from a K–Na alloy under an atmosphere of argon. Compound 1, 2, 3, M1, M2 and M5 were synthesized according to literature procedures or with some modifications (see the ESI†).45–50 Other reagents were obtained from Aldrich and Sinopharm Chemical Reagent Co. (Shanghai, China).
Monomer synthesis
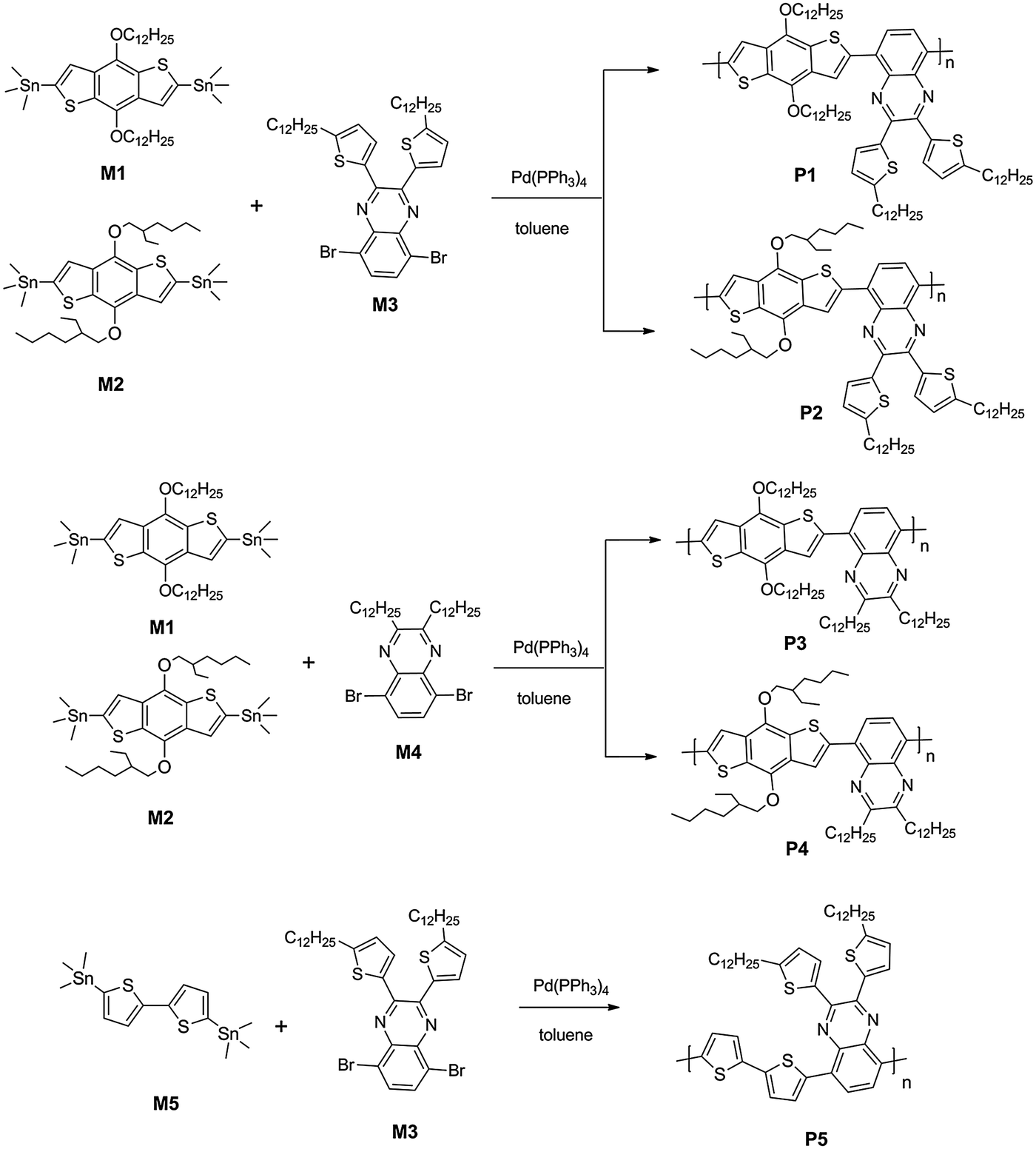
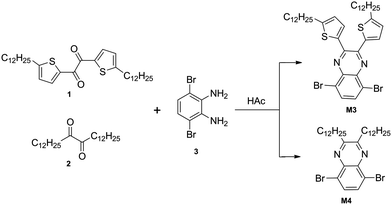
5,8-Dibromo-2,3-bis(5-dodecylthiophen-2-yl)quinoxaline (M3).
Compound 1 (1.31 g, 2.34 mmol) and compound 3 (0.62 g, 2.34 mmol) were mixed in 40 mL of acetic acid and heated to 50 °C under argon for 20 hours. After cooling it to room temperature, the mixture was filtered. The residue was washed with cold ethanol three times. Then the crude product was purified by column chromatography on silica gel with CHCl3/petroleum ether (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) as the eluent to give M3 as yellow powder (1.5 g, 82%). 1H NMR (CDCl3, 300 MHz) δ [ppm]: 7.73 (s, 2H), 7.41 (d, J = 3.3 Hz, 2H), 6.72 (d, J = 3 Hz, 2H), 2.87 (t, J = 7.2 Hz, 4H), 1.77–1.72 (m, 4H), 1.52–1.27 (m, 36H), 0.88 (t, J = 6.6 Hz, 6H). 13C NMR (CDCl3, 75 MHz) δ [ppm]: 152.32, 147.41, 138.66, 138.52, 132.79, 130.57, 125.19, 123.07, 32.17, 31.78, 30.69, 29.90, 29.60, 29.45, 22.94, 14.39. Anal. calcd for C40H56Br2N2S2: C, 60.90; H, 7.16; N, 3.55. Found: C, 60.79; H, 7.17; N, 3.42 (Scheme 1).
