
3-Piperazinyl propenylidene indolone merocyanines: consecutive three-component synthesis and electronic properties of solid-state luminophores with AIE properties†‡
Melanie
Denißen
a,
Nithiya
Nirmalananthan
b,
Thomas
Behnke
b,
Katrin
Hoffmann
b,
Ute
Resch-Genger
 b and
Thomas J. J.
Müller
*a
b and
Thomas J. J.
Müller
*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstrasse 1, D-40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@hhu.de
bBundesanstalt für Materialforschung und -prüfung (BAM), Richard-Willstätter-Strasse 11, D-12489 Berlin, Germany
First published on 8th June 2017
Abstract
A series of twelve 3-piperazinyl propenylidene indolone merocyanines was synthesized in a one-pot fashion using a consecutive three-component insertion-coupling-Michael addition sequence. Physical-organic treatment of the absorption data of a consanguineous series of this library allows semiquantitative Linear Free Energy Relationships (LFERs) to be established and confirmation of the positive absorption solvatochromicity. All Boc-substituted piperazinyl merocyanines display aggregation induced emission (AIE), which was corroborated for two solvent systems. In particular, crystallization-induced emission enhancement (CIEE) induced by ultrasonication could be shown for a model chromophore by confocal laser scanning microscopy (CLSM).
Introduction
Luminophores are functional π-electron systems1 that are essential molecular constituents in modern illumination technologies, such as organic light-emitting diodes (OLEDs),2 and in fluorescent sensing units in environmental or bioanalytics.3 Therefore, exploration of novel fluorophore scaffolds and their efficient and efficacious synthesis with specific photophysical properties has become a peculiar challenge for organic and materials chemists. Particularly advantageous are diversity-oriented syntheses of functional chromophores,4 such as multicomponent5 and domino reactions,6 where fundamental organic reactions are concatenated to a one-pot sequence.7 Multicomponent reactions (MCRs) are founded on a reactivity based concept,8 where a reactive functionality formed in a previous step will be transformed in the subsequent reaction and so on, however, always in the same reaction vessel without intermediary workup. In the past few years, syntheses of functional chromophores by MCR,9 in particular multicomponent syntheses of fluorophores initiated by metal catalysis,10 have become a powerful preparative tool and have considerably inseminated the field of chromophore research.Although the vast field of luminescence has actively been developed over many decades, there are still aspects that either remained challenging or even became known just recently. A hot topical field is the induction and enhancement of fluorescence upon aggregation. Aggregation induced emission (AIE), a phenomenon coined by Tang's group,11 and aggregation induced enhanced emission (AIEE) first reported by Park's group,12 as well as crystallization-induced emission enhancement (CIEE),13 have become particularly attractive and stimulating for many aspects of luminophore research.14 These phenomena are even more remarkable, since the opposite effects, i.e. aggregation caused quenching (ACQ) and concentration quenching (CQ) or self-quenching, have governed for a long time the perception of the fluorescence of organic dyes in solution or bound to surfaces with the exception of some solid-state emitters.15 From a synthetic point of view, the diversity-oriented synthesis of AIE chromophores by MCR is still in its infancy.16 Based upon insertion–alkynylation–isomerization–Diels–Alder sequences furnishing conformationally rigidified, highly solution and solid-state fluorescent butadiene units in a spiro-indolone corset,17 we became particularly interested in developing one-pot three-component access to ω-amino alkenylidene indolones that can be considered as push–pull chromophores 1–3 (Fig. 1).18 Most impressively, besides high yields for the series 1–3 and excellent stereoselectivity for the formation of chromophores 1 and 3, we found all representatives of the series 1 and 2 to display intense orange to red luminescence only in the solid state (crystal, amorphous or film) with huge Stokes shifts (2600–5600 cm−1) and, surprisingly, the absence of solution luminescence for the same chromophores.
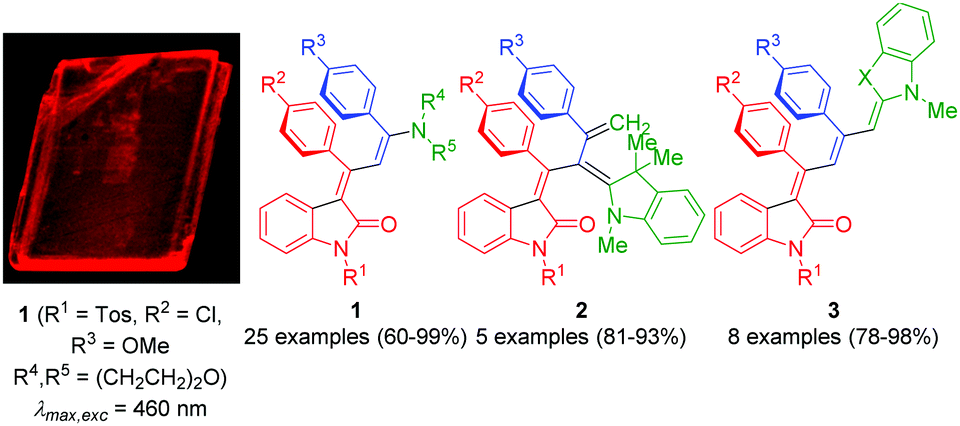 | ||
| Fig. 1 Push–pull chromophores with indolone acceptor moieties 1–3 (right) accessible by consecutive three-component insertion–coupling–addition sequences and red-emissive single crystal (left) of 1 (R1 = Tos, R2 = Cl, R3 = OMe R4,R5 = (CH2CH2)2O), λmax,exc = 460 nm). | ||
From X-ray structural data, a full coplanar arrangement of the push–pull chromophore, additionally stabilized by a parallel π-stacking alignment of aryl substituents with interplanar centroid distances of 3.6 Å, becomes apparent.16,18a Solution solvatochromicity of the longest wavelength absorption band indicates a low polar electronic ground state and the redshift in the crystalline and amorphous solid state supports emissive aggregates of the push–pull chromophores, which, however, clearly differ from J-aggregates with their very narrow and resonant emission.19 For scrutinizing this peculiar emission behavior and for expanding the methodological scope, we reasoned that piperazines as donors could be particularly interesting. Here, we report the consecutive one-pot three-component synthesis of 3-piperazinyl propenylidene indolone merocyanines and their electronic properties.
Results and discussion
Synthesis and structure
The three-component synthesis of 3-aminopropylidene indolones was originally established in THF as a solvent for the initial Pd/Cu-catalyzed insertion-coupling step with the subsequent addition of ethanol as a cosolvent for facilitating the concluding Michael addition.18 A quick orientating screening for alternative single solvent systems revealed that dichloromethane and acetonitrile at elevated temperatures (50–80 °C) furnished equally comparable yields as the THF/EtOH system in the Michael step, however, with considerably shorter reaction times. Acetonitrile as a solvent already required heating in the insertion-coupling step, predominantly due to the solubility issues of the starting 3-aryl propynoyl ortho-bromo anilide. Therefore, in a slight modification to the original protocol,18 upon reaction of 3-aryl propynoyl ortho-bromo anilides 1 and aryl acetylenes 2 under the conditions of the insertion-coupling sequence in dichloromethane at room temperature or acetonitrile at 50 °C, triggered by syn-carbopalladation20,21 and subsequent E,Z-equilibration, the intermediary formed alkynylidene indolones 5 (as monitored by TLC) were subsequently reacted with N-Boc piperazine (3) at 50 (dichloromethane) or 80 °C (acetonitrile) to furnish 3-piperazinyl propenylidene indolones 4 in moderate to excellent yields (Scheme 1 and Fig. 2). The structures of all 3-piperazinyl propenylidene indolones 4 were unambiguously assigned by 1H and 13C NMR spectroscopy and mass spectrometry.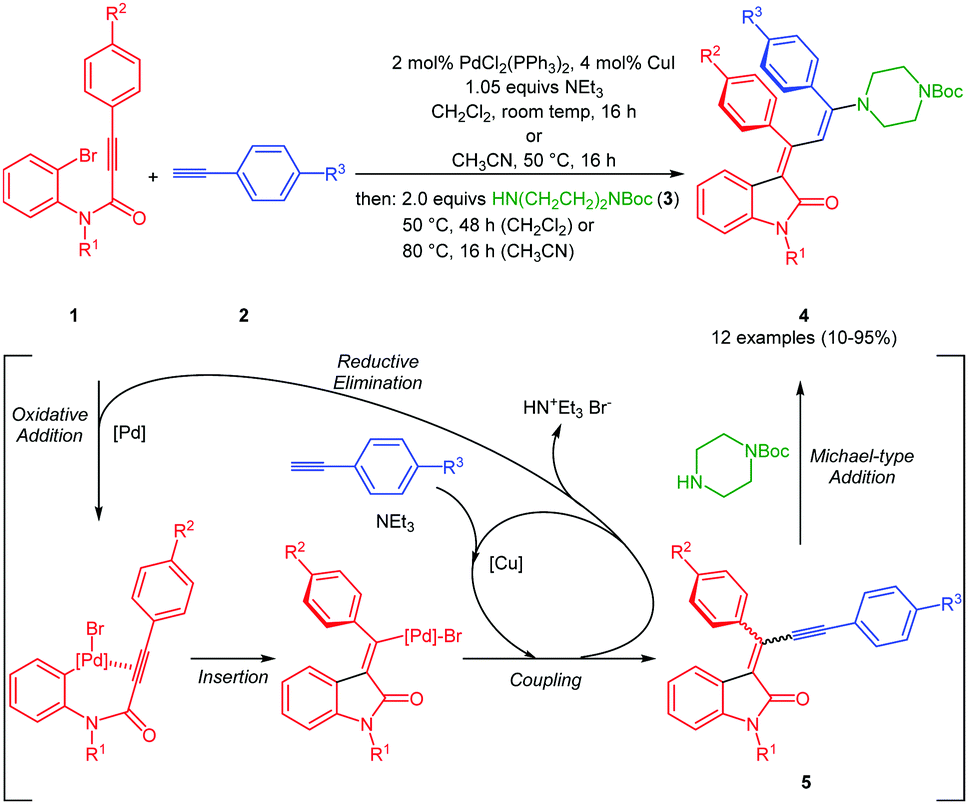 | ||
| Scheme 1 Consecutive one-pot three-component synthesis of 3-piperazinyl propenylidene indolones 4 by an insertion–alkynylation–Michael addition sequence. | ||
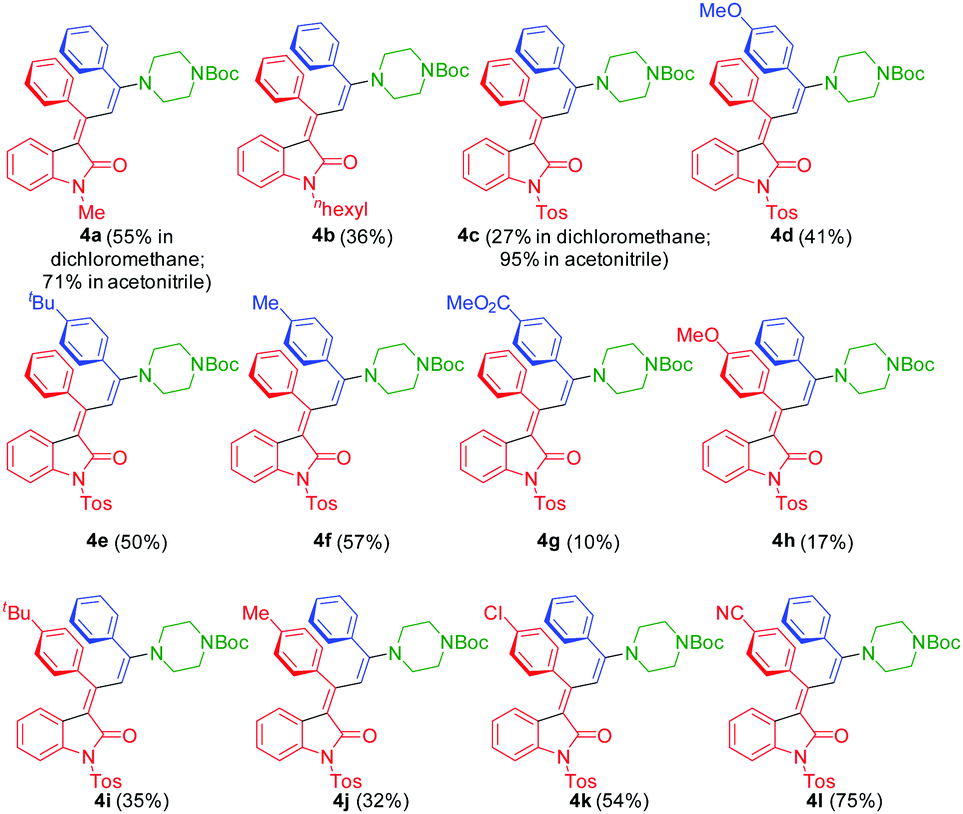 | ||
| Fig. 2 3-Piperazinyl propenylidene indolones 4 synthesized by consecutive three-component synthesis. | ||
The overall stereoselectivity of product formation is additionally supported by the appearance of a single set of signals in the NMR spectra. Expectedly, the proton signals of the substituted piperazine are broadened by rapid ring inversion at room temperature and appear in a narrow range at δ 3.19 to 3.24 and at δ 3.46 to 3.49. In the 13C NMR spectrum for the methylene carbon nuclei only a single resonance at δ 48.8 can be detected. In the 1H NMR spectra, only two protons can be affected by the magnetic anisotropy cones of both aryl substituents on the chromophore, i.e. the H4-benzoproton of the indolone and the only methine proton of the propenylidene sidechain. The indolone H4-benzoprotons appear considerably shifted to a higher field between δ 5.36 and 5.72 as doublets with vicinal coupling constants 3J = 6.8–7.9 Hz. This clearly indicates that they are directly positioned in the anisotropy cone of the neighboring aryl substituents. Interestingly the p-substituent effect of this aryl moiety directly transposes on the shift of this doublet. For the electron releasing methoxy substituent the signal appears at δ 5.72, whereas for the strongly electron-withdrawing cyano substituent the signal is found to be at δ 5.36. Moreover, the distinct upfield shift accounts for a syn-orientation of the adjacent aryl substituent and the H4-benzoproton, i.e. an E-configuration of the α,β-conjugated double bond.
