DOI:
10.1039/C7QM00025A
(Research Article)
Mater. Chem. Front., 2017,
1, 1389-1395
Halogenated conjugated molecules for ambipolar field-effect transistors and non-fullerene organic solar cells†
Received
9th February 2017
, Accepted 22nd February 2017
First published on 23rd February 2017
Abstract
A series of halogenated conjugated molecules, containing F, Cl, Br and I, were easily prepared via Knoevenagel condensation and applied in field-effect transistors and organic solar cells. Halogenated conjugated materials were found to possess deep frontier energy levels and high crystallinity compared to their non-halogenated analogues, which is due to the strong electronegativity and heavy atom effect of halogens. As a result, halogenated semiconductors provide high electron mobilities up to 1.3 cm2 V−1 s−1 in transistors and high efficiencies over 9% in non-fullerene solar cells.
Introduction
Halogens, fluorine (F), chlorine (Cl), bromine (Br) and iodine (I), play important roles in organic electronics.1 Organic semiconductors are usually constructed from aryl halides via metal-catalyzed cross-coupling reactions.2 Halogens are also incorporated into electronic devices, such as dye-sensitized solar cells with triiodide/iodide as the redox couple3 and perovskite solar cells by using crystallized methylammonium lead halides as photoactive layers.4,5 Halogenated conjugated materials also act as charge transport layers in field-effect transistors (FETs) and organic solar cells (OSCs), among which fluorinated derivatives are widely reported to show excellent air-stable electron mobilities6–10 and high power conversion efficiencies (PCEs) in solar cells.11–15 The major benefits are the deep frontier energy levels caused by the strong electron-negative fluorine and good crystalline properties via non-covalent interactions between F and H/C/S.16–18 A similar effect can be found in chlorinated19–25 and brominated conjugated materials,26–28 but these materials are relatively less studied compared to fluorinated derivatives. There are very few reports on the application of iodinated conjugated molecules to organic electronics, although iodine doping is an effective way to improve the conductivity of semiconductors.29,30 The limited number of studies on Cl, Br and I-containing semiconductors is presumably due to steric hindrance effects originating from their large atomic size, consequently lowering the device performance.
