
Supramolecular color-tunable photoluminescent materials based on a chromophore cascade as security inks with dual encryption†
Zhe
Xu
ab,
Dario
Gonzalez-Abradelo
c,
Jun
Li
d,
Cristian A.
Strassert
 c,
Bart Jan
Ravoo
*b and
Dong-Sheng
Guo
*ae
c,
Bart Jan
Ravoo
*b and
Dong-Sheng
Guo
*ae
aCollege of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Key Laboratory of Functional Polymer Materials, Ministry of Education, Nankai University, Tianjin 300071, China. E-mail: dshguo@nankai.edu.cn
bOrganic Chemistry Institute and Center for Nanotechnology (CeNTech), Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: b.j.ravoo@uni-muenster.de
cInstitute of Physics and Center for Nanotechnology (CeNTech), Westfälische Wilhelms-Universität Münster, Heisenbergstrasse 11, 48149 Münster, Germany
dCollege of Life Sciences, Nankai University, Tianjin 300071, China
eCollaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China
First published on 2nd May 2017
Abstract
The design and development of novel broad-spectrum tunable photoluminescent materials for security printing with encrypted information is highly desirable. We construct a supramolecular co-assembly platform of amphiphilic cyclodextrin and calixarene, promoting energy transfer among three chromophores that are loaded by non-covalent interactions. A large matrix of colors was realized by delicately tuning the molar ratios of chromophores, which occupies most of the area in the Commission Internationale de l'Eclairage (CIE) chromaticity diagram. The elaborate supramolecular “cocktail” hides a dual-encryption coding, which exhibits feasible application as fluorescent security inks that are difficult to counterfeit but easy to authenticate.
Introduction
Color-tunable photoluminescent materials have gained considerable attention on account of their delicate tunability in spectrum outputs, exhibiting prosperous applications in light-emitting devices,1 optical sensors,2 as well as security printing.3 One of the most exploited strategies for this purpose is the careful tuning of partial Förster resonance energy transfer (FRET) between donor and acceptor chromophores, which therefore leads to the generation of diverse outputs of photoluminescence.4 In a conventional one-step FRET “cocktail” containing two kinds of chromophores, the color changes are localized in a (almost) linear trajectory with the outputs of individual donor and acceptor as starting and end points, respectively.5 Consequently, developing sophisticated broad-spectrum tunable photoluminescent materials combining a large matrix of fluorescence emissions (colors) still remains a major challenge, where developing two-step (or multi-step) FRET ensembles composed of three or more chromophores is highly demanded.Two-step FRET has been widely engaged in biotechnological applications,6 offering several advantages over one-step FRET: higher efficiency of long-range transfer between initial donor and final acceptor chromophores even beyond the Förster radius and/or in the absence of any spectral overlap,7 larger Stokes shift and better detection sensitivity for acceptor fluorescence.8 More recently, two-step FRET is attracting increasing interest for the preparation of light-emitting materials.9 However, a security ink with broad-spectrum tunable photoluminescence based on the two-step cascade of energy transfer has not been reported yet. A document possesses a high level of security if its encrypted information can be authenticated without being decoded, while also being resistant to counterfeiting. Tunable photoluminescent materials are ideal for applications in security printing technologies. Current stimuli-responsive photoluminescent materials, however, can only provide a small matrix of colors as optical codes.10
Herein we report broad-spectrum tunable photoluminescent materials based on dual-encrypted information, showing feasible application as security inks. The modular platform was successfully obtained by combining two different macrocyclic amphiphiles, that is, the co-assembly of amphiphilic calixarene (CA) and cyclodextrin (CD).11 The obtained co-assembly affords three orthogonal binding sites for chromophores, two macrocyclic cavities and one hydrophobic bilayer. By non-covalently grafting three individual chromophores with spectral overlap, the proximity of the three binding sites facilitates the coupling between the luminescent centers (Scheme 1). Due to the dynamic nature of non-covalent loading, we achieved a large matrix of photoluminescent outputs by varying the chromophore ratios. To our gratifying surprise, the photoluminescence outputs depend non-linearly on the chemical inputs, strongly suggesting not only the host–guest complexation interaction but also multiple energy transfer processes, so that a dual-encryption algorithm was realized. Moreover, the emission outputs are fine-tuned by the excitation inputs, which imparts a facile authentication test without revealing the original color image information. By applying this system in fluorescent security inks, the information encoded in polychromic images can be protected in a way that is difficult to counterfeit, yet easy to verify.