:5) as the eluent to give M3 as yellow powder (1.5 g, 82%). 1H NMR (CDCl3, 300 MHz) δ [ppm]: 7.73 (s, 2H), 7.41 (d, J = 3.3 Hz, 2H), 6.72 (d, J = 3 Hz, 2H), 2.87 (t, J = 7.2 Hz, 4H), 1.77–1.72 (m, 4H), 1.52–1.27 (m, 36H), 0.88 (t, J = 6.6 Hz, 6H). 13C NMR (CDCl3, 75 MHz) δ [ppm]: 152.32, 147.41, 138.66, 138.52, 132.79, 130.57, 125.19, 123.07, 32.17, 31.78, 30.69, 29.90, 29.60, 29.45, 22.94, 14.39. Anal. calcd for C40H56Br2N2S2: C, 60.90; H, 7.16; N, 3.55. Found: C, 60.79; H, 7.17; N, 3.42 (Scheme 1).
 |
| | Scheme 1 The synthetic routes to monomers. | |
5,8-Dibromo-2,3-didodecylquinoxaline (M4).
Compound 2 (0.17 g, 0.43 mmol) and compound 3 (0.114 g, 0.43 mmol) were mixed in 10 mL of acetic acid and heated to 50 °C under argon for 20 hours. After cooling it to room temperature, 50 mL of water was added, and the mixture was extracted with chloroform. The organics were collected and dried over anhydrous Na2SO4. The solvent was concentrated and the crude product was purified by column chromatography on silica gel with CHCl3/petroleum ether (v/v, 1:3) as the eluent to give M4 (0.23 g, 86%). 1H NMR (CDCl3, 300 MHz) δ [ppm]: 7.82 (s, 2H), 3.07 (t, J = 7.8 Hz, 4H), 1.91–1.88 (m, 4H), 1.40–1.26 (m, 36H), 0.88 (t, J = 5.4 Hz, 6H). 13C NMR (CDCl3, 75 MHz) δ [ppm]: 158.50, 139.49, 132.17, 123.55, 34.98, 32.15, 29.87, 29.81, 29.74, 29.58, 28.01, 22.91, 14.33. Anal. calcd for C32H52Br2N2: C, 61.54; H, 8.39; N, 4.49. Found: C, 61.63; H, 8.27; N, 4.45.
Polymer synthesis
Synthesis of P1.
Monomer M1 (0.1985 g, 0.2244 mmol), M3 (0.1770 g, 0.2244 mmol) and Pd(PPh3)4 (8 mg, 3 mol%) were mixed in a Schlenk tube filled full with argon. 10 mL of toluene was added to the tube using a syringe, and the solution was heated to 110 °C for 24 h. Then the mixture was cooled to room temperature and the polymer was precipitated by slowly adding the mixture into CH3OH (200 mL). The precipitated material was collected by filtration through a funnel. Then the crude product was washed using a Soxhlet apparatus with methanol, acetone, hexane, and finally extracted with chloroform. Only the chloroform-soluble portion was collected, concentrated, precipitated from methanol, and dried over vacuum to yield P1 as a black solid (0.23 g, 86%). 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.40–7.57 (m, 6H), 6.81 (br, 2H), 4.40 (br, 4H), 2.94 (br, 4H), 1.82 (br, 8H), 1.27 (br, 72 H), 0.87 (br, 12H) (Scheme 2).
 |
| | Scheme 2 The synthetic route to copolymers. | |
The other copolymers P2, P3, P4 and P5 were all synthesized with the procedure similar to P1 with different starting materials.
Synthesis of P2.
The starting materials were M2 (0.2009 g, 0.2567 mmol), M3 (0.2052 g, 0.2567 mmol) and Pd(PPh3)4 (9 mg, 3 mol%). Black solid, yield: 0.24 g, 85%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.30 (br, 2H), 7.61 (br, 4H), 6.79 (br, 2H), 4.43 (br, 4H), 2.92 (br, 4H), 1.79 (br, 6H), 1.26 (br, 52H), 0.95–0.87 (m, 18H).
Synthesis of P3.
The starting materials were M1 (0.1897 g, 0.2145 mmol), M4 (0.1340 g, 0.2145 mmol) and Pd(PPh3)4 (8 mg, 3 mol%). P3 was obtained as a black solid. Yield: 0.19 g, 85%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.39–8.28 (m, 2H), 7.50–7.39 (m, 2H), 4.43 (br, 4H), 3.22 (br, 4H), 2.16 (br, 4H), 1.98 (br, 4H), 1.28 (br, 72H), 0.87 (br, 12H).
Synthesis of P4.
The starting materials were M2 (0.1611 g, 0.2086 mmol), M4 (0.1303 g, 0.2086 mmol) and Pd(PPh3)4 (7 mg, 3 mol%). P4 was obtained as a black solid. Yield: 0.16 g, 86%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.28 (br, 2H), 7.50 (br, 2H), 4.35 (br, 4H), 3.22 (br, 4H), 2.11 (br, 4H), 1.47 (br, 2H), 1.26 (br, 52H), 1.09–0.87 (m, 18H).
Synthesis of P5.
The starting materials were M5 (0.1311 g, 0.2665 mmol), M3 (0.2102 g, 0.2665 mmol) and Pd(PPh3)4 (9 mg, 3 mol%). P5 was obtained as a black solid. Yield: 0.15 g, 70%. 1H NMR (CDCl3, 300 MHz) δ [ppm]: 8.05 (br, 2H), 7.79 (br, 2H), 7.46 (br, 2H), 7.11 (br, 2H), 6.71 (br, 2H), 2.86 (br, 4H), 1.68 (br, 4H), 1.26 (br, 36H), 0.85 (br, 6H).
Results and discussion
Synthesis and characterization
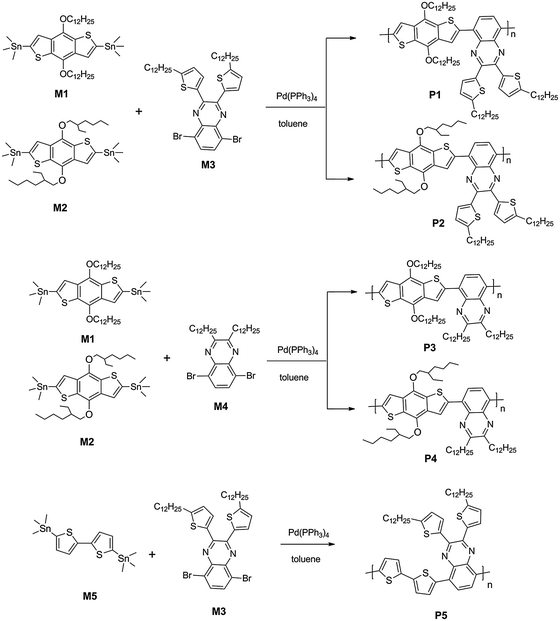
All the copolymers were synthesized by the Stille coupling reaction. The crude products were washed using a Soxhlet apparatus with methanol, acetone, hexane, and finally extracted with chloroform to obtain pure, high molecular weight, solution processable polymers. They showed good solubility in common organic solvents such as chloroform, THF and chlorobenzene. P1, P2, P3 and P4 demonstrated high yields of about 85% after purification whereas P5 was obtained with yield of only 70%. This may be ascribed to the lower solubility of P5 which had fewer sidechains and some high-molecular-weight compositions could not be extracted with chloroform. As a result, P5 had a relatively lower molecular weight than the other copolymers. Molecular weights and polydispersity indices (PDIs) of polymers, as shown in Table 1, were determined by GPC analysis in THF with a polystyrene standard calibration. As the polymer sidechain, a branched alkyl chain may lead to a higher molecular weight than a linear alkyl chain (P2 > P1, P4 > P3), and an alkylthienyl chain also leads to a higher molecular weight than a pure alkyl chain (P1 > P3, P2 > P4). P5 showed the minimum value owing to its worst solubility caused by its fewer sidechains.