We are interested in halogenated semiconductors and their electronic properties for the following reasons: (1) the electronegativity of halogens is decreased as the molar mass increases from F to I, which will influence the energy levels and absorption spectra of conjugated materials; (2) recently, some reports have shown that large heteroatoms, such as silicon,31 germanium,32 selenium33–35 and tellurium,36,37 are able to improve the crystal and charge transport properties of conjugated materials through the planar backbone, the so called “heavy-atom effect”.35 Therefore, high molar mass halogens probably show similar properties that need to be studied.
Herein, a series of conjugated small molecules without halogens and with halogens (F, Cl, Br and I) were developed for application in FETs and OSCs. These conjugated materials contain the same conjugated core, denoted as ITIC (3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene) (Fig. 1a), which also represents the most successful electron acceptor to replace fullerene derivatives in OSCs.38–49 Halogens as the end groups were attached to ITIC, eliminating the steric hindrance. Halogenated ITIC shows improved crystallinity, deep energy levels and red-shifted absorption, resulting in much improved electron mobilities up to 1.3 cm2 V−1 s−1 and PCEs above 9% in non-fullerene solar cells. For comparison, H-ITIC provided an electron mobility of 0.047 cm2 V−1 s−1 and a PCE of 6.4% in solar cells. These mobilities and PCEs are among the highest values reported for Br and I-based organic semiconductors to date.
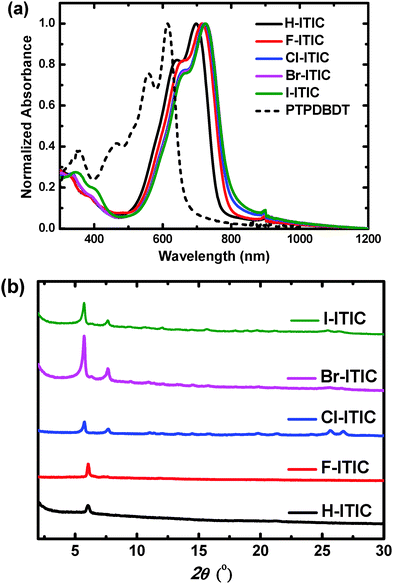 |
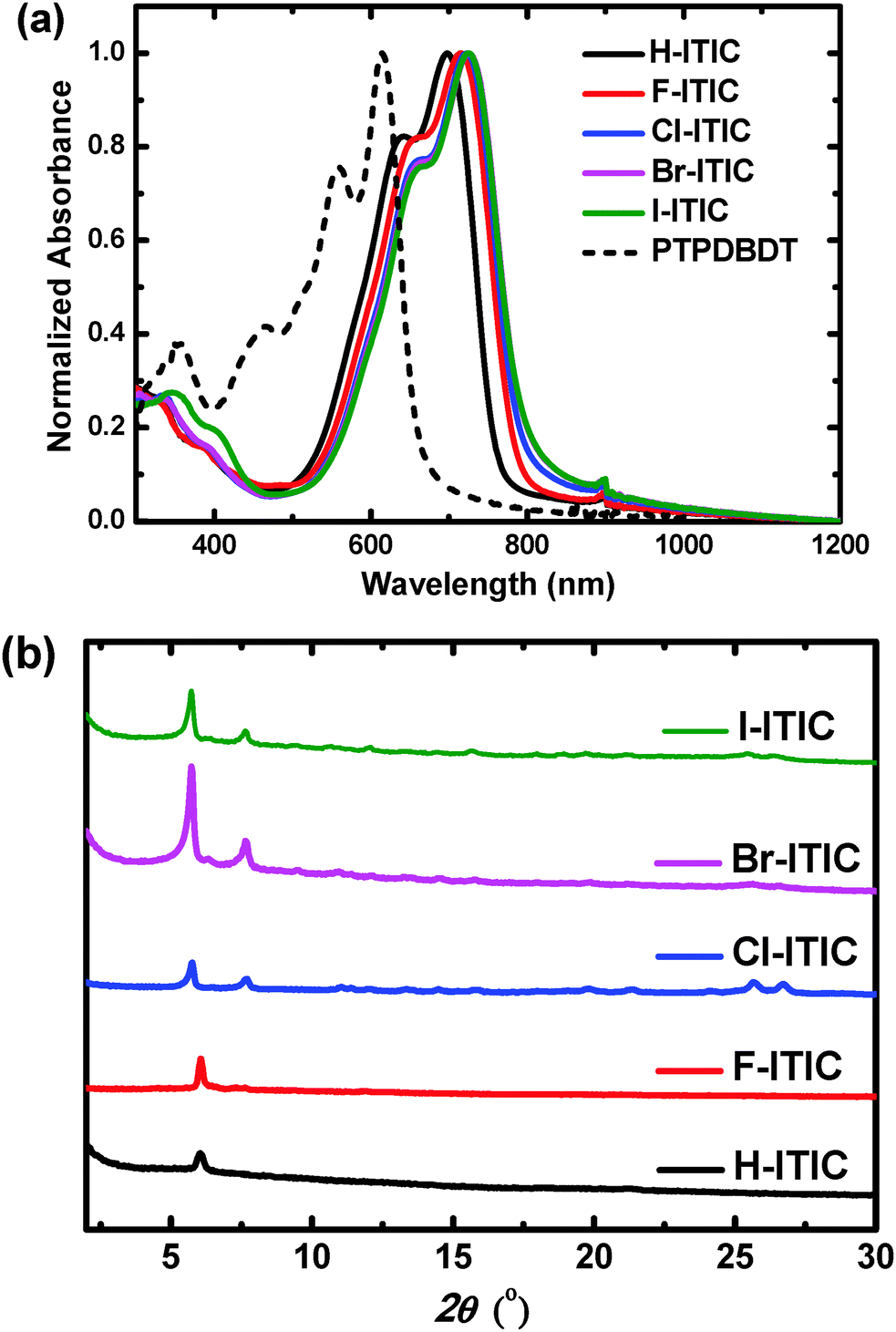
| | Fig. 1 (a) Absorption spectra in thin films, and (b) XRD pattern of the conjugated molecules X-ITIC studied in this work. The absorption of PTPDBDT in thin films was also included. | |
Results and discussion
Synthesis and characterization
Scheme 1 shows the synthetic route of halogenated ITICs. The halogenated 1,3-indanedione derivative (2-X) was obtained by reacting commercially available halogenated phthalic anhydride (1-X) with ethyl acetoacetate in the presence of triethylamine. Halogenated indane derivatives (3-X) were prepared by condensation of malononitrile to halogenated 1,3-indanedione (2-X). The final condensation products were achieved via Knoevenagel condensation reactions between 3-X and IDTT-CHO, respectively. It is worth mentioning that two isomers can be observed due to the synthesis of the precursors 3-X, in which malononitrile can attack one of the two carbonyl groups. Thus, the final condensation products X-ITIC were also a mixture of two isomers, which were difficult to separate using column chromatography. All new compounds were fully characterized using 1H NMR, 13C NMR, and MALDI-TOF mass spectral data (see the ESI†). All these X-ITIC show good solubility in common solvents, such as CHCl3 and chlorobenzene, and good thermal stability with 5% weight loss temperatures above 332 °C (Fig. S1, ESI†).