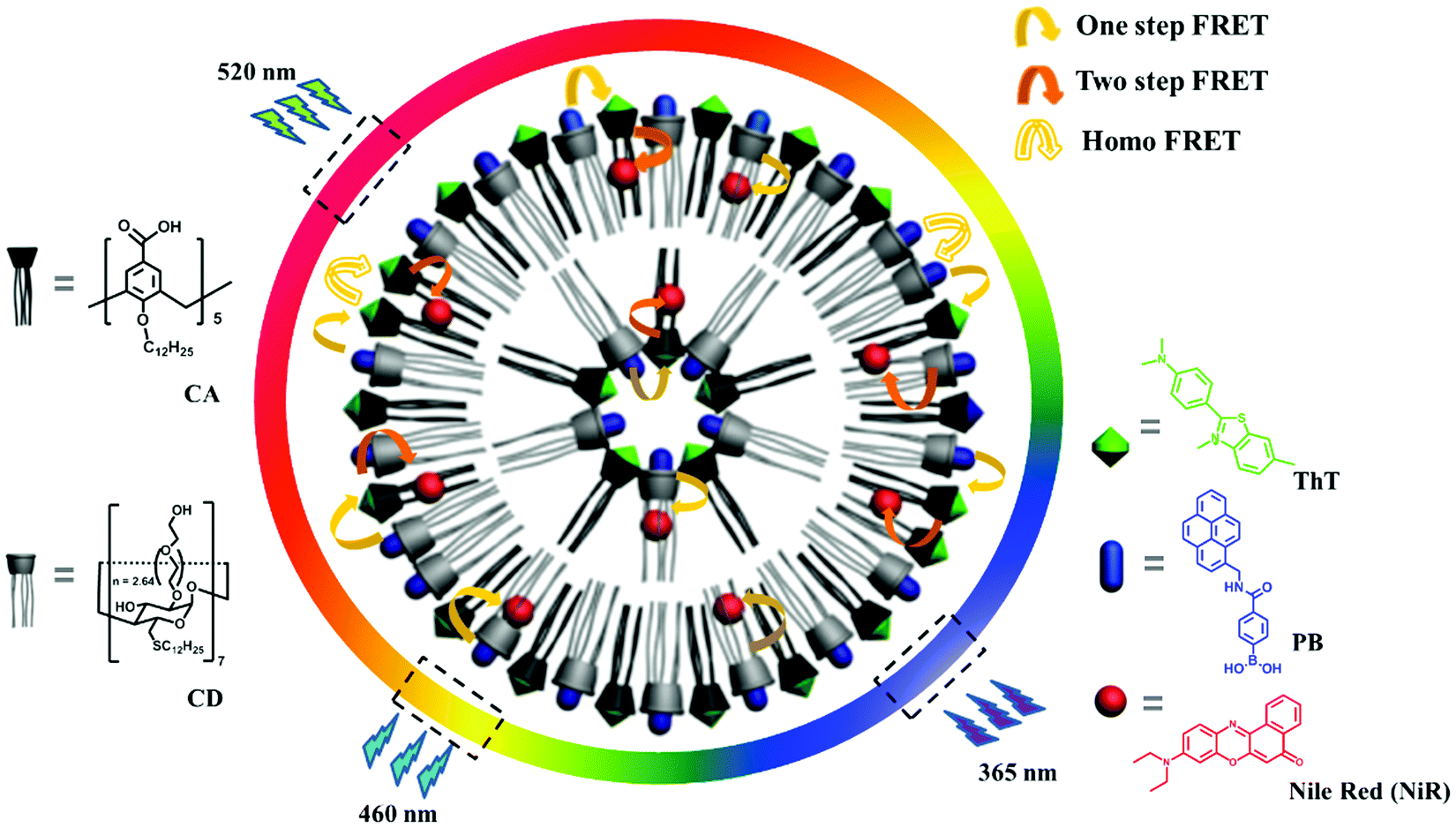 | ||
| Scheme 1 Schematic illustration of the three chromophore cocktail based on the CD/CA co-assembly platform. A broad-spectrum tunable photoluminescence was realized by tuning the multiple energy transfer efficiency. Thus the emission colors also depend on the excitation light. | ||
Results and discussion
The CD/CA co-assembly was prepared by hydrating a mixture of amphiphilic calix[5]arene and β-cyclodextrin, which was characterized by a combination of dynamic light scattering, Zeta potential, scanning electron microscopy and differential scanning calorimetry measurements (see Fig. S2 in the ESI†). The chromophore screening was based on the following design: (1) (4-phenylboronic acid)-1-pyrenemethamide (PB) acts as the first chromophore,12 which is strongly encapsulated in the CD cavity (Ka = 1.1 × 106 M−1), accompanied by pronounced enhancement of the emission of 5 times (Fig. S4, ESI†); (2) thioflavin T (ThT) acts as the second chromophore, which is selectively bound by CA (Ka = 8.3 × 104 M−1), accompanied by extraordinary emission enhancement of 285 times (see Fig. S5, ESI†); (3) Nile red (NiR) acts as the third chromophore, which can be readily entrapped within the hydrophobic bilayer and at the same time shows superior emission in the hydrophobic environment;13 (4) the emission of PB overlaps well with the absorption of ThT, and the emission of ThT overlaps well with the absorption of NiR (Fig. 1a), which promises a feasible two-step cascade energy transfer from PB over ThT to NiR; (5) the emission colors of these three chromophores are diverse and complementary, PB blue, ThT green, and NiR red, which ensures broad-spectrum tunable photoluminescence. Collectively, these design principles enabled us to construct a supramolecular cocktail based on the CD/CA co-assembly platform, and then a broad-spectrum tunable photoluminescent material. We note that based on the concentration of components and the binding constants, the chromophores can be expected to be close enough for efficient energy transfer (see Fig. S10, ESI†).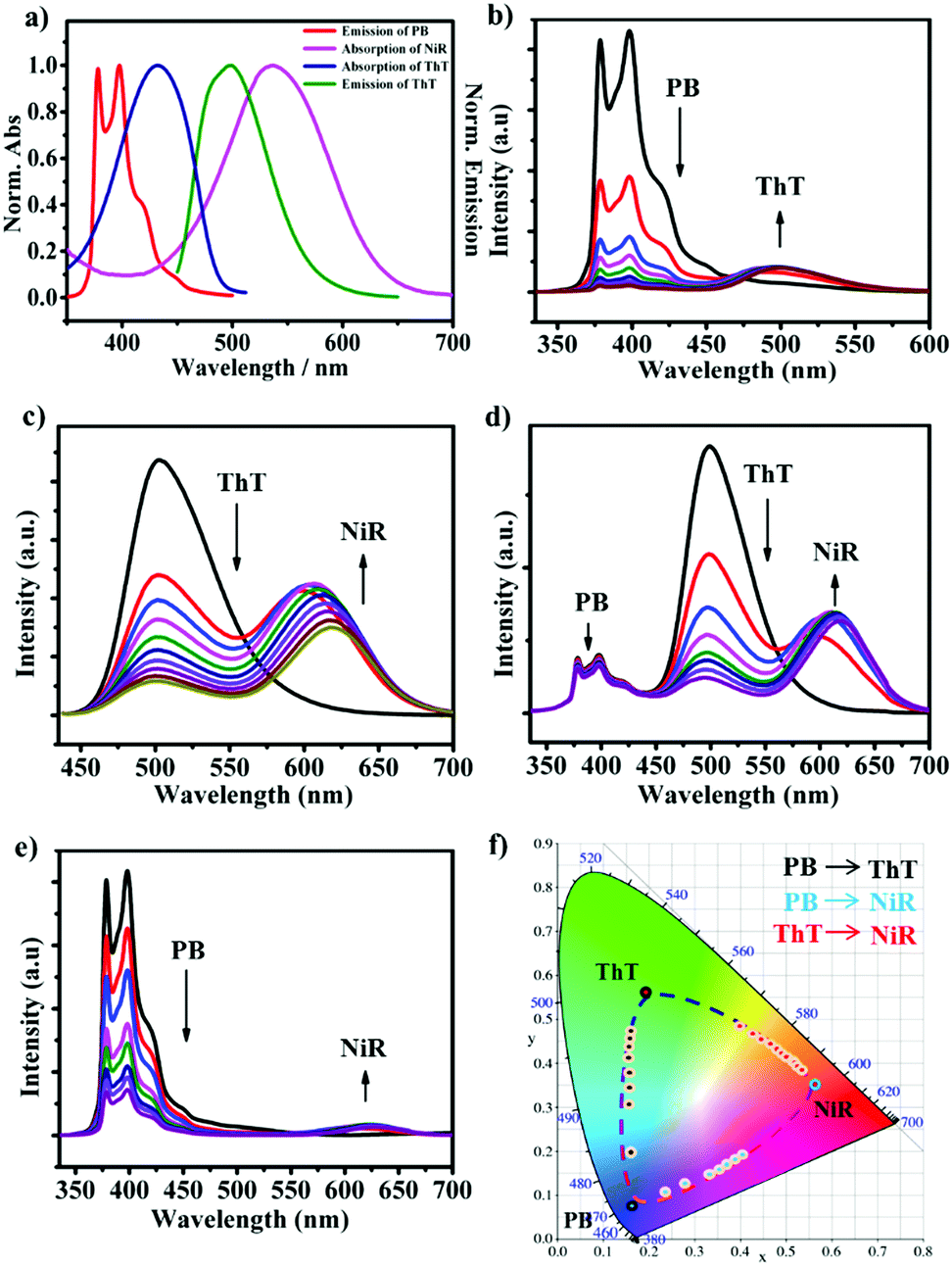 | ||
| Fig. 1 (a) Normalized emission spectrum (PB and ThT) and absorption spectrum (ThT and NiR) in the CD/CA co-assembly in HEPES buffer (pH = 8.0, 10 mM). Fluorescence spectra in the CD/CA co-assembly ([CD] = [CA] = 50 μM) of (b) PB (10 μM) with different concentrations of ThT (0, 2, 4, 6, 8, 10, 12, 14, 16 μM), λex = 326 nm; (c) ThT (50 μM) with different concentrations of NiR (0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5 μM), λex = 430 nm; (d) PB (10 μM)/ThT (16 μM) with different concentrations of NiR (0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5 μM), λex = 326 nm; (e) PB (10 μM) with different concentrations of NiR (0.0, 0.5, 1.0, 2.0, 3.0, 4.0, 5.0, 6.0 μM), λex = 326 nm. (f) The CIE chromaticity diagram showing the photoluminescent color changes of (b) (black dots), (c) (red dots) and (e) (blue dots). | ||
The various fluorescence ratios were monitored individually, as shown in Fig. 1b–e. Titration of ThT gradually lowered the PB emission at 378 and 398 nm while enhancing that of ThT at 500 nm when PB was selectively excited at 326 nm (Fig. 1b). Similarly, titration of NiR gradually lowered the ThT emission at 500 nm while enhancing that of NiR at 620 nm when ThT was selectively excited at 430 nm (Fig. 1c). In the absence of PB and ThT, irradiation of ThT and NiR at the excitation maxima of PB and ThT, respectively, yielded negligible emission since both dyes have negligible absorption at these wavelengths (see Fig. S6, ESI†), which ruled out the possibility for direct excitation. The excited state lifetimes of PB and ThT were significantly shortened upon addition of ThT and NiR, respectively, strongly suggesting a Förster-type energy transfer (see Fig. S9, ESI†). Despite the fact that trivial energy transfer and photoinduced electron transfer processes cannot be excluded, it was abundantly clear that further addition of NiR to the existing one-step energy transfer pair (from PB to ThT) leads to a pronounced decrease of ThT emission accompanied by the appearance of NiR emission when PB was selectively excited (Fig. 1d), suggesting a two-step energy transfer cascade. It should be mentioned that in the absence of ThT, direct coupling from PB to NiR was also observed (Fig. 1e) since there is a marginal overlap between the PB emission and the NiR absorption (Fig. 1a). Consequently, we established a multiple cascade integrating several energy transfer processes, including one-step PB → ThT, one-step PB → NiR, one-step ThT → NiR, and two-step PB → ThT → NiR. Such a coupling between three chromophores endows in principle the self-assembled ensemble with broad-spectrum photoluminescence outputs, which covers a large 2D area in the Commission Internationale de l'Eclairage (CIE) chromaticity diagram (Fig. 1f). In comparison, a one-step energy transfer array between binary chromophores allows for the spectrum tunability merely in a 1D trajectory.