Table 1 Molecular weights and thermal properties of the polymers
| |
M
n
(g mol−1) |
M
w
(g mol−1) |
PDIa (Mw/Mn) |
T
d
(°C) |
|
Number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity indices (PDIs, Mw/Mn) determined by means of GPC with THF as the eluent on the basis of polystyrene calibration.
Temperature at 5% weight loss estimated using TGA under N2.
|
|
P1
|
1.23 × 104 |
2.41 × 104 |
1.96 |
301 |
|
P2
|
2.92 × 104 |
6.79 × 104 |
2.33 |
312 |
|
P3
|
9.91 × 103 |
2.14 × 104 |
2.15 |
320 |
|
P4
|
1.34 × 104 |
3.19 × 104 |
2.38 |
323 |
|
P5
|
5.06 × 103 |
1.39 × 104 |
2.75 |
412 |
Thermal stability
The thermal stability of the polymers was investigated by thermogravimetric analysis (TGA), as shown in Fig. 2. The decomposition temperature (Td) at 5% weight loss for P1–P4 is around 310 °C, while that of P5 is much higher with a value of 412 °C. This may be ascribed to the lack of alkoxy chains on the donor unit of P5 which can affect the thermal stability of polymers. Interestingly, the thermal stability trend is consistent with the photostability trend of a similar class of materials published recently.51 In general, all the Td values are high enough for device fabrication processes.
 |
| | Fig. 2 TGA plots of the copolymers with a heating rate of 10 °C min−1 under an inert atmosphere. | |
Optical properties
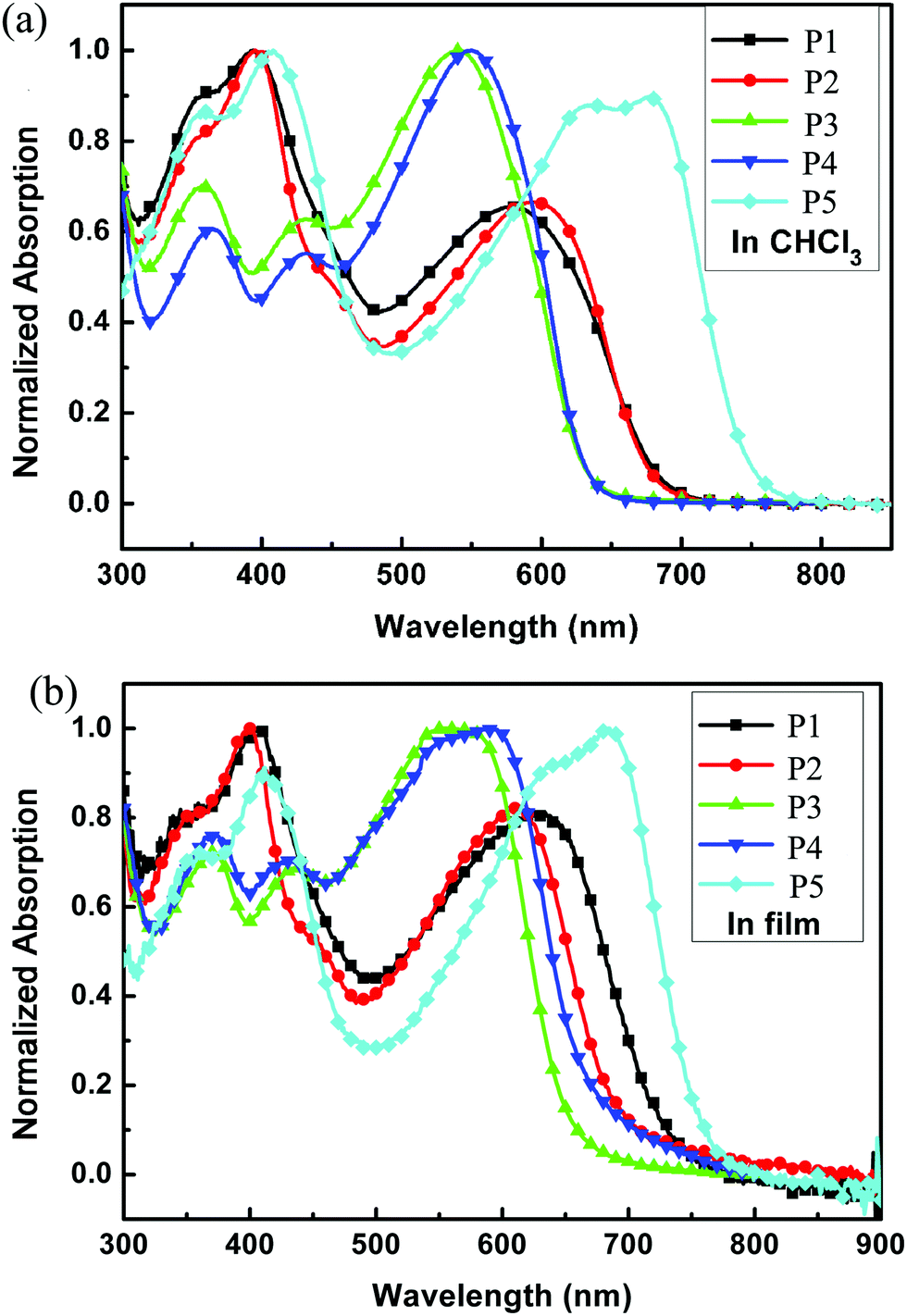
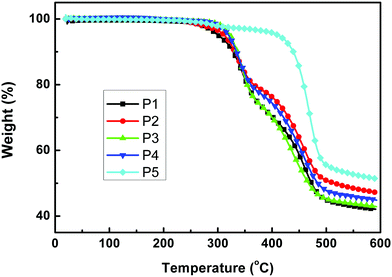
The normalized UV-vis absorption spectra of P1 to P5 were measured in chloroform (ca. 10−5 M) and in thin films spin-coated on quartz substrates (Fig. 3), and the results are summarized in Table 2. All the polymers showed two main absorption areas: one at short wavelengths originating from the π–π* transition and the absorption of sidechain groups, and the longer wavelength absorption should be due to an ICT interaction between the donor and the acceptor along the mainchain. The absorption spectra were quite similar in each group, indicating that the linear or the branched alkyl chain on the donor unit showed little influence on absorption properties when the acceptor units were the same. The absorption intensity of Group 1 at short wavelengths was stronger than that at long wavelengths, and Group 1 showed a wider absorption range than Group 2, indicating that the introduction of the alkylthienyl group into the polymer sidechain can not only enhance the absorption intensity at short wavelengths but also extend the absorption range. Compared to Group 1 and Group 2, P5 manifested a much wider absorption range, which may be because there are more rotatable bonds in the backbone of P5, which will