 |
| | Scheme 1 Synthetic procedures for X-ITIC. (i) Ac2O, triethylamine and ethyl acetoacetate, RT overnight; HCl (aq), 80 °C for 15 min. (ii) NaOAc, malononitrile, RT for 40 min; HCl (aq). (iii) Pyridine, CH2Cl2, RT for 12 h. | |
Optical and electrochemical properties
All the molecules show absorption spectra in the range of 500 nm to 750 nm in CHCl3, in which the absorption peak is red-shifted from H- to F-, Cl-, Br- and I-ITIC (Fig. S2, ESI†). This can be explained by the inductive effect and π-electrons delocalizing onto empty d-orbitals on the halogens.1 In thin films, H-, F-, Cl- and Br-ITIC present red-shifted absorption spectra (Fig. 1a) with an optical band gap (Eg) of 1.59 eV for H-ITIC and 1.53 eV for Br-ITIC, while I-ITIC has a slightly high Eg of 1.55 eV (Table 1). The frontier energy levels of the molecules determined by cyclic voltammetry measurements (Fig. S3, ESI†) are shown in Table 1, in which ITIC shows highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of −5.61 eV and −4.02 eV. The HOMOs and LUMOs of F-, Cl- and Br-ITIC are gradually shifted to a low-lying position, while I-ITIC shows slightly high-lying HOMO and LUMO levels of −5.68 eV and −4.14 eV compared to the other halogen-substituted ITICs. The crystalline properties of these ITIC derivative thin films deposited on Si substrates were investigated by X-ray diffraction (XRD, Fig. 1b). Halogenated ITICs show high and sharp peaks in thin films compared to H-ITIC, indicating their better crystallinity. Among them, Br-ITIC has the highest diffraction peak with the d-spacing of 15.4 Å, and I-ITIC produced reduced crystal peaks. Additional diffraction peaks located at 7.65° corresponding to the d-spacing of 11.5 Å are also observed for halogenated ITICs, which are possibly attributed to the isomers in these materials (Table S1, ESI†). However, currently we have no further evidence to claim that the two diffraction peaks originate from the different crystal structures of two isomers.
Table 1 Optical, electrochemical and field effect mobility of X-ITIC and the donor polymer PTPDBDT
| |
E
g [eV] |
E
HOMO
[eV] |
E
LUMO
[eV] |
μ
h [cm2 V−1 s−1] |
μ
e [cm2 V−1 s−1] |
|
Determined by −4.80 − Eox.
E
LUMO is calculated as EHOMO + Eg.
The FET devices were thermal annealed at 180 °C for 10 min.
The FET devices were thermal annealed at 90 °C for 10 min.
|
| H-ITIC |
1.59 |
−5.61 |
−4.02 |
0.074 |
0.047c |
| F-ITIC |
1.56 |
−5.65 |
−4.09 |
0.0027 |
0.002d |
| Cl-ITIC |
1.56 |
−5.70 |
−4.14 |
0.011 |
0.10c |
| Br-ITIC |