Benefiting from the dynamic nature of non-covalent loading, delicately varying the molar ratio between the PB, ThT and NiR chromophores allows for fine-tuning of the fluorescence ratios, and thus, according to our design, a large matrix of photoluminescence outputs. The CIE chromaticity diagram in Fig. 2a shows the color-tuning map by tailoring the chromophore contents. In the presence of PB, adding ThT changes the color from blue to green. Adding NiR to PB changes the color from blue to red, and adding NiR to ThT changes the color from green to red. More importantly, in a given PB/ThT system (with varied molar ratios), adding NiR can tune the color outputs much more richly, which occupies a large area surrounded by the dashed line. One more feature of the modulated cocktails is that the emission outputs could be tuned by the excitation inputs in a wide range from 326 to 526 nm (Fig. 2e and Fig. S8, ESI†), which imparts a facile authentication test when applying the tunable photoluminescent materials as fluorescent security inks (see the following discussion).
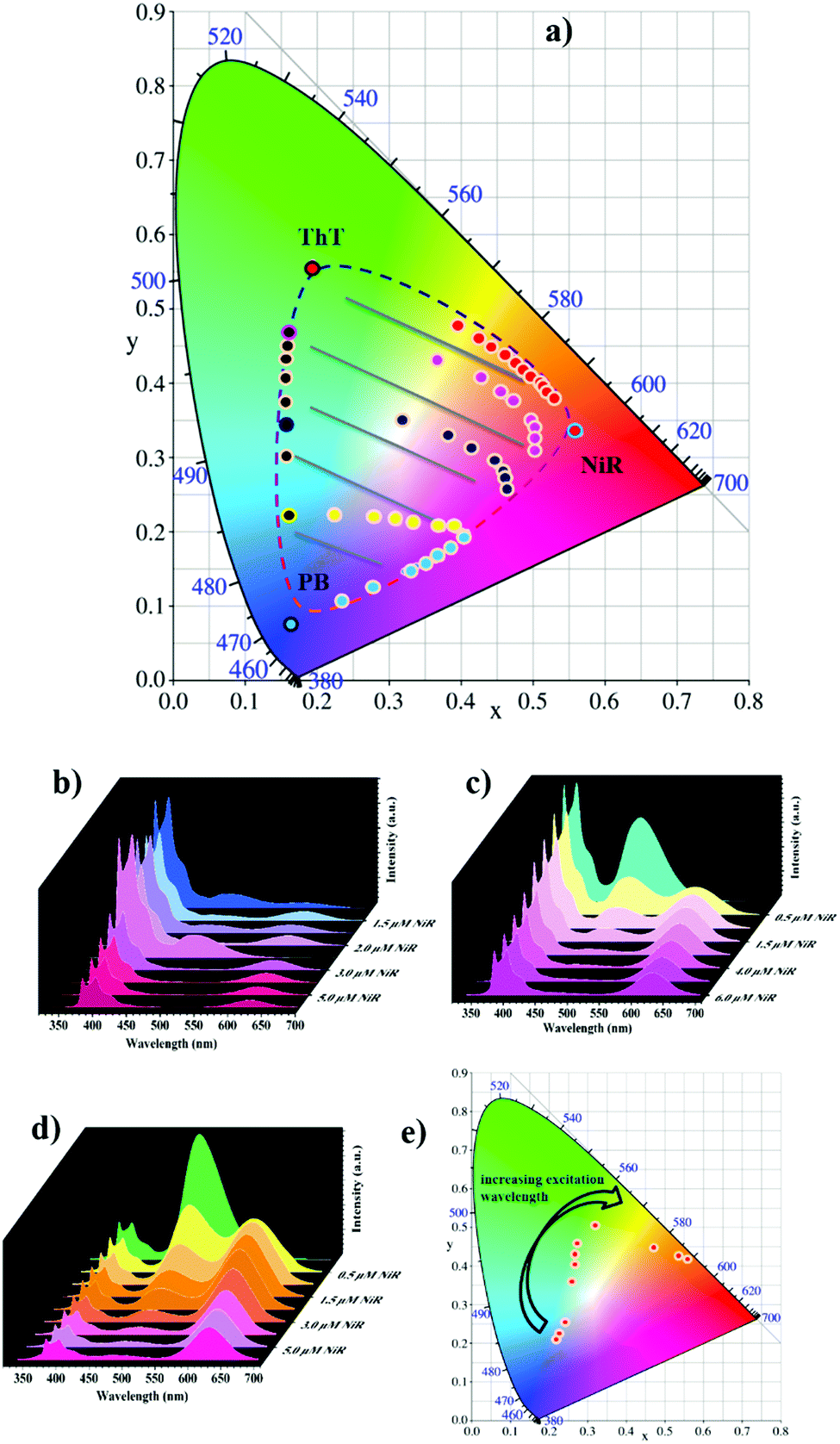 | ||
| Fig. 2 (a) The CIE chromaticity diagram showing the photoluminescent color changes by tuning the molar ratios of chromophores. Fluorescence spectra in the CD/CA co-assembly ([CD] = [CA] = 50 μM) in HEPES buffer (pH = 8.0, 10 mM) of (b) PB (10 μM)/ThT (2 μM) with different concentrations of NiR (0.0, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0 μM), (c) PB (10 μM)/ThT (6 μM) with different concentrations of NiR (0.0, 0.5, 1.0, 1.5, 3.0, 4.0, 5.0, 6.0 μM) and (d) PB (10 μM)/ThT (16 μM) with different concentrations of NiR (0.0, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0 μM), λex = 326 nm. (e) The CIE chromaticity diagram showing the photoluminescent color tunability of the cocktail with 10 μM PB, 2 μM ThT and 0.5 μM NiR upon excitation with different wavelengths (326, 346, 366, 386, 406, 426, 446, 466, 486, 506, 526 nm). | ||
Fig. 3a gives the photographs of the modulated cocktails with varied molar ratios of PB/ThT/NiR under UV light (365 nm), showing rich emission colors that can be clearly seen by the naked eye. Moreover, upon writing the predefined solutions on ordinary filter paper, no appreciable fluorescence and color changes were observed, indicating that the cascade FRET process as well as the corresponding fluorescence was preserved in the dry (solid) state (see Fig. S11, ESI†). Obvious changes of emission colors in the solid state were also observed by varying chromophore ratios (Fig. 3b). This is a prerequisite for application as fluorescent inks. As control experiments, no appreciable emission was observed under irradiation at 365 nm when writing PB, ThT and NiR individually without the CD/CA co-assembly (Fig. 3e). Writing these dyes on the preexisting CD/CA co-assembly gives pronounced emission with different colors (Fig. 3f). These results validated the crucial role of the co-assembly platform in maintaining the solid-sate fluorescence (no aggregation-caused quenching owing to the macrocyclic inclusion) and in accomplishing the spectrum tunability. We further examined the stability of the system in solution and in paper as a function of time, humidity, and exposure to light. As shown in Fig. S12d (ESI†), no appreciable photobleaching in solution was observed under irradiation with different wavelengths for 4 h. More importantly, the colors in paper could be maintained for over 50 days (see Fig. S12a, ESI†), and are inert to humidity (see Fig. S12b, ESI†) and to exposure to light (see Fig. S12c, ESI†). These stability results further validated that the present system is robust to serve as security inks.