lead to a wide distribution of torsion angles and the corresponding wide absorption spectra. Compared to the spectra in solution, the spectra in films were all red-shifted in the longer wavelength region; 52, 17, 18, 39, 7 nm for P1 to P5 respectively. This indicated that the molecules could be well arranged and could aggregate in the solid state owing to the strong interchain interactions, which may facilitate charge transport. All polymers covered a wide absorption range from 300 to around 700 nm and with P5 up to 755 nm in films, indicating that these polymers are potential candidates for application in PSCs.
 |
| | Fig. 3 UV-vis spectra of all polymers (a) in chloroform solution and (b) in solid films. | |
Table 2 Optical and electrochemical data of the polymers
| |
In CHCl3 |
In film |
CV |
|
λ
peak
(nm) |
λ
onset
(nm) |
E
optgc (eV) |
λ
peak
(nm) |
λ
onset
(nm) |
E
optgc (eV) |
E
oxonset/HOMOd (V/eV) |
LUMOe (eV) |
|
Absorption maxima.
The onset edge absorption.
Estimated from the onset edge of absorption in films: Eoptg = 1240/λonset.
Estimated from the onset oxidation, assuming the absolute energy level of ferrocene/ferrocenium to be 4.8 eV below vacuum.
Calculated from the optical bandgap from the film and HOMO.
|
|
P1
|
353, 394, 581 |
682 |
1.82 |
347, 407, 633 |
727 |
1.71 |
0.34/−5.14 |
−3.43 |
|
P2
|
350, 395, 592 |
682 |
1.82 |
347, 399, 610 |
685 |
1.81 |
0.39/−5.19 |
−3.38 |
|
P3
|
359, 431, 539 |
632 |
1.96 |
370, 436, 556 |
650 |
1.91 |
0.34/−5.14 |
−3.23 |
|
P4
|
365, 432, 550 |
632 |
1.96 |
374, 432, 589 |
666 |
1.86 |
0.31/−5.11 |
−3.25 |
|
P5
|
355, 408, 677 |
745 |
1.66 |
360, 411, 684 |
755 |
1.64 |
0.33/−5.13 |
−3.49 |
Electrochemical properties
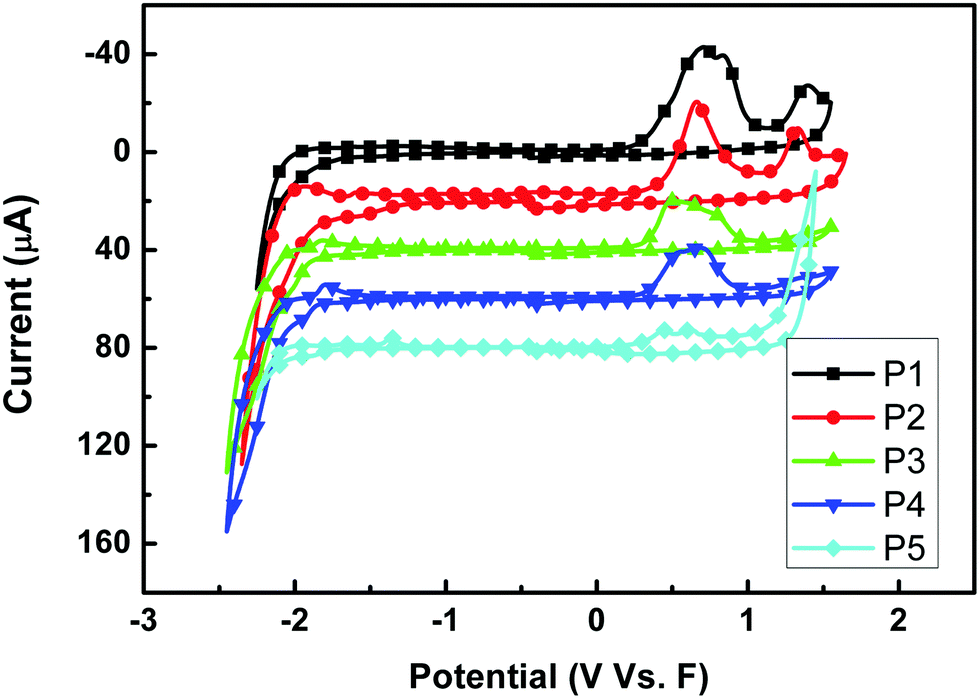
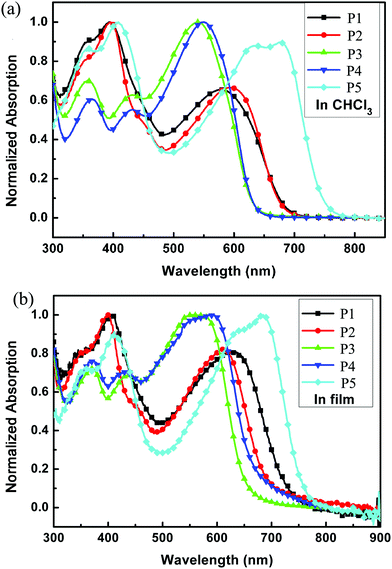
The redox behaviors of P1–P5 were investigated by cyclic voltammetry. Fig. 4 shows the cyclic voltammograms of the five polymer films on a Pt disk electrode in 0.1 mol per L Bu4NPF6 acetonitrile solution. The highest occupied molecular orbital (HOMO) energy levels were estimated from the onset oxidation potentials, assuming the absolute energy level of ferrocene/ferrocenium to be 4.8 eV below vacuum,52–54 and the data are summarized in Table 2. All polymers showed a low HOMO level of about −5.1 eV, indicating that the materials are not susceptible to oxidative doping, which is advantageous when the materials are applied in opto-electronic devices. And different sidechains showed little influence on the HOMO levels when P1 to P4 were compared. However, the alkylthienyl sidechain may lower the LUMO level upon comparing P1 and P3 (or P2 and P4).