1.53 |
−5.73 |
−4.20 |
0.0067 |
1.3c |
| I-ITIC |
1.55 |
−5.68 |
−4.14 |
0.13 |
0.30c |
| PTPDBDT |
1.85 |
−5.60 |
−3.75 |
— |
— |
OFET properties
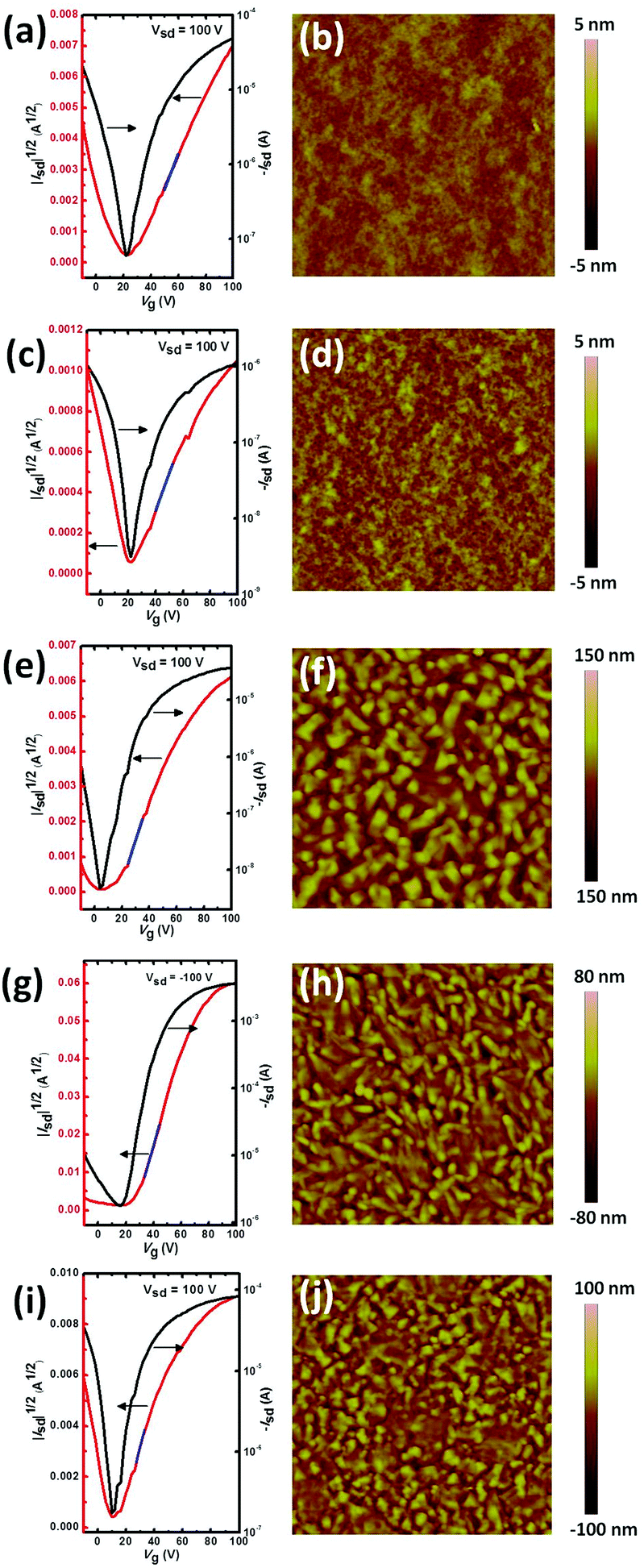
ITIC and its halogenated derivatives were applied in FETs with a bottom gate-bottom contact (BGBC) configuration. The thin films were prepared by spin coating the molecule solution in CHCl3 containing 10% n-hexane as an additive. Hexane was used to improve the wettability of X-ITIC on an OTS modified Si substrate. The thin films were thermally annealed for 10 min at different temperatures, from 25 °C to 90 °C, 150 °C, 180 °C, 210 °C and 250 °C before measurement. Thermal annealing at high temperature is helpful to form crystal domains for Cl, Br- and I-ITIC, but this is not observed in H-ITIC and F-ITIC (Fig. S4, ESI†). The transfer and output curves are summarized in Fig. 2 and Fig. S5, S6 in the ESI.† All the molecules show ambipolar charge transport, in which H-ITIC has balanced hole and electron mobilities (0.074 and 0.047 cm2 V−1 s−1, Table 1). Surprisingly, F-ITIC exhibits very low ambipolar mobilities of 0.0027 and 0.0022 cm2 V−1 s−1. The electron mobilities of Cl-ITIC and Br-ITIC were increased to 0.10 cm2 V−1 s−1 and 1.3 cm2 V−1 s−1, while the hole mobilities of the two molecules were significantly reduced to 0.011 and 0.0067 cm2 V−1 s−1. I-ITIC showed balanced ambipolar mobilities with hole and electron mobilities of 0.13 and 0.30 cm2 V−1 s−1. The distinct hole mobilities in these molecules could be due to the different energy levels and crystal packing structures in the thin films, but the real reason is unclear.
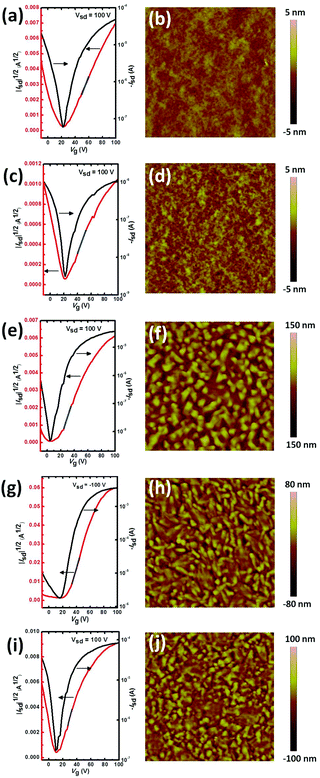 |
| | Fig. 2 n-type transfer characteristics for optimized BGBC FET devices for the ITIC derivatives and the corresponding AFM height images (3 × 3 μm2). (a and b) H-ITIC, (c and d) F-ITIC, (e and f) Cl-ITIC, (g and h) Br-ITIC and (i and j) I-ITIC. The blue lines in (a–e) are used to calculate the electron mobilities. | |
The different electron mobilities of H-ITIC and halogenated ITIC were reflected by atomic force microscopy (AFM) images, as shown in Fig. 2 and Fig. S4 (ESI†). H- and F-ITIC show smooth thin films without micro-phase separation, indicating their poor crystallinity, while crystal domains can be clearly observed in Cl-, Br- and I-ITIC thin films thermal annealed at 180 °C. Therefore, we infer that Cl, Br and I as heavy atoms can improve the crystallinity of conjugated molecules and hence enhance the charge transport properties.