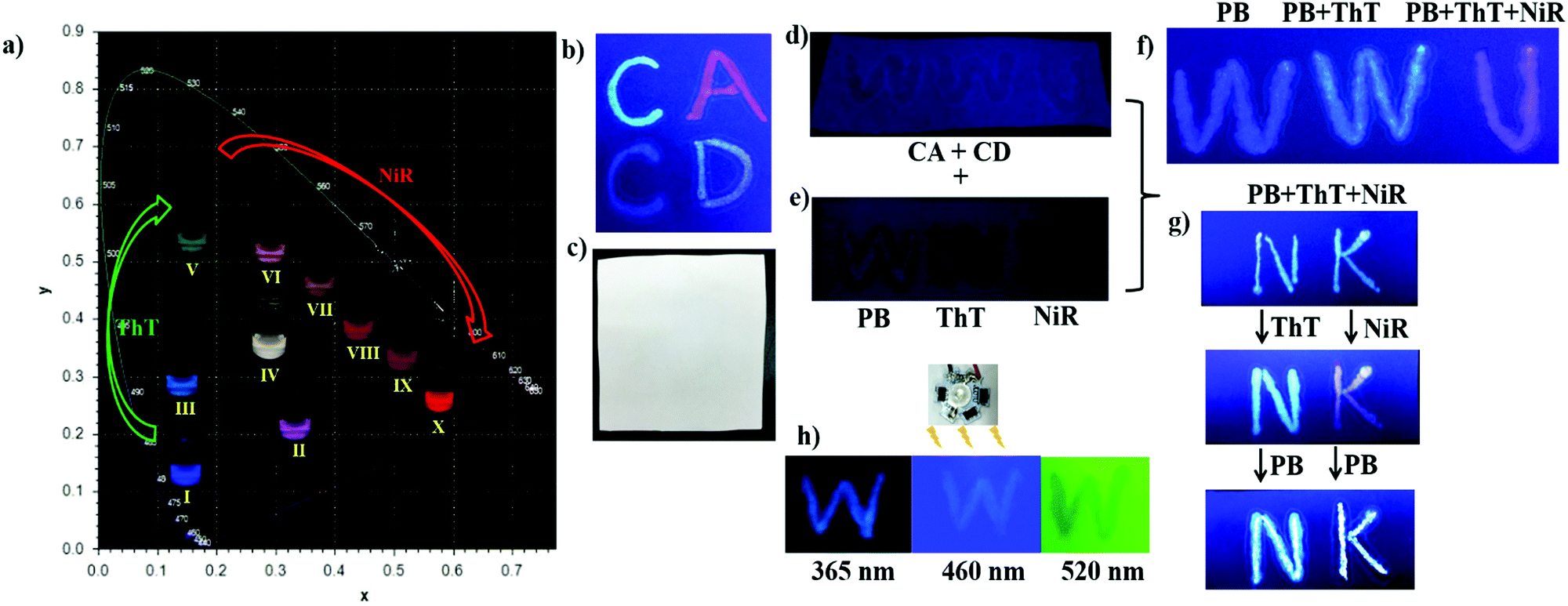 | ||
| Fig. 3 (a) Photographs of the modulated cocktails based on the CD/CA co-assembly ([CD] = [CA] = 50 μM) under 365 nm UV light: (I) PB/ThT/NiR, 10/0/0 μM, CIE coordinate (0.1638, 0.0734); (II) PB/ThT/NiR, 10/2/3 μM, CIE coordinate (0.3726, 0.2150); (III) PB/ThT/NiR, 10/2/0 μM, CIE coordinate (0.1710, 0.2234); (IV) PB/ThT/NiR, 10/6/0.5 μM, CIE coordinate (0.3215, 0.3406); (V) PB/ThT/NiR, 10/16/0 μM, CIE coordinate (0.1723, 0.4683); (VI) PB/ThT/NiR, 10/16/0.5 μM, CIE coordinate (0.3752, 0.4251); (VII) PB/ThT/NiR, 10/16/1 μM, CIE coordinate (0.4269, 0.4090); (VIII) PB/ThT/NiR, 10/16/2 μM, CIE coordinate (0.4621, 0.3809); (IX) PB/ThT/NiR, 10/16/4 μM, CIE coordinate (0.4901, 0.3498); (X) PB/ThT/NiR, 10/16/6 μM, CIE coordinate (0.5012, 0.3176). The writing features of fluorescent security inks based on the CD/CA co-assembly ([CD] = [CA] = 100 μM) platform: photographs under 365 nm UV light (b) and natural light (c) of writing “C” in “CA” with 20/12 μM PB/ThT, “A” with 20/32/12 μM PB/ThT/NiR, “C” in “CD” with 20 μM PB, “D” with 20/32/2 μM PB/ThT/NiR; photographs under 365 nm UV light of (d) writing “WWU” with the CD/CA co-assembly merely, (e) writing the first “W” with 20 μM PB, the second “W” with 50 μM ThT and “U” with 12 μM NiR without the CD/CA co-assembly; (f) rewriting the first “W” with 20 μM PB, the second “W” with 32 μM ThT after 20 μM PB and “U” with 12 μM NiR after 20 μM PB and 32 μM ThT on the preexisting CD/CA co-assembly; (g) from top to bottom, writing “NK” with 20/32/2 μM PB/ThT/NiR, rewriting “N” with 32 μM ThT and “K” with 12 μM NiR, rewriting “N” and “K” with 20 μM PB; (h) photographs of writing “W” with 20/4/1 μM PB/ThT/NiR under 365, 460 and 520 nm, respectively. | ||
Based on the observation that the system outputs depend nonlinearly on the concentrations of components, a dual-encryption coding was implanted as illustrated in Fig. 4. In the first encryption, the complexation-induced fluorescence enhancement of chromophores (PB and ThT) by macrocyclic hosts (CD and CA) depends nonlinearly on the concentrations of hosts and guests. Such a supramolecular encryption principle has recently been demonstrated by Stoddart and co-workers.10 Besides, there is the second encryption in our system. Upon close inspection of the results in Fig. 1, we found that the majority of the blue-shifted fluorescence was quenched by only a small fraction of the red-shifted emitter, indicating an energy trapping process. Even if the photon trapping performance cannot match the efficiency of the natural photosynthesis and some artificial systems,14 the blue-shifted fluorescence still decreases nonlinearly upon gradual addition of the red-shifted emitter (see Fig. S7, ESI†), which is sufficient to realize the proposed encryption. Such a combination of supramolecular encryption settings affords the feasibility of applying our broad-spectrum tunable photoluminescent materials as fluorescent security inks. The information encoded in polychromic images can be protected in such a dual-encryption way that it is close to impossible to reverse engineer.