 |
| | Fig. 4 Cyclic voltammograms of P1–P5 films on a platinum electrode measured in 0.1 mol per L Bu4NPF6 acetonitrile solutions at a scan rate of 100 mV s−1. | |
Theoretical calculation
The geometries of oligomer structures were optimized at the Density Functional Theory (DFT) ri-BP86/def2-SVP level; then the electronic properties were calculated at the B3LYP/6-31g(d) level.55,56 The frontier energy levels (HOMO and LUMO) were characterized for the neutral electronic states. Note that the calculations were performed on model systems of copolymers where all alkyl chain substituents were replaced with methyl groups (this has only a minimal effect on the electronic properties); so three oligomers of Group 1, Group 2 and Group 3 were calculated in fact.
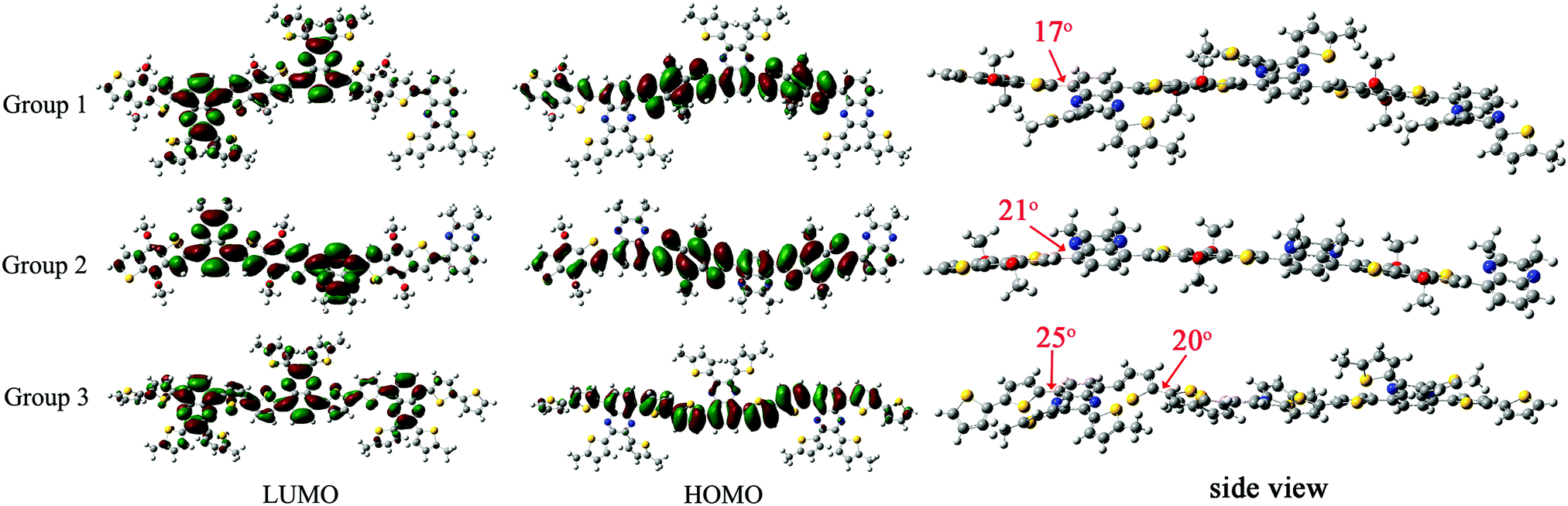
As shown in Fig. 5, the HOMO levels were mainly delocalized on the electron-rich benzodithiophene or bithiophene unit with LUMO levels on the electron-deficient quinoxaline unit. It was worth noting that the LUMO levels in Group 1 were extended to two suspended thiophene groups on the quinoxaline unit, demonstrating that the alkylthienyl group could affect the polymer's energy level when introduced into the molecule as the sidechain. Group 3 showed an obviously narrower bandgap than the other two groups due to its higher HOMO, which was consistent with the experimental data (Table 3). The dihedral angle between adjacent donor and acceptor units in the three sets of polymers are 17°, 21° and 25°, respectively. There are more rotatable bonds in P5 as its donor unit is connected by a single bond with a dihedral angle of 20°, which will lead to a wide distribution of torsion angles and the corresponding wide absorption range.