Expectedly, the N-piperazinyl substituents are too remote for affecting the shift of the methine proton in the central butadiene chromophore. In contrast to the indolone H4-benzoprotone, the only methine proton of the propenylidene sidechain does not experience an upfield shift, and thereby no magnetic anisotropy by adjacent, nearly orthogonally oriented aryl substituents can be deduced, and appears as a singlet between δ 7.33 and 7.80, around δ 7.8 for N-alkyl substituted indolones (4a and 4b) and around δ 7.4–7.6 (4c) for N-tosylated derivatives. Therefore, a Z-configuration of the γ,δ-conjugated double bond can be ruled out indicating an E,E-configuration of the indolone fixed butadiene chromophore, as additionally supported by previous published X-ray structure analyses.18
DFT calculations on the model of compound 4c (B3LYP/6-311G(d,p) using the PCM model with acetonitrile as a solvent)22 clearly indicate that the selectively obtained E,E-configured indolone is by 12.4 kJ mol−1 more stable than the E,Z-configured diastereomer. Therefore, the E,E-configured products are the kinetically and thermodynamically favored stereoisomers under the given reaction conditions.
The substitution pattern on all three reactants is reasonably broad, ranging from electron-donating over electroneutral to electron-withdrawing substituents on the alkynyl aryl substituents. For the Michael addition step, electron-withdrawing substituents at R2 enhance the electrophilicity of the corresponding intermediary alkynylidene indolones 5, thereby favoring the Michael addition, which is reflected by the higher yield of compound 4l (75%) bearing a p-cyano group vs. compound 4h (17%) bearing a p-methoxy group. On the other hand, electron-deficient terminal alkynes are poor coupling partners hampering the initial insertion–alkynylation step (compound 4g furnishing an isolated yield of 10%). It is also noteworthy to mention that ortho-bromo anilides can be equally well transformed with the same Pd catalyst precursor, however, at longer reaction times in comparison to the originally employed ortho-iodo anilides.16
Photophysical properties and AIE
As a consequence of the donor–acceptor substitution pattern, 3-piperazinyl propenylidene indolones 4 can be considered as merocyanines, a class of dyes with multiple applications ranging from photosensitizing over photodynamic anticancer therapy to dye sensitized solar cells, nonlinear optics, laser dyes and nanotechnology.23 As a consequence of their polar nature, merocyanines are usually characterized by absorption and/or emission solvatochromism.24When viewed with the naked eye under daylight (Fig. 3a) or UV light (Fig. 3b), differences of the optical properties of the amorphous solid powders of the three consanguineous 3-piperazinyl propenylidene indolones 4a–c (R2 = R3 = H, R4 = Boc), bearing alkyl (4a and 4b) and tosyl (4c) substituents on the indolone nitrogen atom, can already be detected. While the N-methyl derivative 4a is an orange solid with yellow solid state emission, the N-hexyl derivative 4b displays microcrystalline dark red platelets with a greenish luster with essentially no solid state luminescence. The red N-tosyl indolone 4c also displays an intense yellow emission in the solid state. Apparently, long alkyl chains considerably reduce the longevity of the excited solid state by internal conversion through vibrational and rotational channels. Therefore, it is reasonable to only consider N-methyl or N-tosyl substituted indolones in studies of the solid emission behavior.
 | ||
| Fig. 3 3-Piperazinyl propenylidene indolones 4a–c (a) under daylight and (b) under a handheld UV-lamp (λexc = 365 nm). | ||
The push–pull nature of the underlying merocyanine chromophore is additionally supported by the positive solvatochromicity of the longest wavelength absorption maximum of chromophore 4a, i.e., a red shift of the absorption band upon increasing the solvent polarity from methylcyclohexane over tetrahydrofuran, ethyl acetate, dichloromethane, acetone, dimethylsulfoxide, and acetonitrile to methanol. This can be noticed by the naked eye (Fig. 4a) and originates from the absorption spectra (Fig. 4b).
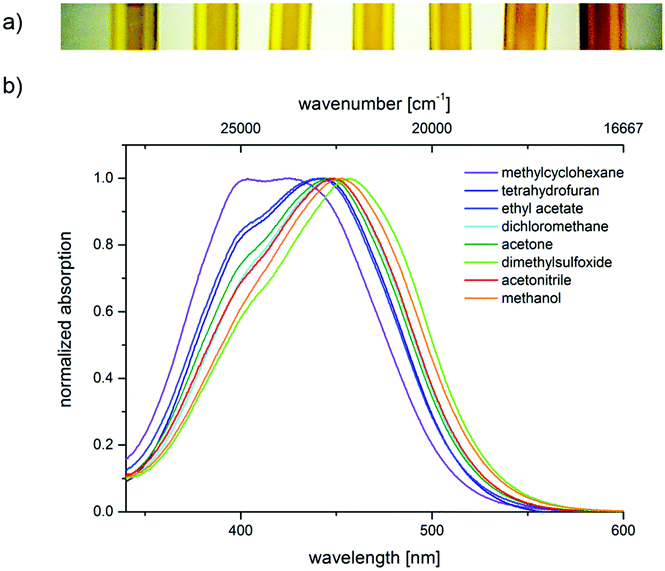 | ||
| Fig. 4 Absorption solvatochromicity of 3-piperazinyl propenylidene indolone 4a (a) under daylight (from left to right: solutions in methylcyclohexane, ethyl acetate, tetrahydrofuran, acetone, acetonitrile, methanol and dimethylsulfoxide) and (b) normalized absorption spectra recorded in methylcyclohexane, tetrahydrofuran, ethyl acetate, dichloromethane, acetone, dimethylsulfoxide, acetonitrile, and methanol at T = 293 K. | ||
The solvatochromicity of a dye is predominantly affected by polarization and dispersion interactions as well as by the underlying mesomeric resonance stabilization.25 A closer inspection of the solvatochromicity of merocyanine 4a reveals distinct solvent effects. In unpolar solvents such as methylcyclohexane, two absorption bands are detected, with the higher energy band gradually disappearing. In contrast, in polar solvents such as methanol, only a broad absorption band is found. While the absorption maximum shifts to the red, here from methylcyclohexane (23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 cm−1) to DMSO (21900 cm−1), the absorption coefficient ε is shifted hyperchromically. This supports the assumption that the longest wavelength absorption band has considerable charge transfer character, whereby the S1 state is associated with a charge shift from the 3-amino donor to the indolone carbonyl acceptor as confirmed by quantum chemical calculations.16 The positive solvatochromicity can be determined quantitatively by a linear Dimroth–Reichardt correlation of the energy of the longest wavelength absorption energy in cm−1 against the molar electronic transition energy of a betain dye for the corresponding solvent in kcal mol−1 (Fig. 5).26
600 cm−1) to DMSO (21900 cm−1), the absorption coefficient ε is shifted hyperchromically. This supports the assumption that the longest wavelength absorption band has considerable charge transfer character, whereby the S1 state is associated with a charge shift from the 3-amino donor to the indolone carbonyl acceptor as confirmed by quantum chemical calculations.16 The positive solvatochromicity can be determined quantitatively by a linear Dimroth–Reichardt correlation of the energy of the longest wavelength absorption energy in cm−1 against the molar electronic transition energy of a betain dye for the corresponding solvent in kcal mol−1 (Fig. 5).26
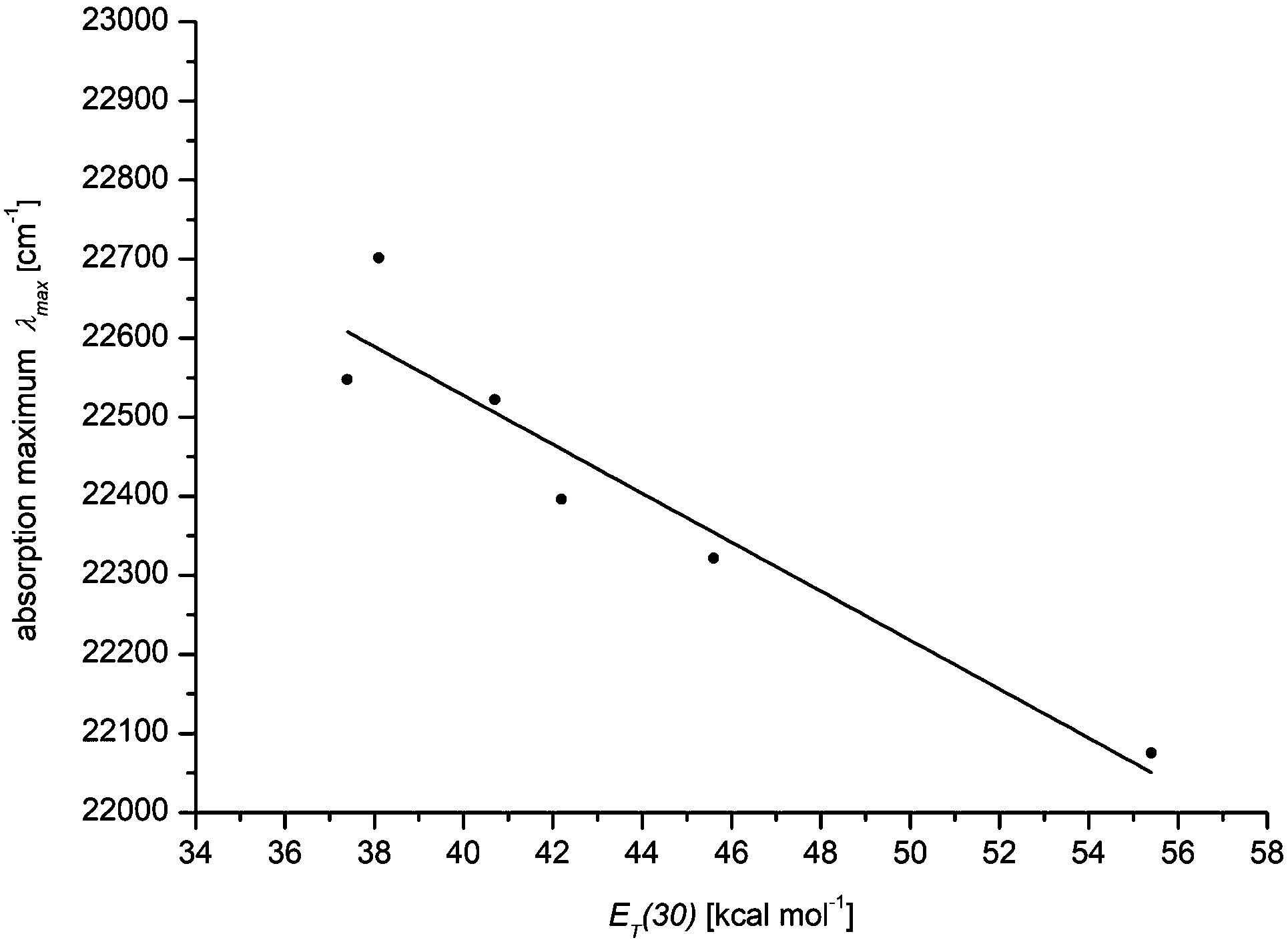 | ||
| Fig. 5 Empirical Dimroth–Reichardt plot of the absorption maxima of merocyanine 4a [cm−1] against the corresponding solvent parameter ET(30) [kcal mol−1] λmax = −30.96·ET(30) + 23766 (r2 = 0.88) (recorded at T = 293 K). | ||
The electronic properties of 3-piperazinyl propenylidene indolones can be altered by the acceptor capacity of the indolone N-substituent. The normalized absorption spectra of merocyanines 4a and 4c clearly show that the electron withdrawing tosyl substituent of merocyanine 4c causes a bathochromic shift of the longest absorption band (λmax = 476 nm, ε = 20550 L mol−1 cm−1) by 1500 cm−1 in comparison to the methyl substituted dye 4a (λmax = 444 nm, ε = 25660 L mol−1 cm−1) (Fig. 6).