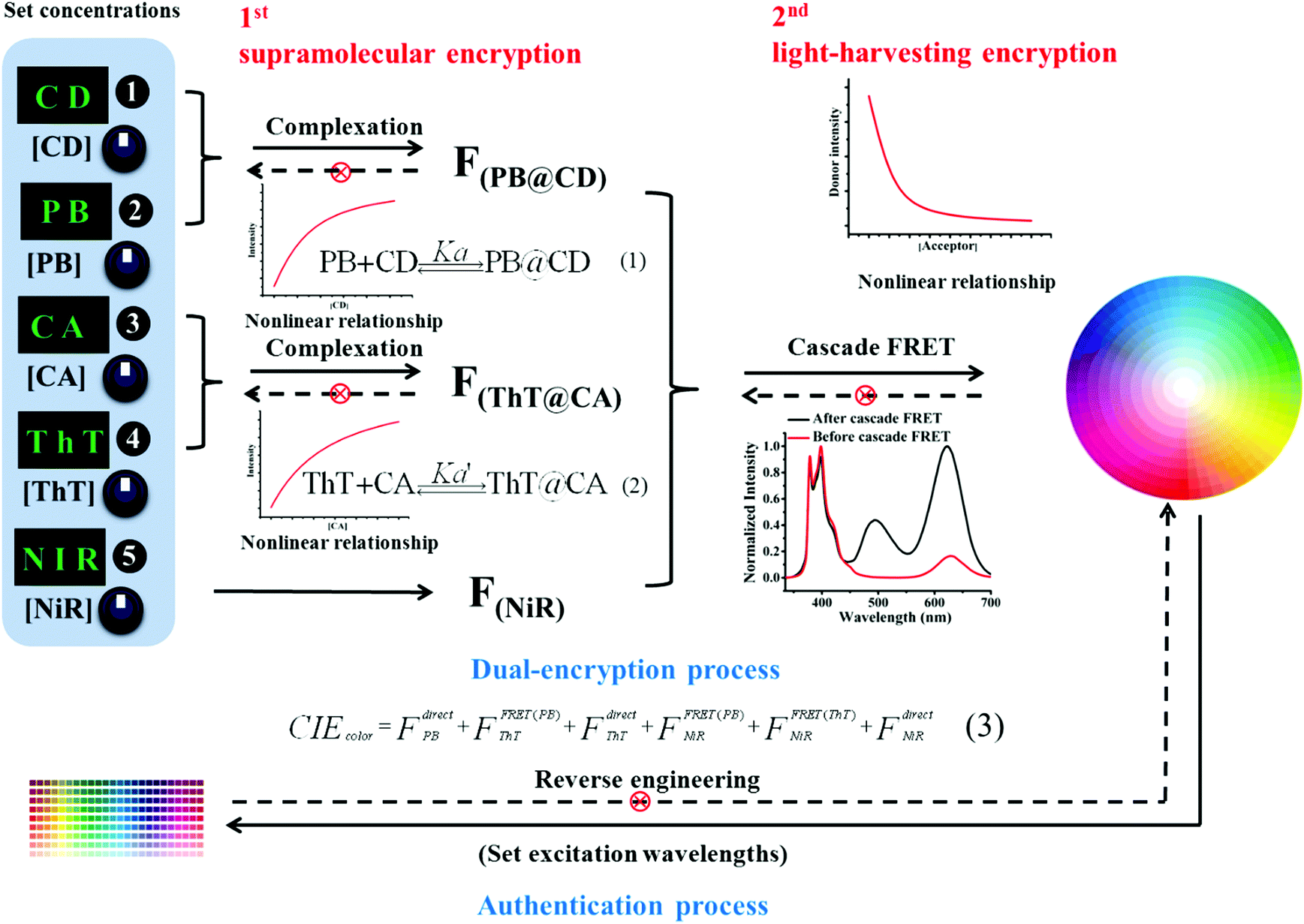 | ||
| Fig. 4 The proposed working principle of security printing with dual-encryption and authentication by applying the self-assembled multicomponent ensembles as fluorescent inks. Fraud detection by varying the excitation wavelengths is based on the authentication process according to eqn (3), where F is the photo emission, direct represents direct excitation, and FRET(dye) represents the energy transfer. | ||
Authentication tests without revealing the original color image information are commonly demanded to prevent counterfeiting. The encrypted information can be verified by chemical authentication. As shown in Fig. 4, post-writing any chromophore (PB, ThT or NiR) generated non-linearly color changes, which cannot be realized by counterfeits made by the conventional cyan-magenta-yellow-dark (CMYK) or red-green-blue (RGB) technology. The CD/CA co-assembly provides a fixed addressability for the chromophores, PB in the CD cavity, ThT in the CA cavity and NiR in the bilayer, which allows for the well-defined reproducibility between parallel experiments, as well as the predictable color change in response to simple chemical stimuli. Moreover, the anti-counterfeit labels made by the present fluorescent security inks can be easily authenticated by varying irradiation sources. The output colors change with the excitation wavelengths. Like the “W” in Fig. 3h, it appears blue, yellow and red under irradiation of 365, 460 and 520 nm, respectively. This is because the CIE color is composed of a sum of three chromophore emissions originated from both sensitization via energy transfer and direct excitation (eqn (3) in Fig. 4, FdirectPB, FFRET(PB)ThT, FdirectThT, FFRET(PB)NiR, FFRET(ThT)NiR, FdirectNiR). Upon 365 nm irradiation, FdirectPB, FFRET(PB)ThT, FFRET(PB)NiR and FFRET(ThT)NiR contribute to the CIE color; upon 460 nm irradiation, FdirectThT and FFRET(ThT)NiR contribute to the CIE color; upon 520 nm irradiation, only FdirectNiR contributes to the CIE color.
Conclusions