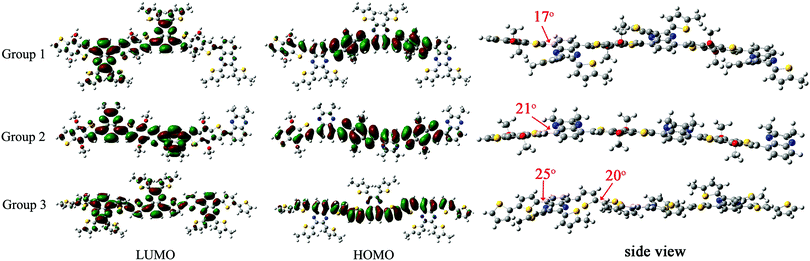 |
| | Fig. 5 LUMO, HOMO levels and dihedral angles between each neighboring unit of the oligomers from theoretical calculation. | |
Table 3 B3LYP/6-31G(d) calculations of the gas-phase HOMO and LUMO levels of the oligomer
| Compound |
HOMO (eV) |
LUMO (eV) |
E
g (eV) |
| Group 1 |
−4.975 |
−2.745 |
2.230 |
| Group 2 |
−4.999 |
−2.768 |
2.231 |
| Group 3 |
−4.865 |
−2.772 |
2.093 |
Charge transport ability
The charge transport ability of the five polymers was evaluated by the OTFT technique under ambient conditions with the most basic architectures: bottom gold contact, bottom gate, and OTS-modified SiO2 dielectric. Devices were annealed at 120 °C under vacuum and tested under ambient air conditions without special precautions. Fig. 6 shows the typical output and transfer curves of the OTFTs using P1–P5 as p-channel semiconductors. The output behaviors demonstrated representative linear and saturation regimes, indicating that the polymers showed typical field-effect transistor performance. The mobility and current on/off ratio estimated in the saturated regime for each polymer are listed in Table 4. When the sidechain group on the acceptor was an alkylthienyl unit in Group 1, P2 with a branched alkyl group on the donor showed higher mobility than P1 with a linear alkyl group on the donor. However, when the sidechain group on the acceptor was an alkyl unit in Group 2, the opposite result was obtained compared to Group 1, as P4 with a branched alkyl group on the donor showed lower mobility than P3. These results revealed that the charge mobility was closely related to the sidechain on both the donor and the acceptor of the polymers. Among the first four polymers, P3 with the minimum molecular weight manifested the highest mobility and the largest current on/off ratio. The sidechains on its donor and acceptor moieties were both linear dodecyl groups, indicating that this choice may give better FET performance when the mainchain was constructed with benzodithiophene–quinoxaline. P5 revealed moderate mobility mainly due to its poor solubility and film-forming ability. As a result, sidechain groups on both donor and acceptor units are of equal importance to gain high mobility, and in specified polymer systems, they should match to give a good molecular arrangement during film formation.
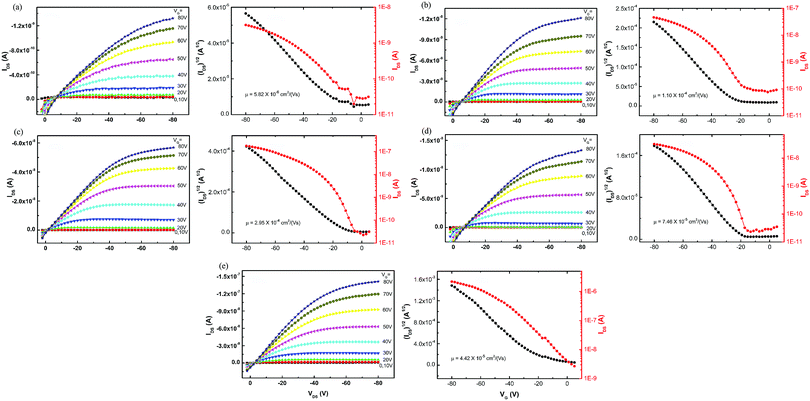 |
| | Fig. 6 The output and transfer curves (VDS = −80 V) of OTFTs using (a) P1, (b) P2, (c) P3, (d) P4 and (e) P5 as semiconductor layers. | |
Table 4 The charge transport ability of polymers evaluated by OTFTs
| Polymer |
μ (cm2 V−1 s−1) |
I
on/off
|
|
P1
|
5.82 × 10−6 |
1.1 × 102 |
|
P2
|
1.10 × 10−4 |
5.4 × 102 |
|
P3
|
2.95 × 10−4 |
5.5 × 103 |
|
P4
|
7.46 × 10−5 |
1.4 × 103 |
|
P5
|
4.42 × 10−5 |
8.4 × 102 |
Film morphology
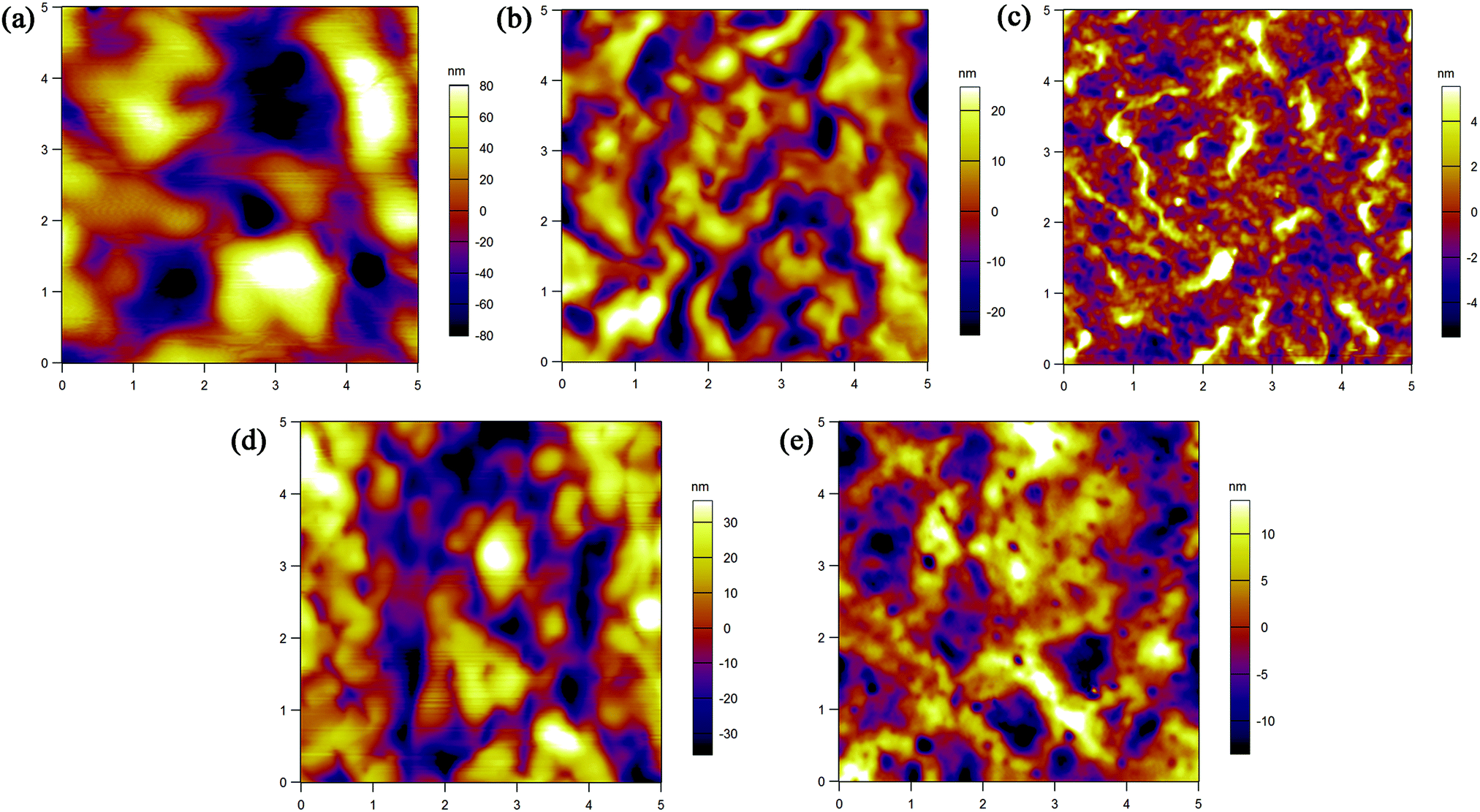
The film morphology of the five polymers was examined using AFM. Fig. 7 shows the tapping-mode AFM height images of spin-coated polymer films on silicon wafers after being annealed at 120 °C for 1 hour. The polymer domains are large with deep boundaries and no obvious crystalline grains are found, which are not advantageous for charge transport. Among the five polymer films, P1 demonstrates a defective arrangement of polymer molecules with big gaps between domains, which may seriously affect the charge transport across polymer domains. P3 shows a relatively uniform surface and small domains, which may cause P3 based FETs to give the best performance. To further evaluate the structural ordering, XRD was carried out (see the ESI†). No Bragg refraction peaks were detected, further verifying the unsatisfactory crystallinity of the films. These results indicate that the structures of both polymer mainchain and sidechain have significant impact on film morphology, and in order to improve the mobility the morphology needs to be carefully optimized.