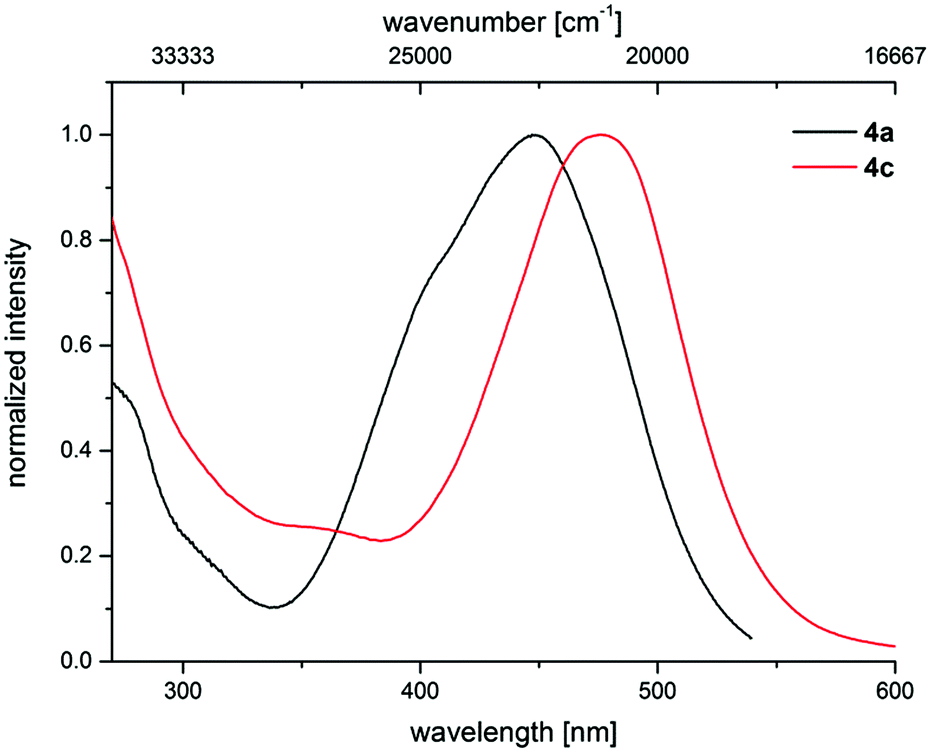 | ||
| Fig. 6 Effect of the acceptor strength varied by N-indolone substitution on the normalized UV/Vis spectra of merocyanines 4a (R1 = Me) and 4c (R1 = Tos) (recorded in CH2Cl2 UVASOL® at T = 293 K). | ||
Although the changes in λmax of the two consanguineous N-tosyl substituted 3-piperazinyl propenylidene indolone series 4c–g, and 4c and 4h–l (Table 1) only occur in a very narrow window, i.e. between 474 and 483 nm, we sought for possible linear structure–property relationships based upon Hammett–Taft correlations, thereby also scrutinizing Dewar's qualitative prediction rules27 in this case.
| Entry | Merocyanine | R2 | R3 | λ max,abs [nm] | ε [L mol−1 cm−1] |
|---|---|---|---|---|---|
| 1 | 4c | H | H | 476.0 | 20550 |
| 2 | 4d | H | OMe | 483.0 | 30580 |
| 3 | 4e | H | t Bu | 482.5 | 29540 |
| 4 | 4f | H | Me | 481.0 | 24240 |
| 5 | 4g | H | CO2Me | 474.0 | 22030 |
| 6 | 4h | OMe | H | 479.0 | 27370 |
| 7 | 4i | t Bu | H | 477.5 | 29380 |
| 8 | 4j | Me | H | 478.0 | 27800 |
| 9 | 4k | Cl | H | 477.5 | 26190 |
| 10 | 4l | CN | H | 482.0 | 25700 |
According to Dewar's rules based upon the first-order perturbation theory, an alternating electron distribution along a merocyanine dipole can be assumed, where substituents placed at marked and unmarked positions of the dipole can quickly predict the energy levels of the HOMO or LUMO. This heuristic predictive concept has previously been applied to the tailored design of merocyanines.28–30 The variation of the para-substituents R2 and R3 of the vicinal phenyl rings will affect the unmarked positions in the conjugated π-system (Fig. 7). Electron releasing substituents R2 and R3 should cause similar increases in the HOMO–LUMO gap and, therefore, a hypsochromic shift of λmax should result. Likewise, electron-withdrawing substituents should lead to bathochromic shifts in both cases.
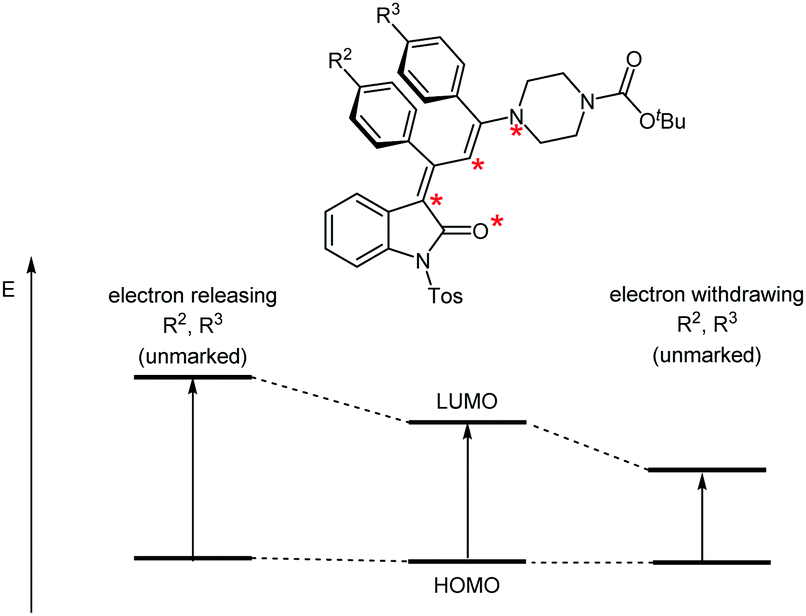 | ||
| Fig. 7 Transposition of Dewar's qualitative rules for controlling the chromogenic properties by the substitution pattern of N-tosyl 3-piperazinyl propenylidene indolone merocyanines 4c–l. | ||
As previously shown, the Reichardt–Hammett correlations (linear free enthalpy relationships) have been successfully applied to quantitatively rationalize the influence of substituent effects on the electronic excitation spectra of pyridinium-N-phenolate betains.
However, correlation studies with various Hammett–Taft parameters31 reveal a quite deviating picture from Dewar's simple first-order perturbation based predictions, and this simple approach fails completely for N-tosyl 3-piperazinyl propenylidene indolone merocyanines. Although λmax in the series 4c–g displays only weak linear correlations with σp (r2 = 0.788), σR (r2 = 0.739), σp+ (r2 = 0.766), and σp− (r2 = 0.665) for the remote substituent R3 (see ESI‡), the positive slopes of the reaction parameters indicate a destabilization of the LUMO by virtue of the blue shift of λmax as a consequence of introducing electron deficient polar substituents, even in the remote position of the substituent R3.
For the remote substituent R2 in the series 4c and 4h–l, another electronic scenario evolves. Even though all attempts to establish linear correlations between λmax and σp, σR, and σp+ for the remote substituent R2 completely fail (for correlation graphs, see ESI‡), interestingly, for the correlation with σp−, a discontinuous behavior at R2 = H can be quickly recognized (Fig. 8).
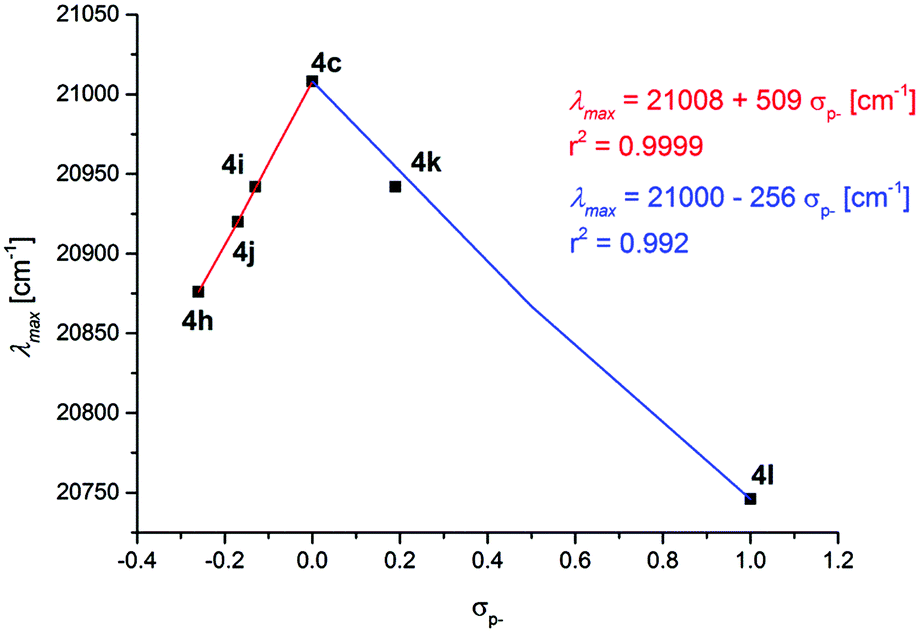 | ||
| Fig. 8 Discontinuous Hammett correlation of λmax [cm−1] vs. σp- for substituents R2 in the consanguineous series 4c and 4h–l (recorded in CH2Cl2 UVASOL® at T = 293 K). | ||
For donor substituents R2 = OMe (4h), tBu (4i), and Me (4j) including the electroneutral substituent R2 = H (4c), an excellent linear correlation is found (r2 = 0.9999) and the positive reaction parameter of 509 indicates a blue shift in the relevant HOMO–LUMO transition with decreasing donor strength. This behavior is comparable to the substituent effect of R3 at the adjacent aryl moiety in the unmarked 3-position of the chromophore. At the electron neutral substitution R2 = H (4c), however, a change of the mechanism for charge stabilization becomes effective. Likewise, for the two acceptor substituents, R2 = Cl (4k) and CN (4l) including the electroneutral substituent R2 = H (4c) an excellent linear correlation is found (r2 = 0.992). The negative reaction parameter of −256 indicates a red shift in the relevant HOMO–LUMO transition with increasing acceptor strength. This finding is generally in agreement with Dewar's rules, however, the overall analyses clearly support the thesis that both electron withdrawing and electron donating substituents can be employed for lowering the HOMO–LUMO gap, yet, with stronger effects resulting for donor substitution. The proximity of the R2-substituted aryl moieties to the merocyanine's indolone acceptor fragment and the σp− correlation exerted by the conjugated indolone acceptor, yet, with a discontinuous behavior, generally rationalizes this stronger influence over R3-substituted aryl moieties in agreement with the findings in the literature for other push–pull chromophores.32 However, it has to be mentioned that N-tosyl 3-piperazinyl propenylidene indolone merocyanines 4 are conformationally dynamic in solution with significant absorption positive solvatochromicity, indicating a highly polar excited state (vide supra).