In summary, we have designed and developed a new type of color-tunable photoluminescent material displaying substantial potential as fluorescent security ink. The present self-assembled multicomponent system exhibits several features: (1) the CD/CA co-assembled platform with full modularity and facile integration of multiple chromophores, (2) broad-spectrum tunability without any additional synthetic effort, (3) solid-state fluorescence bypassing the aggregation-caused quenching by the compartmentalization effect, (4) dual-encryption coding of supramolecular strategies. By applying the smart materials as fluorescent security inks, the encrypted information is highly resistant to counterfeiting, and can be easily authenticated without being decoded. Moreover, the co-assembling strategy of integrating different macrocyclic hosts may be amendable to construct other function-directed materials on demand.Acknowledgements
The Chinese team gratefully acknowledges the support of the NNSFC (21322207 and 21672112) and the Program of Tianjin Young Talents. The German team thanks Dr Till Böckermann for the synthesis of amphiphilic CD and the Alexander von Humboldt Foundation for financial support. We also thank Prof. Dr Monika Schönhoff at the Institute of Physical Chemistry, University of Münster for DSC measurements.Notes and references
- T. Noda, H. Ogawa, N. Noma and Y. Shirota, Adv. Mater., 1997, 9, 720 CrossRef CAS; W. B. Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. Denbaars and R. Seshadri, Adv. Mater., 2011, 23, 2300 CrossRef PubMed; C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059 CrossRef; X.-L. Ni, S. Chen, Y. Yang and Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177 CrossRef PubMed; B. Yoon, D.-Y. Ham, O. Yarimaga, H. An, C. W. Lee and J.-M. Kim, Adv. Mater., 2011, 23, 5492 CrossRef PubMed.
- L. Y. Wang, R. X. Yan, Z. Y. Hao, L. Wang, J. H. Zeng, J. Bao, X. Wang, Q. Peng and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 6054 CrossRef CAS PubMed; H. P. Li and G. C. Bazan, Adv. Mater., 2009, 21, 964 CrossRef; Y. Liu, J. Jin, H. Deng, K. Li, Y. Zheng, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7952 CrossRef PubMed.
- B. Yoon, J. Lee, I. S. Park, S. Jeon, J. Lee and J.-M. Kim, J. Mater. Chem. C, 2013, 1, 2388 RSC; J. Andres, R. D. Hersch, J. E. Moser and A. S. Chauvin, Adv. Funct. Mater., 2014, 24, 5029 CrossRef CAS.
- T. Wang, V. Chirmanov, W. H. M. Chiu and P. V. Radovanovic, J. Am. Chem. Soc., 2013, 135, 14520 CrossRef CAS PubMed; C. Zhang, Y. S. Zhao and J. Yao, New J. Chem., 2011, 35, 973 RSC; Z. Xu, Q. Liao, X. Shi, H. Li, H. Zhang and H. Fu, J. Mater. Chem. B, 2013, 1, 6035 RSC; J. Chen, P. Zhang, G. Fang, P. Yi, F. Zeng and S. Wu, J. Phys. Chem. B, 2012, 116, 4354 CrossRef PubMed; X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623 CrossRef PubMed; K. S. Sanju, P. P. Neelakandan and D. Ramaiah, Chem. Commun., 2011, 47, 1288 RSC; P. Bairi, B. Roy and A. K. Nandi, Chem. Commun., 2012, 48, 10850 RSC; X. Zhang, D. Görl and F. Würthner, Chem. Commun., 2013, 49, 8178 RSC.