Photovoltaic properties
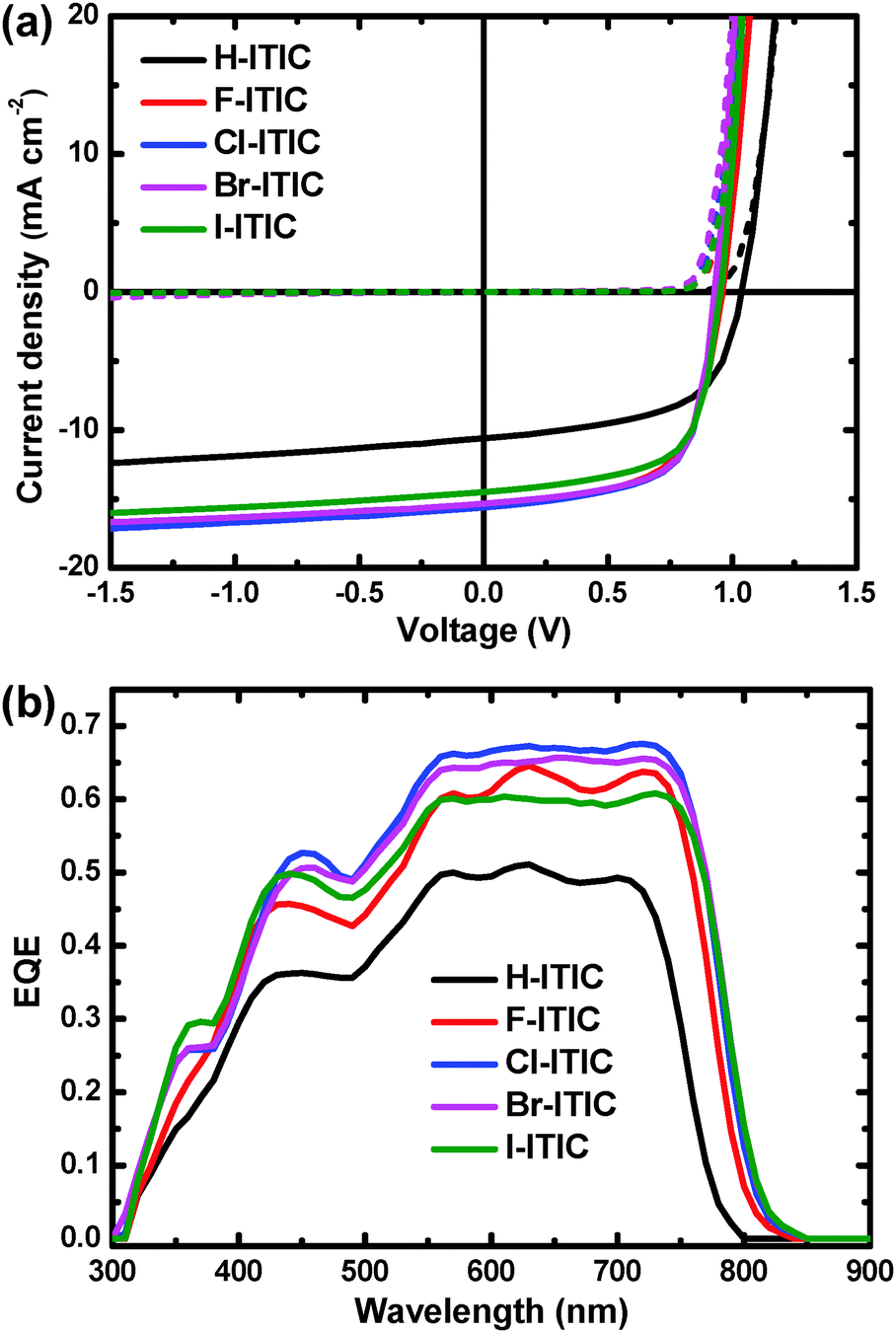
These ITIC derivatives were also applied as a non-fullerene electron acceptor in bulk-heterojunction solar cells. We selected a wide band gap polymer, PTPDBDT with thieno[3,4-c]pyrrole-4,6-dione alternated with two-dimensional benzodithiophene, as the electron donor. Notably, we used a modified synthetic procedure to prepare this polymer (Scheme S1, ESI†). It is also interesting to see that the HOMO offset between H-ITIC and PTPDBDT is only 0.01 eV, which is enhanced to 0.05–0.13 eV based on halogenated ITIC (Table 1). The photovoltaic performance of PTPDBDT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :X-ITIC (1:1) cells was evaluated by using an inverted device configuration, in which photoactive layers were spin coated from CHCl3 with 0.5% DIO as an additive. For halogenated ITIC based cells, the active layers thermally annealed at 150 °C can improve the device performance (Table S2, ESI†). The J–V characteristics are shown in Fig. 3a and the photovoltaic parameters are summarized in Table 2.
:X-ITIC (1:1) cells was evaluated by using an inverted device configuration, in which photoactive layers were spin coated from CHCl3 with 0.5% DIO as an additive. For halogenated ITIC based cells, the active layers thermally annealed at 150 °C can improve the device performance (Table S2, ESI†). The J–V characteristics are shown in Fig. 3a and the photovoltaic parameters are summarized in Table 2.
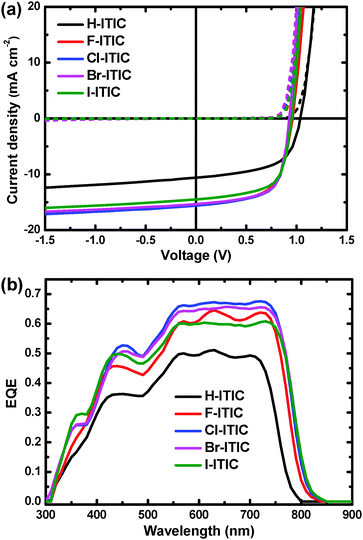 |
| | Fig. 3 (a) J–V characteristics of optimized PTPDBDT:X-ITIC solar cells in the dark (dashed lines) and under simulated solar illumination (solid lines). (b) EQE of the solar cells. | |
Table 2 Characteristics of optimized inverted solar cells of PTPDBDT:X-ITIC (1:1) fabricated from CHCl3/DIO (0.5%) as an additive
| Acceptor |
J
sc
[mA cm−2] |
V
oc [V] |
FF |
PCE [%] |
E
loss [eV] |
|
J
sc values are calculated by integrating the EQE spectrum with the AM1.5G spectrum.