As already reported for the initial series of 3-amino propenylidene indolones 118, all N-tosyl 3-piperazinyl propenylidene indolone merocyanines 4 are nonluminescent in dichloromethane solution but are luminescent in the solid state (vide supra). This suggests that upon aggregation, intramolecular motions, such as rotations and vibrations of aryl substituents, favoring radiationless deactivation, become considerably restricted.33 This is the operating mechanisms of AIE or AIEE, inducing emission from the lowest vibrational level of the excited singlet state.15
In order to confirm the AIE effect, aggregation studies have been performed with a dilution series of merocyanines 4c, 4i, and 4l in different acetonitrile/water as well as THF/water mixtures. Increasing the water content from 0% to a maximum of 98% in the mixtures reduces the solubility of the hydrophobic dyes and induces the formation of dye aggregates. By recording optical spectroscopic properties like absorption and emission spectra, quantum yields, and fluorescence lifetimes, the properties of the aggregated fluorophores were studied. Here we present exemplarily the absorption and emission spectra of merocyanine 4c in different mixtures of water and acetonitrile (Fig. 9). The results obtained for the chromophores 4i and 4l are shown in the ESI.‡
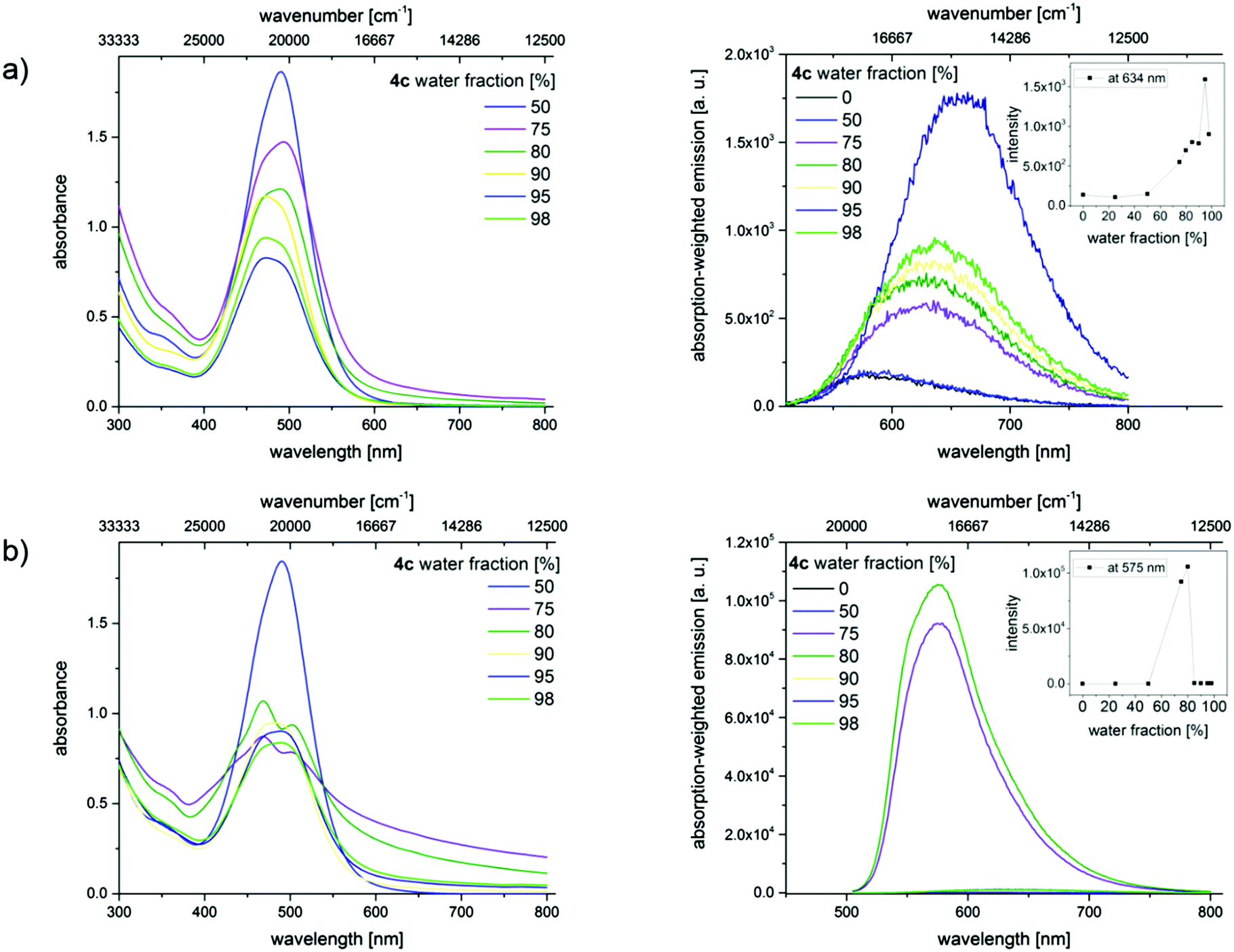 | ||
| Fig. 9 Absorption (left) and absorption-weighted emission spectra (right) of merocyanine 4c in acetonitrile/water mixtures containing different water fractions (a) without sonication and (b) with sonication at T = 293 K (λexc = 484 nm). The insets depict the change of the emission intensity with different water fractions. Absorption-weighted emission spectra, which consider the absorption factor at the excitation wavelength, correlate directly with Φf and present hence a measure for this quantity. | ||
These changes in solvent composition lead to a slight broadening of the absorption band at 484 nm, a decrease in absorbance, and an enhancement in the intensity of the red-shifted emission band with increasing water content. All of these point to the restriction of intramolecular rotations. A rather unusual fluorescence behavior was noticed upon sonication, whereby energy is introduced into the system. Similar ultrasound-sensitive fluorescence features were reported by Sun et al. for a different dye class, here diphenylamine (DPA)-derived dyes.34
The absorption spectra of the dyes in the solvent mixtures with water contents of 75% and 80% show a scattering background, which provides a first hint for the formation of larger aggregates. The broadening of the non- or barely shifted absorption bands indicates an increasing heterogeneity of the dye's micro-environment. Additionally, the absorption band shows a shoulder which may indicate the presence of a second species. Simultaneously, significant changes in the emission characteristics become evident. This comprises blue shifts in emission and a strong fluorescence enhancement. Lower or higher water contents do not lead to dye aggregation. Similar results were obtained for 4i and 4l (see ESI‡).
The optical properties of the dye aggregates were subsequently studied in more detail. The fluorescence quantum yields (Φf) and fluorescence lifetimes (τ) of the different dyes in various solvent mixtures excited at the respective absorption maxima 484 nm (4c), 475 nm (4i), and 484 nm (4l) are shown in Table 2. In pure acetonitrile, the Φf and τ of the three dyes are too small to be measured within a reasonable time frame. As follows from Table 2, the aggregates derived from acetonitrile with a water content of 75% show the highest Φf for 4l followed by 4c and 4i, respectively. A similar trend is observed for τ. The structure with the electron acceptor substituent (4l, R3 = CN) shows the highest charge increment and the derivative with the electron donating (4i, R3 = tBu) the lowest one, respectively.
| Water fraction [%] | Φ f | τ [ns] | |
|---|---|---|---|
| 4c | 0 | <0.01 | <0.1 |
| 75 | 0.026 | 0.315 | |
| 4i | 0 | <0.01 | <0.1 |
| 75 | 0.01 | 0.943 | |
| 4l | 0 | <0.01 | <0.1 |
| 75 | 0.038 | 0.145 | |
Since aggregate morphology can be affected by ultrasonic treatment,34 we monitored the aggregation of the ultrasound-sensitive dye 4c in acetonitrile/water mixtures with confocal laser scanning microscopy (CLSM). After dye addition, amorphous non-emissive aggregates were observed. Ultrasound treatment for 30 min, respectively, led to the formation of ordered crystalline needles with sizes of about 5 μm and also affected the fluorescence properties of these dye aggregations as confirmed by spectrally resolved emission measurements shown in panel b of Fig. 10. The strong fluorescence induced by the ultrasound treatment indicates the restriction of molecular rotations in these aggregates.
 | ||
| Fig. 10 (a) CLSM images of merocyanine 4c derived from an acetonitrile/water mixture with a water content of 75% (left panel: transmission image of amorphous aggregates; right panel: fluorescence image of 5 μm fluorescent crystals) and (b) CLSM emission spectra. | ||
A similar study was performed with merocyanines 4c, 4i, and 4l using less polar THF. The corresponding absorption and emission spectra of 4c are shown in Fig. 11 and the results obtained for 4i and 4l are given in the ESI.‡ Spectroscopic measurements performed with dye 4c without ultrasound treatment provide clear hints for the presence of aggregates, i.e., a scattering background (apparent absorption) that increases at shorter wavelengths and a broadening and red-shift of the absorption band, which decreases in intensity. An enhancement of the emission was, however, not observed. A high amplification of the fluorescence occurs solely upon treatment with ultrasound, which induces the formation of crystals as follows from the CLSM images depicted in Fig. 10. Apparently, extra energy is required for the induction of crystal formation and such crystalline structures are a prerequisite for a strong fluorescence enhancement, and not just aggregate formation. This behavior presents a striking example of crystallization-induced emission enhancement (CIEE),13 a phenomenon recently reported also for other systems like for the BF2-complex 1-phenyl-3-(3,5-dimethoxyphenyl)-propane-1,3-dione. In the case of dye 4c, Φf and τ are thus higher and slightly longer compared to the fluorescence properties of the dye aggregates derived from the acetonitrile/water mixture without ultrasound treatment and exceed those of merocyanines 4i and 4l.
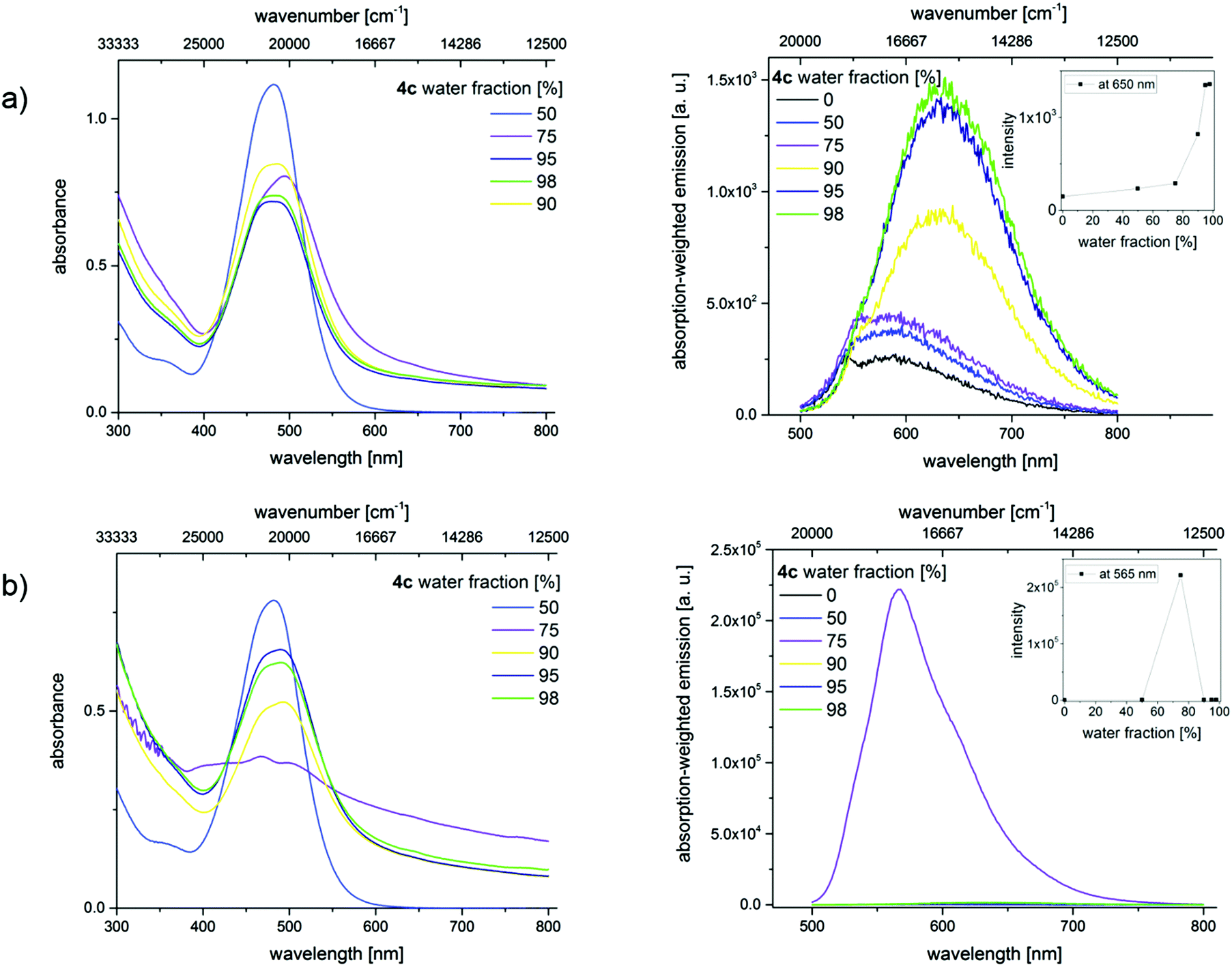 | ||
| Fig. 11 Absorption (left) and absorption-weighted emission spectra (right) of dye 4c in THF/water mixtures containing different water fractions (a) without sonication and (b) with sonication at T = 293 K (λexc = 466 nm). The insets in the different panels depict the change in emission intensity accompanying the different water fractions. | ||
The results from testing of other solvent mixtures regarding the formation of amorphous and crystalline aggregates are shown in the ESI‡ (see Fig. SI-10–SI-13). In THF–water mixtures, merocyanine 4l revealed CIEE, although such effects are absent in acetonitrile–water mixtures. For dye 4i, we did not observe CIEE in any of the solvent mixtures assessed. These observations are summarized in Table 3 for the three dyes 4c, 4i, and 4l in two different THF–water mixtures. This underlines the superior behavior of dye 4c compared to dye 4l and particularly dye 4i, which does not show CIEE under the conditions used by us.
| Water fraction [%] | Φ f | τ [ns] | |
|---|---|---|---|
| 4c | 0 | <0.01 | <0.1 |
| 75 | 0.026 | 0.315 | |
| 4i | 0 | <0.01 | <0.1 |
| 75 | 0.01 | 0.943 | |
| 4l | 0 | <0.01 | <0.1 |
| 75 | <0.01 | <0.1 | |
Exemplarily for compound 4c, we compared Φf and τ of the dye aggregates with the fluorescence properties of dye aggregates derived from an acetonitrile/water mixture with ultrasound treatment. The results are shown in Fig. 11. A comparison with the spectroscopic features of 4i and 4l shown in the ESI‡ (Fig. SI-10–SI-13) reveals the superior features of this dye under the conditions used in this study. These observations are highlighted in Table 3 for 4c, 4i, and 4l in two different THF–water mixtures. The comparison of Φf and τ underlines the favorable properties of merocyanine 4c compared to dye 4l, revealing smaller Φf and τ in a THF/water mixture than in an acetonitrile/water mixture after ultrasonication, and particularly compared to compound 4i. Although ultrasonication of 4i leads to a transformation of amorphous aggregates into more stable crystals, we did not observe CIEE under the conditions used by us. Only for the unpolar tBu-substituted dye 4i in the THF/water mixture, no ultrasound-induced rearrangement could be observed. A possible explanation could be here the higher stability of the initially formed amorphous aggregates.