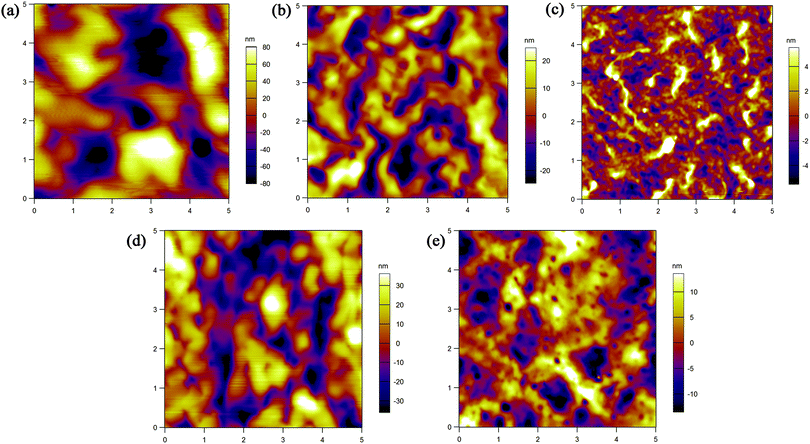 |
| | Fig. 7 AFM height images (5 × 5 μm) of spin-coated (a) P1, (b) P2, (c) P3, (d) P4 and (e) P5 films on silicon wafers after annealing them at 120 °C. | |
Conclusion
We have successfully synthesized five D–A copolymers containing benzodithiophene or bithiophene as the donor and quinoxaline as the acceptor in the mainchain with various sidechains on each unit. All the polymers showed high thermal stability and good solubility and could form films by a solution-processed method. UV-vis spectra revealed that the polymers covered a wide range of absorption in solution, from 300 nm to 682 nm for P1 and P2, to 632 nm for P3 and P4, and to 745 nm for P5. And the absorption peaks were obviously red-shifted in films. The UV-vis measurement also demonstrated that the alkylthienyl group as the sidechain on the acceptor can enhance the short-wavelength absorption and expand the absorption range. P5 with a bithiophene unit as the donor could show a wider absorption range. Electrochemical properties revealed that the sidechains showed little influence on the HOMO level of the polymers among P1–P4, but can affect their LUMO levels. The charge transport ability of the five polymers was evaluated by OTFTs, and P3 with a dodecyl group on both donor and acceptor showed the best FET performance, indicating that a linear alkyl sidechain may be preferable in this system with the polymer mainchain constructed with benzodithiophene and quinoxaline units. In addition, their wide absorption range and moderate charge mobility also suggest their potential application in PSCs.
Acknowledgements
This work was supported by the National Science Foundation of China (no. 20772094). The authors gratefully acknowledge the assistance of Prof. Jinping Zhou from Wuhan University with AFM measurements and Dr Zhengran Yi from Huazhong University of Science and Technology with XRD measurements.
Notes and references
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS PubMed.
- D. J. Harkin, K. Broch, M. Schreck, H. Ceymann, A. Stoy, C. Yong, M. Nikolka, I. McCulloch, N. Stingelin, C. Lambert and H. Sirringhaus, Adv. Mater., 2016, 28, 6378–6385 CrossRef CAS PubMed.
- J. Yu, X. Yin, Z. Xu, P. Deng, Y. Han, B. Zhou and W. Tang, Dyes Pigm., 2017, 136, 312–320 CrossRef CAS.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS PubMed.
- R. Xu, Y. Li and J. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- O. A. Melville, B. H. Lessard and T. P. Bender, ACS Appl. Mater. Interfaces, 2015, 7, 13105–13118 CAS.
- G. Li, X. Gong, J. Zhang, Y. Liu, S. Feng, C. Li and Z. Bo, ACS Appl. Mater. Interfaces, 2016, 8, 3686–3692 CAS.
- Y. Ma, Z. Kang and Q. Zheng, J. Mater. Chem. A, 2017, 5, 1860–1872 CAS.
- D. Chen, C. Zhong, X. Dong, Z. Liu and J. Qin, J. Mater. Chem., 2012, 22, 4343–4348 RSC.
- D. Chen, Y. Yang, C. Zhong, Z. Yi, F. Wu, L. Qu, Y. Li, Y. Li and J. Qin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3852–3862 CrossRef CAS.
- M. K. Pola, K. M. Boopathi, H. Padhy, P. Raghunath, A. Singh, M. Lin, C. Chu and H. Lin, Dyes Pigm., 2017, 139, 349–360 CrossRef CAS.
- M. L. Keshtov, A. R. Khokhlov, S. A. Kuklin, S. A. Osipov, N. A. Radychev, D. Y. Godovskiy, I. O. Konstantinov and G. D. Shrama, Org. Electron., 2017, 43, 268–276 CrossRef CAS.