Active layers without thermal annealing.
Active layers were thermal annealed at 150 °C for 30 min before metal deposition. The thicknesses of the active layers are 60–70 nm. PCEs in the parentheses show the average performance from eight devices.
|
| H-ITICb |
10.6 |
1.04 |
0.58 |
6.4 (6.1) |
0.55 |
| F-ITICc |
14.1 |
0.94 |
0.66 |
8.8 (8.3) |
0.62 |
| Cl-ITICc |
15.6 |
0.94 |
0.65 |
9.5 (9.0) |
0.62 |
| Br-ITICc |
15.4 |
0.93 |
0.66 |
9.4 (9.1) |
0.60 |
| I-ITICc |
14.5 |
0.95 |
0.65 |
8.9 (8.3) |
0.60 |
PTPDBDT:ITIC cells provide a PCE of 6.4% with a short-circuit current density (Jsc) of 10.6 mA cm−2, a high open-circuit voltage (Voc) of 1.04 V and a fill factor (FF) of 0.58. When using F-, Cl-, Br- and I-ITIC as electron acceptors, the PCEs were significantly enhanced to 8.8%, 9.5%, 9.4% and 8.9%. The improvement was mainly due to enhanced Jsc of 14.1–15.6 mA cm−2 and FF of 0.66, although the Vocs dropped to 0.93–0.95 V. The enhanced Jscs were reflected by their external quantum efficiencies (EQEs), as shown in Fig. 3b. Solar cells based on halogenated ITICs show a broad photo-response from 300 to 850 nm with maximum EQE above 0.60, while H-ITIC based cells have a relatively narrow photo-response from 300 to 800 nm and low EQE below 0.50. It is also interesting to observe that Br-ITIC and I-ITIC based cells show good stability in an N2 filled glove box (Fig. S7, ESI†).
It is desirable to discuss the improved PCEs of solar cells based on halogenated ITICs. As mentioned above, PTPDBDT:X-ITIC systems provide a slightly higher HOMO offset than that of H-ITIC based system. High HOMO offset as a driving force is beneficial for hole transfer processes from acceptors to donors, and hence enhances the photocurrent of cells.50 This is also reflected by the different energy loss (Eloss) in these cells, which is calculated as the difference between Eg and eVoc (Table 2).51–54 PTPDBDT:H-ITIC cells provide a very low Eloss of 0.55 eV, while Eloss is increased to 0.60–0.62 eV for halogenated ITIC based cells. High Eloss corresponds to the increased driving force for exciton dissociation into free charges, explaining the high photocurrent in these cells. Besides, we also note that halogenated ITIC based cells have high FF, indicating the better charge transport in these cells. We further applied a space charge limit current (SCLC) method to determine the hole and electron mobility in blended thin films (Table 3 and Fig. S8, ESI†), in which we found relatively balanced hole and electron mobility for all the blends. In addition, the morphology of blended thin films was also studied using atom force microscopy, but the roughness of these films is similar and therefore it is hard to get useful information (Fig. S9, ESI†). From these aspects, we are unable to explain the enhanced FF in halogenated ITIC based cells.
Table 3 Hole and electron mobility of the pure polymer, X-ITIC and blended thin films
| |
μ
h [cm2 V−1 s−1] |
μ
e [cm2 V−1 s−1] |
μ
h/μe |
μ
e
[cm2 V−1 s−1] |
|
Electron mobility of the pure X-ITIC fabricated from CHCl3/DIO (0.5%) as an additive. The hole mobility of PTPDBDT was 3.8 × 10−4 cm2 V−1 s−1 by SCLC measurement.
|
| PTPDBDT:H-ITIC |
1.1 × 10−4 |
1.4 × 10−4 |
0.79 |
2.7 × 10−4 |
| PTPDBDT:F-ITIC |
5.7 × 10−5 |
3.1 × 10−4 |
0.18 |
1.1 × 10−4 |
| PTPDBDT:Cl-ITIC |
7.8 × 10−5 |
5.2 × 10−4 |
0.15 |
1.9 × 10−4 |
| PTPDBDT:Br-ITIC |
9.3 × 10−5 |
5.1 × 10−4 |
0.18 |
1.1 × 10−4 |
| PTPDBDT:I-ITIC |
7.1 × 10−5 |
4.1 × 10−4 |
0.17 |
1.2 × 10−4 |
Experimental
Material synthesis
Halogenated phthalic anhydride 1-F, 1-Cl and 1-Br were purchased from TCI and 1-I was synthesized according to literature procedures.55 5-Chloro-1,3-indanedione (2-Cl),56 5-bromo-1,3-indanedione (2-Br),57IDTT-CHO38 and H-ITIC38 were synthesized according to literature procedures.