Experimental
General considerations
All reactions were performed in flame-dried Schlenk tubes under a nitrogen atmosphere. The reaction process was qualitatively monitored by thin layer chromatography using silica gel layered aluminium foil (60 F254 Merck, Darmstadt). For detection, UV light of wavelengths of 254 and 366 nm were employed. Column chromatography was performed using silica gel 60 (Macherey Nagel), mesh 230–400. All commercially available chemicals were purchased from Sigma Aldrich, Alfa Aesar, Fluorochem, and ACROS and were used as received without any further purification. 1H and 13C NMR spectra were measured using a Bruker Avance III-300 or Bruker Avance III-600 spectrometer. Chemical shifts are given in ppm (δ) and were referenced to the internal solvent signal: CDCl3 (1H δ 7.26, 13C δ 77.2) or acetone-d6 (1H δ 2.50, 13C δ 39.5). Multiplicities are stated as: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublet), ddd (doublet of doublet of doublet), and m (multiplet). Broad signals are assigned as br. Coupling constants (J) are given in Hz. The assignment of primary (CH3), secondary (CH2), tertiary (CH) and quaternary carbon nuclei (Cquat) was made using the DEPT-135 spectra. Mass spectrometric investigations were carried out in the Department of Mass Spectrometry of the Institute of Inorganic and Structural Chemistry, Heinrich-Heine-Universität Düsseldorf. IR spectra were recorded using a Shimadzu IRAffinity-1. The intensities of the IR bands are abbreviated as w (weak), m (medium), and s (strong). Absorption spectra were recorded in various spectroscopy grade solvents at 293 K using a PerkinElmer UV/VIS/NIR Lambda 19 spectrometer. Emission spectra of drop-casted films were recorded at 293 K using a Perkin Elmer LS55 spectrometer. Emission spectra in the solution were recorded at 293 K using a Hitachi F7000 spectrometer. The molar extinction coefficients were carried out in a multipoint setup. Melting points (uncorrected) were measured using a Büchi Melting Point B-540. Combustion analyses were measured on a Perkin Elmer Series II Analyser 2400 in the Institute of Pharmaceutical and Medicinal Chemistry, Heinrich-Heine University, Düsseldorf.Instrumentation
Absorption spectra were recorded on a two-beam spectrometer from BRUINS INSTRUMENTS (OMEGA 10) in a 0°/180° geometry. The scan range was set to 300 nm to 800 nm and the slit width and step width were set to 1 nm.Fluorescence spectra were measured on the fluorometer from Edinburgh Instruments (FSP 920) in 0°/90° excitation–emission geometry. The fluorescence spectra were recorded with an excitation slit width of 2 nm, an emission slit width of 4 nm, an integration time of 0.2 s, a step width of 1 nm and a scan range of 500 nm to 800 nm and while three scans for each sample were taken.
The fluorescence lifetime (τ), the average time in which the fluorophore is in an excited state before it relaxes to the ground state, was recorded on a fluorometer FLS 920 (Edinburgh Instruments) equipped with a MCP-PMT as a detector and a supercontinuum laser (Fianium SC400-2-PP) at a frequency of 10 MHz. All of the samples were excited at their absorption maxima, while the emission was detected at the emission maximum of each sample employing a spectral bandwidth of the excitation and emission monochromator of 8 nm, applying a 4096 channel setting and a time range of 20 ns, 50 ns or 100 ns, respectively. All the measurements were performed with air saturated dye solutions at T = 298 K using 10 mm × 10 mm quartz cuvettes from Hellma GmbH always filled with 3 mL of solvent or dye solution. All the dye aggregate measurements were done immediately after sample preparation. The lifetime measurements were analyzed with the FAST program and fitted with a reconvolution fit. All the lifetimes could be evaluated mono, bi- or tri-exponentially with a reduced χ2 between 0.8 and 1.2. From the multi-exponential decays, the intensity-weighted average lifetimes were calculated.
Photoluminescence quantum yields (Φf), the ratio of the number of emitted photons to the number of absorbed photons, were determined with an integrating sphere setup from Hamamatsu (Quantaurus-QY C11347-11).35 All the Φf measurements were performed with specific long neck quartz cuvettes from Hamamatsu at T = 298 K.36
For confocal laser scanning microscopy (CLSM) measurements, crystals formed in the suspension were transferred onto a 24 × 65 mm coverslip and analyzed with an Olympus FluoView™ FV1000 CLSM system (Olympus GmbH, Hamburg, Germany). A multiline argon ion laser (30 mW) was applied as an excitation source (excitation wavelength of 488 nm), which was reflected by a dichroic mirror (DM405/488) and focused onto the sample through an Olympus objective UPLSAPO 40× (numerical aperture N.A. 0.9). The fluorescence emission was collected between 540 nm and 640 nm by the same objective and focused into the photomultiplier tube (PMT).
General procedure (GP) for the consecutive three-component synthesis of 3-piperazinyl prop-2-enylidene indolones 4
In a dry screw-cap Schlenk tube with a magnetic stir bar, PdCl2(PPh3)2 (14.2 mg, 0.02 mmol) and CuI (7.63 mg, 0.04 mmol) were dissolved in dry dichloromethane (5 mL) or dry acetonitrile (4 mL) under nitrogen. Then, the corresponding ortho-bromo anilide 1 (1.00 mmol) and the aryl acetylene 2 (1.00 mmol) were successively added (for experimental details, see Table 4) and stirred for 10 min at room temperature or at 50 °C. After addition of the terminal alkyne, triethylamine (106 mg, 1.05 mmol) was added to the reaction mixture. The mixture turned brown and was then stirred at the temperatures (T1) and times (t1) indicated. Then, piperazine 3 (2.00 mmol) was added under nitrogen and the reaction mixture was stirred at the temperatures (T2) and times (t2) indicated. After cooling to room temperature, saturated aqueous ammonium chloride solution (10 mL) and dichloromethane (10 mL) were added to the red solution. The organic layer was separated and the aqueous phase was extracted with dichloromethane (3 × 30 mL). The combined organic layers were extracted with brine (15 mL) and dried (anhydrous magnesium sulfate). After filtration the filtrate was absorbed on Celite® and purified by flash chromatography on silica gel to give the analytically pure merocyanines 4 as red solids. Deviating workup procedures are given in the details of the analytics of the compounds 4.| Entry | ortho-Bromo anilide 1 [mg] (mmol) | Aryl acetylene 2 [mg] (mmol) | T 1, t1 | Piperazine 3 [mg] (mmol) | T 2, t2 | Merocyanine 4 [mg] (%) |
|---|---|---|---|---|---|---|
| a In dichloromethane as a solvent and with an induction period of 10 min at RT prior to starting the insertion–coupling step by the addition of NEt3. b The reaction was performed on a 1.6 mmol scale with respect to 1b (22.8 mg of PdCl2(PPh3)2, 14.0 mg of CuI, and 169 mg (1.67 mmol) of NEt3. c In acetonitrile as a solvent and the induction period of 10 min at 50 °C prior to starting the insertion–coupling step by the addition of NEt3. | ||||||
| 1a | 314 (1.00) of 1a (R1 = Me, R2 = H) | 113 (1.10) of 2a (R3 = H) | RT, 16 h | 377 (2.00) | 50 °C, 48 h | 289 (55) of 4a |
| 2a,b | 607 (1.60) of 1b (R1 = nhexyl, R2 = H) | 169 (1.70) of 2a | RT, 16 h | 591 (3.20) | 50 °C, 48 h | 337 (36) of 4b |
| 3a | 457 (1.00) of 1c (R1 = Tos, R2 = H) | 113 (1.10) of 2a | RT, 16 h | 378 (2.00) | 50 °C, 48 h | 179 (27) of 4c |
| 4c | 454 (1.00) of 1c | 118 (1.16) of 2a | 50 °C, 16 h | 378 (2.00) | 80 °C, 16 h | 628 (95) of 4c |
| 5c | 454 (1.00) of 1c | 144 (1.09) of 2b (R3 = MeO) | 50 °C, 16 h | 378 (2.00) | 80 °C, 16 h | 284 (41) of 4d |
| 6c | 420 (0.92) of 1c | 158 (1.00) of 2c (R3 = tBu) | 50 °C, 16 h | 344 (1.85) | 80 °C, 16 h | 332 (50) of 4e |
| 7c | 457 (1.01) of 1c | 132 (1.14) of 2d (R3 = Me) | 50 °C, 16 h | 378 (2.00) | 80 °C, 16 h | 385 (57) of 4f |
| 8c | 452 (0.99) of 1c | 177.1 (1.11) of 2e (R3 = COOCH3) | 50 °C, 16 h | 378 (2.00) | 80 °C, 16 h | 70 (10) of 4g |
| 9c | 419 (0.87) of 1d (R1 = Tos, R2 = MeO) | 109 (1.07) of 2a | 50 °C, 16 h | 329 (1.77) | 80 °C, 16 h | 116 (17) of 4h |
| 10c | 508 (1.00) of 1e (R1 = Tos, R2 = tBu) | 115 (1.13) of 2a | 50 °C, 16 h | 378 (2.00) | 80 °C, 16 h | 251 (35) of 4i |
| 11c | 352 (0.75) of 1f (R1 = Tos, R2 = Me) | 86 (0.84) of 2a | 50 °C, 16 h | 280 (1.50) | 80 °C, 16 h | 162 (32) of 4j |
| 12c | 487 (1.00) of 1g (R1 = Tos, R2 = Cl) | 115 (1.13) of 2a | 50 °C, 16 h | 378 (2.00) | 80 °C, 16 h | 373 (54) of 4k |
| 13c | 130 (0.27) of 1h (R1 = Tos, R2 = CN) | 31.5 (0.31) of 2a | 50 °C, 16 h | 101 (0.54) | 80 °C, 16 h | 140 (75) of 4l |
tert-Butyl-4-((E)-3-((E)-1-methyl-2-oxindolin-3-yliden)-1,3-diphenylprop-1-en-1-yl)piperazin-1-carboxylate (4a)
According to the GP and after chromatography on silica gel (n-hexane/acetone 10:1), compound 4a (289 mg, 55%) was obtained as a bright orange amorphous solid, mp 212 °C, Rf (n-hexane/ethyl acetate 5:1): 0.09. 1H NMR (300 MHz, CDCl3): δ 1.46 (s, 9H), 3.23 (m, br, 4H), 3.32 (s, 3H), 3.46 (m, br, 4H), 5.52 (d, J = 7.5 Hz, 1H), 6.43–6.49 (m, 1H), 6.72 (d, J = 7.5 Hz, 1H), 6.81 (dd, J = 8.0, 1.5 Hz, 2H), 6.87 (dd, J = 8.2, 1.4 Hz, 2H), 7.05–6.90 (m, 7H), 7.75 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 25.8 (CH3), 28.5 (CH3), 48.8 (CH2), 80.4 (Cquat), 106.7, 107.3, 115.9 (Cquat), 120.7, 121.7, 124.5 (Cquat), 125.5, 127.2, 127.87, 127.94, 128.4, 129.4, 130.4, 136.7 (Cquat), 140.0 (Cquat), 140.8 (Cquat), 154.7 (Cquat), 155.7 (Cquat), 161.2 (Cquat), 168.5 (Cquat). IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 3010 (w), 2999 (w), 2976 (w), 2901 (w), 1701 (s), 1668 (s), 1609 (m), 1545 (s), 1483 (m), 1470 (m), 1445 (m), 1408 (s), 1400 (s), 1373 (m), 1350 (m), 1335 (m), 1300 (m), 1285 (m), 1250 (m), 1225 (s), 1171 (s), 1155 (s), 1115 (m), 1076 (s), 1051 (s), 1022 (s), 999 (m), 924 (m), 878 (m), 775 (s), 745 (m), 727 (s), 714 (s), 700 (s), 689 (m), 646 (m), 606 (m). MALDI-TOF: m/z 522.2 (M + H+). Anal. calcd for C33H35N3O3 (521.7): C 75.98, H 6.76, N 8.06; found: C 75.92, H 6.63, N 7.97.