- M. Wakioka, R. Takahashi, N. Ichihara and F. Ozawa, Macromolecules, 2017, 50, 927–934 CrossRef CAS.
- S. Zhang, N. E. Bauer, I. Y. Kanal, W. You, G. R. Hutchison and T. Y. Meyer, Macromolecules, 2017, 50, 151–161 CrossRef CAS.
- M. L. Keshtov, D. V. Marochkin, Y. Fu, Z. Xie, Y. Geng, V. S. Kochurov and A. R. Khokhlov, Chin. J. Polym. Sci., 2014, 32, 844–853 CrossRef CAS.
- S. Subramaniyan, H. Xin, F. S. Kim, N. M. Murari, B. A. E. Courtright and S. A. Jenekhe, Macromolecules, 2014, 47, 4199–4209 CrossRef CAS.
- J. Yuan, J. Ouyang, V. Cimrová, M. Leclerc, A. Najari and Y. Zou, J. Mater. Chem. C, 2017, 5, 1858–1879 RSC.
- Z. Yi, L. Ma, B. Chen, D. Chen, X. Chen, J. Qin, X. Zhan, Y. Liu, W. J. Ong and J. Li, Chem. Mater., 2013, 25, 4290–4296 CrossRef CAS.
- X. Long, N. Wang, Z. Ding, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2016, 4, 9961–9967 RSC.
- I. Jeong, S. Chae, A. Yi, J. Kim, H. H. Chun, J. H. Cho, H. J. Kim and H. Suh, Polymer, 2017, 109, 115–125 CrossRef CAS.
- X. Gong, G. Li, S. Feng, L. Wu, Y. Liu, R. Hou, C. Li, X. Chen and Z. Bo, J. Mater. Chem. C, 2017, 5, 937–942 RSC.
- D. Liu, Q. Zhu, C. Gu, J. Wang, M. Qiu, W. Chen, X. Bao, M. Sun and R. Yang, Adv. Mater., 2016, 28, 8490–8498 CrossRef CAS PubMed.
- T. A. Shastry, I. Balla, H. Bergeron, S. H. Amsterdam, T. J. Marks and M. C. Hersam, ACS Nano, 2016, 10, 10573–10579 CrossRef CAS PubMed.
- Y. Li, J. Wang, Y. Liu, M. Qiu, S. Wen, X. Bao, N. Wang, M. Sun and R. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 26152–26161 CAS.
- B. Nketia-Yawson, H. Lee, D. Seo, Y. Yoon, W. Park, K. Kwak, H. J. Son, B. Kim and Y. Noh, Adv. Mater., 2015, 27, 3045–3052 CrossRef CAS PubMed.
- Y. Wang, H. Masunaga, T. Hikima, H. Matsumoto, T. Mori and T. Michinobu, Macromolecules, 2015, 48, 4012–4023 CrossRef CAS.
- Y. Deng, J. Quinn, B. Sun, Y. He, J. Ellard and Y. Li, RSC Adv., 2016, 6, 34849–34854 RSC.
- Y. J. Jeong, E. J. Yoo, L. H. Kim, S. Park, J. Jang, S.
H. Kim, S. W. Lee and C. E. Park, J. Mater. Chem. C, 2016, 4, 5398–5406 RSC.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- D. Gedefaw, M. Tessarolo, W. Zhuang, R. Kroon, E. Wang, M. Bolognesi, M. Seri, M. Muccini and M. R. Andersson, Polym. Chem., 2014, 5, 2083–2093 RSC.
- M. Bolognesi, D. Gedefaw, D. Dang, P. Henriksson, W. Zhuang, M. Tessarolo, E. Wang, M. Muccini, M. Seri and M. R. Andersson, RSC Adv., 2013, 3, 24543–24552 RSC.
- R. Hansson, C. Lindqvist, L. K. E. Ericsson, A. Opitz, E. Wang and E. Moons, Phys. Chem. Chem. Phys., 2016, 18, 11132–11138 RSC.
- S. Chen, K. C. Lee, Z. Zhang, D. S. Kim, Y. Li and C. Yang, Macromolecules, 2016, 49, 527–536 CrossRef CAS.
- H. Liu, P. Wu, S. Kuo, C. Chen, E. Chang, C. Wu and Y. Chan, J. Am. Chem. Soc., 2015, 137, 10420–10429 CrossRef CAS PubMed.
- C. Bathula, C. E. Song, W. Lee, J. Lee, S. Badgujar, R. Koti, I. Kang, W. S. Shin, T. Ahn, J. Lee, S. Moon and S. K. Lee, Thin Solid Films, 2013, 537, 231–238 CrossRef CAS.
- Y. Kim, H. R. Yeom, J. Y. Kim and C. Yang, Energy Environ. Sci., 2013, 6, 1909–1916 CAS.
- J. Lee, W. Shin, J. Haw and D. Moon, J. Mater. Chem., 2009, 19, 4938–4945 RSC.
- X. Wang, Y. Zhou, T. Lei, N. Hu, E. Chen and J. Pei, Chem. Mater., 2010, 22, 3735–3745 CrossRef CAS.
- D. D. Kenning, K. A. Mitchell, T. R. Calhoun, M. R. Funfar, D. J. Sattler and S. C. Rasmussen, J. Org. Chem., 2002, 67, 9073–9076 CrossRef CAS PubMed.
- Y. Tsubata, T. Suzuki, T. Miyashi and Y. Yamashita, J. Org. Chem., 1992, 57, 6749–6755 CrossRef CAS.
- R. Sheng, Q. Liu, M. Xiao, C. Gu, T. Hu, J. Ren, M. Sun and R. Yang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1558–1566 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364–10367 CrossRef CAS PubMed.
- D. Gedefaw, M. Tessarolo, M. Prosa, M. Bolognesi, P. Henriksson, W. Zhuang, M. Seri, M. Muccini and M. R. Andersson, Sol. Energy Mater. Sol. Cells, 2016, 144, 150–158 CrossRef CAS.
- J. L. Bredas, R. Silbey, D. S. Boudreaux and R. R. Chance, J. Am. Chem. Soc., 1983, 105, 6555–6559 CrossRef CAS.
- D. W. De Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bssler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00229g |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Cheng
Zhong
b,
Yan
Zhao
c,
Yunqi
Liu
*ab,
Cheng
Zhong
b,
Yan
Zhao
c,
Yunqi
Liu