5-fluoro-1,3-indanedione (2-F).
To a solution of 4-fluorophthalic anhydride (1-F) (1.5 g, 9.03 mmol) in Ac2O (8 mL) and triethylamine (2.75 g 27.09 mmol), ethyl acetoacetate (2.43 g, 18.06 mmol) was quickly added. The solution was stirred at room temperature for 24 h, followed by adding 5 M HCl (15 mL). The mixture was stirred at 80 °C for 15 min and cooled to room temperature. The mixture was extracted with CH2Cl2, washed by water and brine, and dried by evaporation. The resulting solid was subjected to column chromatography (silica, eluent CH2Cl2) to afford 2-F as a crude product. Pure 2-F (0.80 g, 55%) was obtained by recrystallization from n-hexane. 1H NMR δ (ppm): 7.99–8.03 (m, 1H), 7.49–7.61 (m, 2H), 3.28 (s, 2H). 13C NMR δ (ppm): 196.24, 196.21, 195.63, 169.23, 165.78, 146.25, 146.13, 139.85, 139.82, 126.10, 125.97, 123.85. 123.53, 109.96, 109.65, 45.44.
5-Iodo-1,3-indanedione (2-I).
The same procedure was used as for 2-F, but now 4-iodophthalic anhydride (1-I) (1.67 g, 6.1 mmol) was used as the precursor. Yield: 0.80 g (48%). 1H NMR δ (ppm): 8.34 (d, 1H), 8.15–8.18 (m, 1H), 7.67–7.70 (d, 1H), 3.22 (s, 2H). 13C NMR δ (ppm): 196.36, 195.74, 144.42, 144.05, 142.11, 132.45, 124.24, 103.84, 44.55.
3-F
.
To a solution of 2-F (0.74 g, 4.5 mmol) and malononitrile (0.60 g, 9.1 mmol) in ethanol (15 mL), NaOAc (1.11 g, 13.5 mmol) was added. The solution was stirred at room temperature for 40 min and poured into water (200 mL). 5 M HCl was added to adjust the pH of the solution to 1–2, and the precipitate was filtered. The resulting solid was subjected to column chromatography (silica, eluent CH2Cl2) to afford 3-F (0.24 g, 21%). 1H NMR δ (ppm): 8.67–8.69 (m, 0.27H), 8.29–8.30 (d, 0.67H), 8.00–8.02 (m, 0.67H), 7.57–7.62 (m, 1.23H), 3.75–3.77 (d, 2H). 13C NMR δ (ppm): 193.54, 192.96, 168.08, 167.89, 166.35, 166.14, 164.96, 164.94, 164.80, 144.72, 144.66, 143.63, 143.57, 138.73, 137.03, 137.01, 128.69, 128.63, 127.20, 127.13, 124.20, 124.04, 123.90, 123.75, 112.93, 112.76, 112.22, 111.96, 111.69, 111.56, 111.41, 80.66, 78.93, 43.74, 43.46.
3-Cl
.
The same procedure was used as for 3-F, but now 2-Cl (0.80 g, 4.4 mmol) was used as the precursor. Yield: 0.30 g (30%). 1H NMR δ (ppm): 8.58–8.60 (d, 1H), 7.91–7.93 (d, 1H), 7.80–7.84 (m, 1H), 3.75 (s, 2H). 13C NMR δ (ppm): 193.38, 164.77, 143.65, 143.30, 143.04, 141.88, 140.62, 138.77, 136.35, 136.11, 127.16, 125.94, 125.82, 124.88, 112.11, 111.92, 111.74, 80.57, 79.61, 43.47, 43.37.
3-Br
.
The same procedure was used as for 3-F, but now 2-Br (1.11 g, 4.9 mmol) was used as the precursor. Yield: 0.25 g (19%). 1H NMR δ (ppm): 8.77 (s, 0.52H), 8.49–8.51 (d, 0.44H), 8.10 (s, 0.43H), 7.96–7.99 (m, 1H), 7.83–7.84 (d, 0.55H), 3,72–3.74 (d, 2H). 13C NMR δ (ppm): 193.65, 193.49, 165.09, 164.73, 143.81, 141.85, 141.08, 139.30, 139.22, 139.08, 131.95, 131.73, 129.06, 128.14, 127.22, 125.90, 112.19, 112.02, 111.81, 80.63, 79.81, 43.39, 43.35.
3-I
.
The same procedure was used as for 3-F, but now 2-I (1.38 g, 5.1 mmol) was used as the precursor. Yield: 0.32g (20%). 1H NMR δ (ppm): 8.98 (s, 0.32H), 8.33–8.36 (t, 1.26H), 8.17–8.21 (m, 0.97H), 7.67–7.69 (d, 0.34H), 3.69–3.71 (d, 2H). 13C NMR δ (ppm): 193.94, 193.41, 165.26, 164.55, 145.06, 144.86, 143.62, 141.51, 141.33, 135.02, 134.23, 126.86, 125.58, 112.13, 111.99, 111.80, 111.77, 104.50, 104.27, 80.37, 79.77, 43.08, 43.03.