3010 (w), 2999 (w), 2976 (w), 2901 (w), 1701 (s), 1668 (s), 1609 (m), 1545 (s), 1483 (m), 1470 (m), 1445 (m), 1408 (s), 1400 (s), 1373 (m), 1350 (m), 1335 (m), 1300 (m), 1285 (m), 1250 (m), 1225 (s), 1171 (s), 1155 (s), 1115 (m), 1076 (s), 1051 (s), 1022 (s), 999 (m), 924 (m), 878 (m), 775 (s), 745 (m), 727 (s), 714 (s), 700 (s), 689 (m), 646 (m), 606 (m). MALDI-TOF: m/z 522.2 (M + H+). Anal. calcd for C33H35N3O3 (521.7): C 75.98, H 6.76, N 8.06; found: C 75.92, H 6.63, N 7.97.
tert-Butyl-4-((E)-3-((E)-1-hexayl-2-oxindolin-3-yliden)-1,3-diphenylprop-1-en-1-yl)piperazin-1-carboxylate (4b)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1) compound 4b (337 mg, 36%) was obtained as dark red platelets with a green luster, mp 172 °C, Rf (n-hexane/ethyl acetate 4:1): 0.21. 1H NMR (300 MHz, CDCl3): δ 0.91 (t, J = 6.7 Hz, 3H), 1.27–1.35 (m, 6H), 1.46 (s, 9H), 1.67–1.77 (m, 2H), 3.19 (m, br, 4H), 3.45 (m, br, 4H), 3.81 (t, J = 7.5 Hz, 2H), 5.49 (d, J = 7.3 Hz, 1H), 6.41–6.52 (m, 1H), 6.73 (d, J = 7.5 Hz, 1H), 6.81 (dd, J = 8.0, 1.5 Hz, 2H), 6.87 (dd, J = 8.2, 1.5 Hz, 2H), 6.91–7.03 (m, 7H), 7.77 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 14.2 (CH3), 22.8 (CH2), 27.0 (CH2), 28.1 (CH2), 28.5 (CH3), 31.8 (CH2), 39.8 (CH2), 48.7 (CH2), 80.2 (Cquat), 106.7, 107.2, 116.0 (Cquat), 120.4, 121.8, 124.6 (Cquat), 125.5, 127.1, 127.9, 128.0, 128.3, 129.4, 130.4, 136.8 (Cquat), 140.0 (Cquat), 140.3 (Cquat), 154.7 (Cquat), 155.5 (Cquat), 161.3 (Cquat), 168.2 (Cquat). MALDI-TOF: m/z 592.4 (M + H+).
tert-Butyl-4-((E)-3-((E)-2-oxo-1-tosylindolin-3-yliden)-1,3-diphenylprop-1-en-1-yl)-piperazin-1-carboxylate (4c)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1) compound 4c (179 mg, 27%) was obtained as an orange red amorphous solid, mp 182 °C, Rf (n-hexane/ethyl acetate 2:1): 0.49. 1H NMR (300 MHz, CDCl3): δ 1.47 (s, 9H), 2.43 (s, 3H), 3.24 (m, br, 4H), 3.34–3.54 (m, 4H), 5.43 (d, J = 7.5 Hz, 1H), 6.50–6.52 (m, 1H), 6.72–6.76 (m, 2H), 6.80–6.82 (m, 2H), 6.91–7.03 (m, 6H), 7.32 (d, J = 8.0 Hz, 2H), 7.31–7.42 (m, 1H), 7.44 (s, 1H), 7.88 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.0 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 21.8 (CH3), 28.5 (CH3), 48.8 (CH2), 80.4 (Cquat), 107.3, 111.8 (Cquat), 112.57, 121.3, 123.0, 125.5, 126.0 (Cquat), 127.9, 128.0, 128.2, 128.9 (Cquat), 129.0 (Cquat), 129.3, 129.7, 130.2, 135.6 (Cquat), 136.7 (Cquat), 139.7 (Cquat), 144.7 (Cquat), 154.7 (Cquat), 164.1 (Cquat), 165.9 (Cquat).§1H NMR (600 MHz, CDCl3): δ 1.47 (s, 9H), 2.43 (s, 3H), 3.23 (m, br, 4H), 3.45 (m, br, 4H), 5.43 (d, J = 7.6 Hz, 1H), 6.50–6.55 (m, 1H), 6.72 (d, J = 7.5 Hz, 2H), 6.80 (d, J = 6.7 Hz, 2H), 6.88–7.05 (m, 6H), 7.32 (d, J = 8.1 Hz, 2H), 7.41–7.48 (m, 2H), 7.88 (d, J = 7.9 Hz, 1H), 8.05 (d, J = 8.3 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 21.8 (CH3), 28.5 (CH3), 48.8 (CH2), 80.4 (Cquat), 107.3, 111.8 (Cquat), 112.57, 121.3, 123.0, 125.5, 126.0 (Cquat), 127.9, 128.0, 128.1, 128.2, 128.9 (Cquat), 129.0 (Cquat), 129.26, 129.29, 129.7, 130.2, 135.6 (Cquat), 136.7 (Cquat), 139.7 (Cquat), 144.7 (Cquat), 154.7 (Cquat), 164.1 (Cquat), 165.9 (Cquat). IR (ATR): 3065 (w), 2976 (w), 1697 (m), 1672 (m), 1634 (w), 1508 (m), 1489 (m), 1456 (w), 1418 (m), 1389 (w), 1360 (m), 1346 (m), 1321 (m), 1285 (w), 1248 (m), 1227 (s), 1200 (m), 1163 (s), 1121 (m), 1080 (s), 993 (m), 962 (m), 924 (w), 905 (w), 868 (m), 772 (m), 748 (w), 735 (m), 689 (s), 660 (s). MALDI-TOF: m/z 662.3 (M + H+), 614.9 (M+ − CO2), 508.2 (M+ − Tos). Anal. calcd for C39H39N3O5S (661.8): C 70.78, H 5.94, N 6.35, S 4.85; found: C 70.65, H 6.18, N 6.20, S 4.81.
tert-Butyl-4-(1-(4-methoxyphenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-3-phenylprop-1-en-1-yl)-piperazin-1-carboxylate (4d)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4d (284 mg, 41%) was obtained as an orange red amorphous solid, mp 202 °C, Rf (n-hexane/ethyl acetate 3:1): 0.16. 1H NMR (300 MHz, CDCl3): δ 1.48 (s, 9H), 2.42 (s, 3H), 3.25 (m, br, 4H), 3.45 (m, br, 4H), 3.70 (s, 3H), 5.45 (d, J = 7.6 Hz, 1H), 6.46 (d, J = 8.1 Hz, 2H), 6.51–6.56 (m, 1H), 6.73–6.76 (m, 4H), 6.96 (t, J = 7.8 Hz, 4H), 7.32 (d, J = 8.1 Hz, 2H), 7.37 (br, 1H), 7.88 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 21.8 (CH3), 28.5 (CH3), 49.0 (CH2), 55.4 (CH3), 80.4 (Cquat), 107.5, 111.4 (Cquat), 112.6, 113.6, 121.2, 123.0, 125.3, 126.1 (Cquat), 127.6, 127.9, 128.1, 128.5 (Cquat), 129.3, 129.7, 131.7, 135.5 (Cquat), 136.8 (Cquat), 139.9 (Cquat), 144.7 (Cquat), 154.7 (Cquat), 159.1 (Cquat), 160.1 (Cquat), 164.4 (Cquat), 165.9 (Cquat). IR (ATR): 3060 (w), 2970 (w), 2911 (w), 2859 (w), 2837 (w), 1740 (w), 1697 (s), 1678 (m), 1597 (w), 1576 (w), 1533 (m), 1514 (s), 1491 (m), 1472 (m), 1454 (m), 1443 (m), 1400 (m), 1364 (s), 1348 (s), 1319 (s), 1298 (m), 1281 (m), 1229 (s), 1159 (s), 1138 (m), 1109 (s), 1065 (s), 1045 (s), 1024 (m), 991 (s), 955 (m), 916 (m), 903 (m), 876 (m), 860 (m), 837 (m), 816 (m), 775 (m), 748 (m), 683 (s), 660 (s), 633 (m). MALDI-TOF: m/z 691.2 (M+), 645.0 (M+ − CO2), 538.4 (M+ − Tos). Anal. calcd for C40H41N3O6S (691.8): C 69.44, H 5.97, N 6.07, S 4.63; found: C 69.23, H 6.00, N 5.82, S 4.38.
tert-Butyl-4-(1-(4-(tert-butyl)phenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-3-phenylprop-1-en-1-yl)-piperazin-1-carboxylate (4e)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4e (332 mg, 50%) was obtained as an orange red amorphous solid, mp 240 °C, Rf (n-hexane/ethyl acetate 3:1): 0.39. 1H NMR (600 MHz, CDCl3): δ 1.20 (s, 9H), 1.47 (s, 9H), 2.42 (s, 3H), 3.26 (m, br, 4H), 3.46 (m, br, 4H), 5.39 (d, J = 7.7 Hz, 1H), 6.50–6.53 (m, 1H), 6.72 (m, br, 3H), 6.85–7.00 (m, 6H), 7.32 (d, J = 8.2 Hz, 2H), 7.36 (s (breit), 1H), 7.45–7.48 (m, 1H), 7.88 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 21.8 (CH3), 28.5 (CH3), 31.2 (CH3), 34.6 (Cquat), 48.8 (CH2), 80.4 (Cquat), 107.1, 111.4 (Cquat), 112.5, 121.2, 123.0, 124.9, 125.2, 126.2 (Cquat), 127.5, 127.9, 128.1, 128.6, 128.7, 129.1, 129.3, 129.7, 130.0, 133.0 (Cquat), 135.5 (Cquat), 136.8 (Cquat), 139.7 (Cquat), 144.7 (Cquat), 151.7 (Cquat), 154.7 (Cquat), 159.1 (Cquat), 164.5 (Cquat), 165.9 (Cquat). IR (ATR): 3006 (w), 2961 (w), 2905 (w), 2866 (w), 1692 (s), 1595 (w), 1533 (m), 1516 (s), 1491 (m), 1456 (m), 1443 (m), 1406 (m), 1395 (m), 1368 (s), 1350 (s), 1323 (m), 1287 (m), 1248 (m), 1231 (s), 1203 (s), 1171 (s), 1163 (s), 1140 (m), 1121 (m), 1072 (s), 1049 (m), 1024 (m), 995 (s), 961 (s), 918 (m), 905 (m), 876 (s), 779 (m), 762 (m), 741 (s), 700 (m), 689 (s), 660 (s). MALDI-TOF: m/z 718.3 (M + H+), 670.7 (M+ − CO2), 662.3 (C39H40N3O5S+), 564.3 (C36H42N3O3+). HR-MS (ESI) calcd for C43H47N3O5S + H+: 718.3309; found: 718.3309.
tert-Butyl-4-(3-(2-oxo-1-tosylindolin-3-yliden)-3-phenyl-1-(4-tolyl)prop-1-en-1-yl)-piperazin-1-carboxylate (4f)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4f (385 mg, 57%) was obtained as an orange red amorphous solid, mp 204 °C, Rf (n-hexane/ethyl acetate 5:1): 0.15. 1H NMR (600 MHz, CDCl3): δ 1.47 (s, 9H), 2.18 (s, 3H), 2.43 (s, 3H), 3.25 (m, br, 4H), 3.45 (m, br, 4H), 5.41 (d, J = 6.8 Hz, 1H), 6.51–6.54 (m, 1H), 6.63–6.72 (m, 5H), 6.68–6.71 (m, 4H), 7.32 (d, J = 7.8 Hz, 2H), 7.33–7.42 (m, 1H), 7.43 (br, 1H), 7.88 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 7.4 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 21.3 (CH3), 21.8 (CH3), 28.5 (CH3), 48.8 (CH2), 80.4 (Cquat), 107.2, 111.4 (Cquat), 112.6, 121.2, 123.0, 125.3, 126.1 (Cquat), 127.3, 127.9, 128.0, 128.7, 129.0, 129.3, 129.7, 130.1, 131.0, 132.5, 133.2 (Cquat), 135.5 (Cquat), 136.8 (Cquat), 138.8 (Cquat), 139.8 (Cquat), 144.7 (Cquat), 154.7 (Cquat), 159.1 (Cquat), 164.5 (Cquat), 165.9 (Cquat). 1H NMR (300 MHz, CD2Cl2): δ 1.45 (s, 9H), 2.19 (s, 3H), 2.44 (s, 3H), 3.28 (m, br, 4H), 3.43–3.46 (m, 4H), 5.40 (d, J = 6.4 Hz, 1H), 6.49–6.54 (m, 1H), 6.70–6.75 (m, 6H), 6.91–7.05 (m, 5H), 7.36 (d, J = 8.1 Hz, 2H), 7.84 (d, J = 8.0 Hz, 1H). 7.98 (d, J = 8.3 Hz, 2H). 13C NMR (75 MHz, CD2Cl2): δ 21.4 (CH3), 22.0 (CH3), 28.6 (CH3), 49.3 (CH2), 80.4 (Cquat), 107.1, 110.9 (Cquat), 112.8, 121.5, 123.2, 125.4, 126.8 (Cquat), 128.2, 128.5, 129.1, 129.2, 129.3, 129.7, 130.1, 130.5, 131.5 (Cquat), 132.9 (Cquat), 135.8 (Cquat), 137.2 (Cquat), 140.3 (Cquat), 145.6 (Cquat), 154.9 (Cquat), 165.5 (Cquat).¶ IR (ATR): 3008 (w), 2976 (w), 2922 (w), 2857 (w), 1722 (w), 1692 (s), 1678 (m), 1638 (w), 1593 (m), 1530 (m), 1512 (s), 1491 (m), 1454 (m), 1406 (m), 1364 (s), 1346 (s), 1321 (s), 1300 (m), 1287 (m), 1231 (s), 1161 (s), 1121 (s), 1070 (s), 1047 (m), 1011 (m), 995 (s), 962 (m), 918 (m), 903 (m), 876 (s), 812 (s), 773 (m), 746 (s), 704 (m), 692 (s), 660 (s). MALDI-TOF: m/z 676.3 (M + H+), 628.7 (M+ − CO2), 620.3 (C36H34N3O5S+), 522.3 (C33H36N3O3+).