- Z. Xu, S. Peng, Y.-Y. Wang, J.-K. Zhang, A. I. Lazar and D.-S. Guo, Adv. Mater., 2016, 28, 7666 CrossRef CAS PubMed; W.-C. Geng, Y.-C. Liu, Y.-Y. Wang, Z. Xu, Z. Zheng, C.-B. Yang and D.-S. Guo, Chem. Commun., 2017, 53, 392 RSC; M. Bälter, S. Li, M. Morimoto, S. Tang, J. Hernando, G. Guirado, M. Irie, F. M. Raymo and J. Andréasson, Chem. Sci., 2016, 7, 5867 RSC; Y. Ner, J. G. Grote, J. A. Stuart and G. A. Sotzing, Angew. Chem., Int. Ed., 2009, 48, 5134 CrossRef PubMed; C. Vijayakumar, K. Sugiyasu and M. Takeuchi, Chem. Sci., 2011, 2, 291 RSC.
- X. Zhang, J. Yu, Y. Rong, F. Ye, D. T. Chiu and K. Uvdal, Chem. Sci., 2013, 4, 2143 RSC; X. Hu, Y. Li, T. Liu, G. Zhang and S. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 15551 Search PubMed; B. L. Nguyen, J.-E. Jeong, I. H. Jung, B. Kim, V. S. Le, I. Kim, K. Kyhm and H. Y. Woo, Adv. Funct. Mater., 2014, 24, 1748 CrossRef CAS; P. A. Bhat, O. A. Chat and A. A. Dar, ChemPhysChem, 2016, 17, 2360 CrossRef PubMed.
- S. Kawahara, T. Uchimaru and S. Murata, Chem. Commun., 1999, 563 RSC; J. K. Hannestad, P. Sandin and B. Albinsson, J. Am. Chem. Soc., 2008, 130, 15889 CrossRef CAS PubMed; M. Cotlet, T. Vosch, S. Habuchi, T. Weil, K. Müllen, J. Hofkens and F. D. Schryver, J. Am. Chem. Soc., 2005, 127, 9760 CrossRef PubMed.
- G. Chadha, Q.-Z. Yang and Y. Zhao, Chem. Commun., 2015, 51, 12939 RSC; X. Song, J. Shi, J. Nolan and B. Swanson, Anal. Biochem., 2001, 291, 133 CrossRef CAS PubMed.
- T.-H. Kim, H. K. Lee, O. O. Park, B. D. Chin, S.-H. Lee and J. K. Kim, Adv. Funct. Mater., 2006, 16, 611 CrossRef CAS; P. Bairi, B. Roy, P. Chakraborty and A. K. Nand, ACS Appl. Mater. Interfaces, 2013, 5, 5478 Search PubMed; K.-P. Tseng, F.-C. Fang, J.-J. Shyue, K.-T. Wong, G. Raffy, A. D. Guerzo and D. M. Bassani, Angew. Chem., Int. Ed., 2011, 50, 7032 CrossRef PubMed; B.-B. Du, Y.-X. Zhu, M. Pan, M.-Q. Yue, Y.-J. Hou, K. Wu, L.-Y. Zhang, L. Chen, S.-Y. Yin, Y.-N. Fan and C.-Y. Su, Chem. Commun., 2015, 51, 12533 RSC; Q.-Y. Yang, M. Pan, S.-C. Wei, K. Li, B.-B. Du and C.-Y. Su, Inorg. Chem., 2015, 54, 5707 CrossRef PubMed.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef CAS PubMed; Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef PubMed; A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef PubMed; H. Tian and S. J. Yang, Chem. Soc. Rev., 2004, 33, 85 RSC; Y. Liu, K.-R. Wang, D.-S. Guo and B.-P. Jiang, Adv. Funct. Mater., 2009, 19, 2230 CrossRef.
- P. Falvey, C. W. Lim, R. Darcy, T. Revermann, U. Karst, M. Giesbers, A. T. M. Marcelis, A. Lazar, A. W. Coleman, D. N. Reinhoudt and B. J. Ravoo, Chem. – Eur. J., 2005, 11, 1171 CrossRef CAS PubMed; A. Mazzaglia, R. Donohue, B. J. Ravoo and R. Darcy, Eur. J. Org. Chem., 2001, 1715 CrossRef; B. J. Ravoo and R. Darcy, Angew. Chem., Int. Ed., 2000, 39, 4324 CrossRef.
- R. Ozawa, T. Hashimoto, A. Yamauchi, I. Suzuki, B. D. Smith and T. Hayashita, Anal. Sci., 2008, 24, 207 CrossRef CAS PubMed.
- P. Greenspan, E. P. Mayer and S. T. Fowler, J. Cell Biol., 1985, 100, 965 CrossRef CAS PubMed.
- H.-Q. Peng, Y.-Z. Chen, Y. Zhao, Q.-Z. Yang, L.-Z. Wu, C.-H. Tung, L.-P. Zhang and Q.-X. Tong, Angew. Chem., Int. Ed., 2012, 51, 2088 CrossRef CAS PubMed; H.-Q. Peng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Rev., 2015, 115, 7502 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterisation data. See DOI: 10.1039/c7qm00091j |
| This journal is © the Partner Organisations 2017 |