tert-Butyl-4-(1-(4-(methoxycarbonyl)phenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-3-phenyl-prop-1-en-1-yl)piperazin-1-carboxylate (4g)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4g (70 mg, 10%) was obtained as an orange amorphous solid, mp 201 °C, Rf (n-hexane/ethyl acetate 5:1): 0.08. 1H NMR (600 MHz, CDCl3, 298 K): δ 1.47 (s, 9H), 2.44 (s, 3H), 3.18 (m, br, 4H), 3.45 (m, br, 4H), 3.89 (s, 3H), 5.48 (d, J = 7.7 Hz, 1H), 6.51–6.56 (m, 1H), 6.73 (d, J = 7.1 Hz, 2H), 7.04–6.84 (m, 6H), 7.33 (d, J = 8.1 Hz, 2H), 7.47 (br, 1H), 7.60 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H). 1H NMR (600 MHz, CDCl3, 263 K): δ 1.46 (s, 9H), 2.43 (s, 3H), 3.19–3.44 (m, br, 8H), 3.88 (s, 3H), 5.42 (d, J = 7.9 Hz, 1H), 6.50–6.58 (m, 1H), 6.70 (dd, J = 8.1, 1.2 Hz, 2H), 6.87–6.93 (m, 4H), 6.97–7.03 (m, 2H), 7.33 (d, J = 8.1 Hz, 2H), 7.44 (br, 1H), 7.57–7.64 (m, 2H), 7.89 (dd, J = 8.3, 1.1 Hz, 1H), 8.00–8.06 (m, 2H). 13C NMR (151 MHz, CDCl3, 298 K): δ 21.9 (CH3), 28.5 (CH3), 48.8 (CH2), 52.4 (CH3), 80.5 (Cquat), 107.8, 112.7, 112.9 (Cquat), 121.5, 123.2, 125.6 (Cquat), 126.0, 127.9, 128.0, 128.3, 129.2, 129.5, 129.8, 130.1 (Cquat), 130.3, 135.9 (Cquat), 136.6 (Cquat), 139.5 (Cquat), 141.0 (Cquat), 144.9 (Cquat), 154.6 (Cquat), 157.5 (Cquat), 162.0 (Cquat), 165.9 (Cquat), 166.5 (Cquat). IR (ATR): 3007 (w), 2978 (w), 2947 (w), 2853 (w), 1717 (s), 1690 (s), 1647 (w), 1593 (w), 1570 (w), 1535 (m), 1518 (m), 1491 (m), 1477 (w), 1456 (m), 1435 (m), 1406 (m), 1371 (s), 1348 (s), 1317 (m), 1275 (s), 1246 (m), 1229 (s), 1213 (m), 1190 (s), 1161 (s), 1134 (m), 1115 (s), 1103 (s), 1070 (s), 1016 (m), 995 (s), 957 (m), 920 (w), 903 (m), 880 (s), 864 (m), 814 (m), 800 (w), 773 (s), 750 (s), 735 (m), 704 (s), 687 (s), 658 (s). MALDI-TOF: m/z 720.2 (M + H+), 672.6 (M+ − CO2), 566.2 (M+ − Tos). Anal. calcd C41H41N3O7S (719.9): C 68.41, H 5.74, S 4.45; found: C 68.23, H 5.55, S 4.29.
tert-Butyl-4-(3-(4-methoxyphenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-1-phenylprop-1-en-1-yl)-piperazin-1-carboxylate (4h)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4h (116 mg, 17%) was obtained as a yellow-orange amorphous solid, mp 190 °C, Rf (n-hexane/ethyl acetate 5:1): 0.13. 1H NMR (300 MHz, CDCl3): δ 1.47 (s, 9H), 2.43 (s, 3H), 3.22 (m, br, 4H), 3.45 (m, br, 4H), 3.72 (s, 3H), 5.72 (d, J = 7.7 Hz, 1H), 6.46 (d, J = 8.1 Hz, 2H), 6.56–6.61 (m, 1H), 6.68 (d, J = 8.2 Hz, 2H), 6.83 (d, J = 6.8 Hz, 2H), 6.96 (d, J = 8.1 Hz, 4H), 7.29–7.33 (m, 3H), 7.89 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 21.9 (CH3), 28.5 (CH3), 48.9 (CH2), 55.4 (CH3), 80.4 (Cquat), 108.1, 111.8 (Cquat), 112.6, 113.7, 121.2, 123.0, 125.4, 126.2 (Cquat), 127.9, 128.0, 128.7, 129.7, 130.2, 131.0, 132.2 (Cquat), 135.6 (Cquat), 136.4 (Cquat), 136.8 (Cquat), 144.7 (Cquat), 154.7 (Cquat), 158.7 (Cquat), 159.3 (Cquat), 164.1 (Cquat), 165.9 (Cquat). IR (ATR): 3010 (w), 2978 (w), 2926 (w), 2864 (w), 1730 (w), 1694 (s), 1601 (w), 1578 (w), 1516 (s), 1501 (s), 1454 (m), 1412 (m), 1369 (s), 1350 (s), 1319 (m), 1287 (m), 1231 (s), 1202 (m), 1159 (s), 1136 (m), 1117 (s), 1094 (w), 1065 (s), 1038 (m), 993 (s), 961 (m), 941 (w), 922 (m), 908 (m), 872 (m), 841 (m), 822 (m), 772 (s), 752 (s), 729 (m), 712 (m), 685 (s), 662 (s), 642 (m). MALDI-TOF: m/z 692.3 (M + H+), 644.9 (M+ − CO2), 538.2 (M+ − Tos). Anal. calcd for C40H41N3O6S (691.8): C 69.44, H 5.97, N 6.07, S 4.63; found: C 69.15, H 6.04, N 5.78, S 4.40.
tert-Butyl-4-(3-(4-(tert-butyl)phenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-1-phenylprop-1-en-1-yl)-piperazin-1-carboxylate (4i)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4i (162 mg, 35%) was obtained as a red amorphous solid, mp 185 °C, Rf (n-hexane/ethyl acetate 5:1): 0.11. 1H NMR (300 MHz, CDCl3): δ 1.22 (s, 9H), 1.47 (s, 9H), 2.43 (s, 3H), 3.22 (m, br, 4H), 3.45 (m, br, 4H), 5.52 (d, J = 7.9 Hz, 1H), 6.50–6.55 (m, 1H), 6.64 (d, J = 8.0 Hz, 2H), 6.80 (d, J = 7.1 Hz, 2H), 6.92 (d, J = 7.6 Hz, 4H), 6.97 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.43 (br, 1H), 7.88 (d, J = 7.9 Hz, 1H), 8.05 (d, J = 8.3 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 21.8 (CH3), 28.5 (CH3), 31.3 (CH3), 34.6 (Cquat), 48.8 (CH2), 80.4 (Cquat), 107.5, 111.6 (Cquat), 112.5, 121.3, 123.0, 125.0, 125.3, 126.2 (Cquat), 127.9, 128.6, 129.1, 129.7, 130.2, 135.5 (Cquat), 136.3 (Cquat), 136.6 (Cquat), 136.8 (Cquat), 144.7 (Cquat), 150.5 (Cquat), 154.7 (Cquat), 159.1 (Cquat), 164.1 (Cquat), 165.9 (Cquat). IR (ATR): 3011 (w), 2947 (w), 2927 (w), 2860 (w), 1695 (s), 1684 (s), 1599 (w), 1516 (s), 1504 (s), 1474 (s), 1456 (m), 1400 (m), 1389 (m), 1375 (s), 1348 (s), 1319 (s), 1302 (m), 1283 (m), 1229 (s), 1200 (m), 1161 (s), 1138 (m), 1109 (s), 1063 (s), 1047 (s), 1005 (s), 993 (s), 957 (m), 922 (m), 910 (m), 862 (m), 837 (m), 814 (m), 775 (m), 752 (m), 731 (m), 702 (m), 683 (s), 662 (s). MALDI-TOF: m/z 717.3 (M+), 670.8 (M+ − CO2), 564.3 (M+ − Tos). Anal. calcd for C43H47N3O5S (717.9): C 71.94, H 6.60, N 5.85, S 4.47; found: C 71.72, H 6.71, N 5.67, S 4.32.
tert-Butyl-4-(3-(2-oxo-1-tosylindolin-3-yliden)-1-phenyl-3-(p-tolyl)prop-1-en-1-yl)-piperazin-1-carboxylate (4j)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4j (162 mg, 32%) was obtained as an orange-red amorphous solid, mp 199 °C, Rf (n-hexane/ethyl acetate 5:1): 0.09. 1H NMR (300 MHz, CDCl3): δ 1.47 (s, 9H), 2.20 (s, 3H), 2.43 (s, 3H), 3.22 (m, br, 4H), 3.45 (m, br, 4H), 5.63 (d, J = 7.8 Hz, 1H), 6.53–6.61 (m, 3H), 6.71 (d, J = 7.2 Hz, 2H), 6.79 (d, J = 6.7 Hz, 2H), 6.90–7.04 (m, 4H), 7.32 (d, J = 8.1 Hz, 2H), 7.38 (br, 1H), 7.88 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 8.3 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 21.3 (CH3), 21.9 (CH3), 28.5 (CH3), 48.8 (CH2), 80.4 (Cquat), 107.7, 111.7 (Cquat), 112.6, 121.3, 123.0, 125.4, 126.1 (Cquat), 127.9, 127.9, 128.4, 128.8, 129.3, 129.7, 130.2, 135.5 (Cquat), 136.4 (Cquat), 136.7 (Cquat), 136.8 (Cquat), 137.4 (Cquat), 144.7 (Cquat), 154.7 (Cquat), 159.1 (Cquat), 164.1 (Cquat), 165.9 (Cquat). IR (ATR): 2978 (w), 2920 (w), 2901 (w), 2868 (w), 1694 (s), 1595 (w), 1522 (s), 1501 (s), 1491 (s), 1452 (m), 1418 (s), 1366 (s), 1350 (s), 1319 (m), 1287 (w), 1233 (s), 1202 (w), 1163 (s), 1157 (s), 1136 (m), 1121 (m), 1069 (s), 1042 (w), 1018 (w), 995 (s), 961 (m), 920 (m), 908 (m), 872 (m), 843 (w), 816 (s), 772 (m), 748 (s), 729 (m), 687 (s), 660 (s). MALDI-TOF: m/z 675.3 (M+), 628.9 (M+ − CO2), 522.3 (M+ − Tos). Anal. calcd for C40H41N3O5S (675.8): C 71.09, H 6.11, N 6.22, S 4.74; found: C 70.83, H 6.15, N 6.02, S 4.63.
tert-Butyl-4-(3-(4-chlorphenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-1-phenylprop-1-en-1-yl)piperazin-1-carboxylate (4k)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4k (373 mg, 54%) was obtained as a bright red amorphous solid, mp 204 °C, Rf (n-hexane/ethyl acetate 5:1): 0.05. 1H NMR (600 MHz, CDCl3): δ 1.47 (s, 9H), 2.43 (s, 3H), 3.23 (m, br, 4H), 3.45 (m, br, 4H), 5.60 (d, J = 7.7 Hz, 1H), 6.57–6.60 (m, 1H), 6.66 (d, J = 7.6 Hz, 2H), 6.79 (d, J = 6.9 Hz, 2H), 6.89 (d, J = 7.8 Hz, 2H), 6.98–7.00 (m, 3H), 7.10–7.16 (m, 1H), 7.32 (d, J = 8.2 Hz, 2H), 7.42 (br, 1H), 7.89 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.2 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 21.8 (CH3), 28.5 (CH3), 48.8 (CH2), 80.5 (Cquat), 107.1, 111.73 (Cquat), 112.7, 121.1, 123.1, 125.66 (Cquat), 125.72, 127.9, 128.2, 128.3, 128.9, 129.7, 130.1, 130.8, 133.6 (Cquat), 135.7 (Cquat), 136.3 (Cquat), 136.7 (Cquat), 138.2 (Cquat), 144.8 (Cquat), 154.6 (Cquat), 156.9 (Cquat), 163.9 (Cquat), 165.8 (Cquat). IR (ATR): 3008 (w), 2976 (w), 1694 (s), 1595 (w), 1533 (m), 1508 (s), 1485 (s), 1452 (m), 1420 (m), 1371 (s), 1350 (s), 1321 (s), 1298 (w), 1287 (w), 1233 (s), 1202 (m), 1159 (s), 1136 (w), 1121 (m), 1093 (m), 1069 (s), 1042 (m), 1013 (s), 995 (s), 961 (m), 920 (m), 907 (m), 872 (m), 841 (m), 818 (m), 773 (m), 750 (s), 723 (m), 687 (s), 662 (s), 631 (w). MALDI-TOF: m/z 696.2 (M + H+), 648.8 (M+ − CO2), 542.6 (M+ − Tos). Anal. calcd for C39H38ClN3O5S (696.3): C 67.28, H 5.50, N 6.04, S 4.61; found: C 67.30, H 5.59, N 5.74, S 4.48.
tert-Butyl-4-(3-(4-cyanophenyl)-3-(2-oxo-1-tosylindolin-3-yliden)-1-phenylprop-1-en-1-yl)piperazin-1-carboxylate (4l)
According to the GP and after chromatography on silica gel (n-hexane/ethyl acetate 20:1, n-hexane/dichloromethane 5:1, dichloromethane/ethyl acetate 4:1) and trituration with n-hexane or pentane, compound 4l (140 mg, 75%) was obtained as an orange-red amorphous solid, mp 144 °C, Rf (n-hexane/ethyl acetate 5:1): 0.03. 1H NMR (600 MHz, CDCl3): δ 1.47 (s, 9H), 2.43 (s, 3H), 3.24 (m, br, 4H), 3.46 (m, br, 4H), 5.36 (d, J = 7.9 Hz, 1H), 6.54–6.57 (m, 1H), 6.78 (d, J = 7.3 Hz, 2H), 6.86 (d, J = 7.8 Hz, 2H), 6.98–7.02 (m, 3H), 7.09–7.17 (m, 1H), 7.23 (d, J = 7.8 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 7.48 (br, 1H), 7.90 (d, J = 8.2 Hz, 1H), 8.04 (d, J = 8.2 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 21.9 (CH3), 28.5 (CH3), 48.8 (CH2), 80.6 (Cquat), 106.3, 111.1 (Cquat), 111.8 (Cquat), 112.9, 118.7 (Cquat), 120.9, 123.2, 125.1 (Cquat), 126.2, 128.0, 128.4, 129.3, 129.8, 130.2, 130.2, 131.8, 135.9 (Cquat), 136.2 (Cquat), 136.5 (Cquat), 144.7 (Cquat), 145.0 (Cquat), 154.6 (Cquat), 155.3 (Cquat), 163.6 (Cquat), 165.7 (Cquat). IR (ATR): 3103 (w), 2972 (w), 2928 (w), 2862 (w), 2228 (m), 1690 (s), 1595 (m), 1508 (s), 1491 (s), 1454 (m), 1406 (m), 1366 (s), 1346 (s), 1319 (m), 1296 (m), 1287 (m), 1231 (s), 1198 (m), 1159 (s), 1138 (m), 1119 (m), 1070 (s), 995 (s), 959 (m), 922 (m), 908 (m), 872 (m), 841 (m), 812 (m), 772 (s), 745 (m), 729 (m), 683 (m), 658 (s), 611 (m). MALDI-TOF: m/z 686.2 (M+), 639.8 (M+ − CO2), 533.2 (M+ − Tos). HR-MS (ESI) calcd for C40H38N4O5S + H+: 687.2627; found: 687.2633.
Conclusions
In summary, we have established efficient consecutive three-component syntheses of 3-piperazinyl propenylidene indolone merocyanines, a novel class of functional chromophores displaying AIE behavior and containing a nonconjugating bridge for ligation of additional chromophores. The stereochemistry of the underlying merocyanine chromophore was unambiguously assigned by comprehensive NMR studies and quantum chemical calculation, assigning an overall E,E-butadiene configuration. By comprehensive physical-organic treatment, the positive solvatochromicity of the absorption in the consanguineous series of 3-piperazinyl propenylidene indolone merocyanines was confirmed. LFER correlation studies support an unusual discontinuous behavior of the longest wavelength absorption bands with respect to donor and acceptor substitution at the aryl substituent in the 1-position of the propenylidene chromophore. Furthermore, the AIE characteristics were corroborated for a model system in two different aqueous solvent systems. For one derivative, the crystallization-induced emission enhancement (CIEE) induced by ultrasonication could be shown by confocal laser scanning microscopy (CLSM). In the near future, we will design bichromophore systems to expand this modular consecutive multicomponent approach to the synthesis of dually emissive bichromophores.Acknowledgements
The authors cordially thank the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (Mu 1088/9-1) for financial support.Notes and references
- Functional Organic Materials. Syntheses, Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007 Search PubMed.
- (a) Organic Light-Emitting Materials and Devices, ed. Z. R. Li, CRC Press, 2nd edn, 2015 Search PubMed; (b) N. Thejo Kalayani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef; (c) Organic Light-Emitting Diodes – Synthesis, Properties, and Applications, ed. K. Müllen and U. Scherf, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (d) For a review on flexible AM-OLED, see e.g., J.-S. Park, H. Chae, H. K. Chung and S. I. Lee, Semicond. Sci. Technol., 2011, 26, 034001 CrossRef.
- (a) D. Nilsson, T. Kugler, P.-O. Svensson and M. Berggren, Sens. Actuators, B, 2002, 86, 193–197 CrossRef CAS; (b) C.-T. Chen, H. Wagner and W. C. Still, Science, 1998, 279, 851–853 CrossRef CAS PubMed; (c) For selected reviews in biophysical applications, see e.g., E. Kim and S. B. Park, Chem. – Asian J., 2009, 4, 1646–1658 CrossRef CAS PubMed; (d) C. W. Cairo, J. A. Key and C. M. Sadek, Curr. Opin. Chem. Biol., 2010, 14, 57–63 CrossRef CAS PubMed; (e) H.-A. Wagenknecht, Ann. N. Y. Acad. Sci., 2008, 1130, 122–130 CrossRef CAS PubMed.
- For reviews on diversity-oriented syntheses of π-systems, see: (a) C. A. Briehn and P. Bäuerle, Chem. Commun., 2002, 1015–1023 RSC; (b) T. J. J. Müller and D. M. D'Souza, Pure Appl. Chem., 2008, 80, 609–620 CrossRef; (c) T. J. J. Müller, in Functional Organic Materials. Syntheses, Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007, pp. 179–223 Search PubMed.
- For reviews, see e.g. (a) Multi-component Reactions in Organic Synthesis, ed. J. Zhu, Q. Wang and M.-X. Wang, Wiley-VCH, Weinheim, 2015 Search PubMed; (b) in Multicomponent Reactions, Science of Synthesis Series, ed. T. J. J. Müller, Georg Thieme Verlag KG, Stuttgart, 2014 Search PubMed; (c) J. D. Sunderhaus and S. F. Martin, Chem. – Eur. J., 2009, 15, 1300–1308 CrossRef CAS PubMed; (d) N. Isambert and R. Lavilla, Chem. – Eur. J., 2008, 14, 8444–8454 CrossRef CAS PubMed; (e) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (f) Multi-component Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, Weinheim, 2005 Search PubMed; (g) R. V. A. Orru and M. de Greef, Synthesis, 2003, 1471–1499 CrossRef CAS; (h) H. Bienaymé, C. Hulme, G. Oddon and P. Schmitt, Chem. – Eur. J., 2000, 6, 3321–3329 CrossRef; (i) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef; (j) I. Ugi, A. Dömling and B. Werner, J. Heterocycl. Chem., 2000, 37, 647–658 CrossRef CAS; (k) L. Weber, K. Illgen and M. Almstetter, Synlett, 1999, 366–374 CrossRef CAS; (l) R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123–131 CrossRef CAS; (m) I. Ugi, A. Dömling and W. Hörl, Endeavour, 1994, 18, 115–122 CrossRef CAS.
- For monographs, see e.g. (a) Domino Reactions in Organic Synthesis, ed. L. F. Tietze, G. Brasche and K. M. Gericke, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (b) Domino Reactions: Concepts for Efficient Organic Synthesis, ed. L. F. Tietze, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed; (c) For reviews, see e.g., L. F. Tietze, J. Heterocycl. Chem., 1990, 27, 47–69 CrossRef CAS.
- For reviews on one-pot methodologies, see e.g. (a) G. H. Posner, Chem. Rev., 1986, 86, 831–844 CrossRef CAS; (b) L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131–163 CrossRef; (c) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed; (d) B. Ganem, Acc. Chem. Res., 2009, 42, 463–472 CrossRef CAS PubMed; (e) S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
- T. J. J. Müller, in Multicomponent Reactions 1. General Discussion and Reactions Involving a Carbonyl Compound as Electrophilic Component, ed. T. J. J. Müller, Science of Synthesis Series, Georg Thieme Verlag KG, Stuttgart, 2014, pp. 5–27 Search PubMed.
- L. Levi and T. J. J. Müller, Chem. Soc. Rev., 2016, 45, 2825–2846 RSC.
- L. Levi and T. J. J. Müller, Eur. J. Org. Chem., 2016, 2907–2918 Search PubMed.
- For leading reviews on AIE, see e.g. (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (c) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- T. J. J. Müller, in Aggregation Induced Emission: Materials and Applications, ed. M. Fujiki, B. Z. Tang and B. Liu, ACS Symposium Series e-book, 2016, ch. 6, pp. 85–112 Search PubMed.
- (a) D. M. D'Souza, F. Rominger and T. J. J. Müller, Angew. Chem., Int. Ed., 2005, 44, 153–158 CrossRef PubMed; (b) D. M. D'Souza, A. Kiel, D.-P. Herten and T. J. J. Müller, Chem. – Eur. J., 2008, 14, 529–547 CrossRef PubMed; (c) J. Schönhaber and T. J. J. Müller, Org. Biomol. Chem., 2011, 9, 6196–6199 RSC.
- (a) D. M. D'Souza, C. Muschelknautz, F. Rominger and T. J. J. Müller, Org. Lett., 2010, 12, 3364–3367 CrossRef PubMed; (b) C. Muschelknautz, B. Mayer, F. Rominger and T. J. J. Müller, Chem. Heterocycl. Compd., 2013, 49, 922–934 CrossRef; (c) C. Muschelknautz, R. Visse, J. Nordmann and T. J. J. Müller, Beilstein J. Org. Chem., 2014, 10, 599–612 CrossRef PubMed.
- For a review, see e.g., D. Möbius, Adv. Mater., 1995, 7, 437–444 CrossRef.
- L. F. Tietze, T. Hungerland, C. Depken and D. Stalke, Synlett, 2012, 2516–2520 CrossRef CAS.
- J. Wallbaum, R. Neufeld, D. Stalke and D. B. Werz, Angew. Chem., Int. Ed., 2013, 52, 13243–13246 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) V. Z. Shirinian and A. A. Shimki, Top. Heterocycl. Chem., 2008, 14, 75–105 CAS; (b) M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781–805 CrossRef CAS; (c) A. A. Ishchenko, Russ. Chem. Rev., 1991, 60, 865–884 CrossRef; (d) A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2011 CrossRef CAS PubMed; (e) M. Levitu and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123–151 CrossRef PubMed.
- (a) L. G. S. Brooker, G. H. Keyes, R. H. Sprague, R. H. VanDyke, E. VanLare, G. VanZandt and F. L. White, J. Am. Chem. Soc., 1951, 73, 5326–5332 CrossRef CAS; (b) C. Reichardt, Solvent and solvent effects in organic chemistry, Weinheim VCH, 2nd edn, 1990 Search PubMed.
- S. Hünig and H. Hermann, Liebigs Ann. Chem., 1960, 636, 32–41 CrossRef.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- M. J. S. Dewar, Constitution and Colour Part 1. Basic Dyes, 1950, pp. 2329–2334 Search PubMed.
- S. R. Marder, C. B. Gorman, F. Meyers, J. W. Perry, G. Bourhill, J.-L. Bredas and B. M. Pierce, Science, 1994, 265, 632–635 CAS.
- M. Blanchard-Desce, V. Alain, P. V. Bedworth, S. R. Marder, A. Fort, C. Runser, M. Barzoukas, S. Lebus and R. Wortmann, Chem. – Eur. J., 1997, 3, 1091–1104 CrossRef CAS.
- Y.-J. Cheng, J. Luo, S. Huang, X. Zhou, Z. Shi, T.-D. Kim, D. H. Bale, S. Takahashi, A. Yick, B. M. Polishak, S.-H. Jang, L. R. Dalton, P. J. Reid, W. H. Steier and A. K. Y. Jen, Chem. Mater., 2008, 20, 5047–5054 CrossRef CAS.
- For a compilation of different σ-parameters, see e.g., C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. Hung, W. Liang, J. Luo, Z. Shi, A. K. Y. Jen and X. Li, J. Phys. Chem. C, 2010, 114, 22284–22288 CAS.
- J. Luo, K. Song, F. L. Gu and Q. Miao, Chem. Sci., 2011, 2, 2029–2034 RSC.
- H. Sun, Y. Zhang, W. Yan, W. Chen, Q. Lan, S. Liu, L. Jiang, Z. Chi, X. Chen and J. Xu, J. Mater. Chem. C, 2014, 2, 5812–5817 RSC.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- C. Würth, C. Lochmann, M. Spieles, J. Pauli, K. Hoffmann, T. Schuttrigkeit, T. Franzl and U. Resch-Genger, Appl. Spectrosc., 2010, 64, 733–741 CrossRef PubMed.
Footnotes |
| † Dedicated to Professor Dr Dr. h. c. Lutz F. Tietze on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Preparation and analytical data of starting materials 1 and 3, 1H and 13C NMR spectra of compounds 4; solvatochromism data of compound 4a; Hammett correlations of compounds 4c–l; computed xyz coordinates and energies of structures E,E-4c and E,Z-4c; AIE studies of compounds 4i and 4l in acetonitrile/water and THF/water mixtures. See DOI: 10.1039/c7qm00198c |
| § Superposition of two signals at δ 128.2 and 129.3. |
| ¶ Superposition of two signals. |
| This journal is © the Partner Organisations 2017 |