F-ITIC
.
To a solution of 3-F (60 mg, 0.28 mmol) and IDTT-CHO (100 mg, 0.09 mmol) in chloroform (20 mL), pyridine (1 mL) was added. The solution was stirred at room temperature for 12 h and evaporated. The resulting solid was subjected to column chromatography (silica, eluent petroleum ether/CH2Cl2) to afford F-ITIC as a crude product. Pure F-ITIC (106 mg, 78%) was obtained by precipitation into methanol for organic electronic devices. 1H NMR δ (ppm): 8.85 (s, 1.88H), 8.69–8.71 (t, 0.56H), 8.36–8.37 (d, 2H), 8.21–8.23 (d, 1.79H), 7.90–7.92 (t, 1.37H), 7.65 (s, 1.92H), 7.54–7.55 (d, 0.56H), 7.41–7.42 (d, 1.92H), 7.14–7.23 (m, 16H), 2.56–2.58 (t, 8H), 1.59–1.61 (m, 7H), 1.28–1.34 (t, 25H), 0.85–0.87 (t, 12H). 13C NMR δ (ppm): 186.72, 186.66, 167.59, 167.17, 165.58, 165.45, 159.25, 158.94, 155.77, 153.29, 153.24, 147.74, 147.71, 147.46, 147.30, 143.81, 142.56, 142.36, 142.29, 139.98, 139.93, 139.54, 138.87, 138.45, 138.33, 137.14, 137.00, 135.91, 133.10, 128.89, 128.73, 127.88, 125.96, 125.89, 122.59, 122.50, 121.93, 121.77, 118.62, 114.41, 114.25, 114.18, 112.87, 112.69, 110.86, 110.71, 70.18, 69.17, 63.27, 35.61, 31.70, 31.25, 29.18, 22.58, 14.08. MS (MALDI): calculated: 1463.92, found: 1464.2 (M+).
Cl-ITIC
.
The same procedure was used as for F-ITIC, but now 3-Cl (73 mg, 0.32 mmol) and IDTT-CHO (120 mg, 0.11 mmol) were used as the precursors. Yield: 138 mg (83%). 1H NMR δ (ppm): 8.86 (s, 1.91H), 8.64 (s, 0.52H), 8.59–8.61 (d, 1.38H), 8.23 (s, 1.80H), 7.84 (s, 1.88H), 7.66–7.68 (d, 3.93H), 7.15–7.23 (m, 16H), 2.56–2.59 (t, 8H), 1.56–1.61 (m, 7H), 1.34 (s, 25H), 0.85–0.86 (d, 12H). 13C NMR δ (ppm): 186.72, 186.62, 159.12, 158.76, 155.70, 153.33, 147.64, 147.47, 147.37, 143.80, 142.49, 141.76, 141.15, 139.55, 138.74, 138.56, 138.23, 137.94, 137.09, 136.94, 134.93, 134.84, 134.46, 128.80, 127.77, 126.35, 125.26, 124.62, 123.78, 122.16, 118.55, 114.42, 114.29, 114.17, 114.09, 70.00, 69.39, 63.16, 35.50, 31.59, 31.14, 29.08, 22.48, 13.98. MS (MALDI): calculated: 1496.83, found: 1497.3 (M+).
Br-ITIC
.
The same procedure was used as for Br-ITIC, but now 3-Br (64 mg, 0.2 mmol) and IDTT-CHO (85 mg, 0.08 mmol) were used as the precursors. Yield: 80 mg (64%). 1H NMR δ (ppm): 8.80–8.86 (d, 2.68H), 8.51–8.52 (d, 1.07H), 8.21–8.22 (d, 1.82H), 8.00 (s, 1.07H), 7.81–7.84 (t, 1.94H), 7.74–7.52 (d, 0.83H), 7.65–7.66 (d, 1.96H), 7.15–7.23 (m, 16H), 2.56–2.59 (t, 8H), 1.57–1.61 (m, 7H), 1.34 (s, 24H), 0.86 (s, 12H). 13C NMR δ (ppm): 186.92, 186.53, 159.21, 158.65, 155.71, 153.36, 147.65, 147.49, 147.41, 143.81, 142.49, 141.19, 139.60, 139.57, 138.73, 138.58, 138.53, 138.34, 138.11, 137.75, 137.39, 137.10, 136.94, 135.30, 130.28, 129.64, 128.80, 128.15, 127.77, 126.86, 126.38, 124.66, 122.01, 121.96, 118.56, 114.42, 114.32, 114.16, 114.08, 69.98, 69.40, 63.16, 35.50, 31.59, 31.14, 29.07, 22.48, 13.98. MS (MALDI): calculated: 1585.73, found: 1586.5 (M+).
I-ITIC
.
The same procedure was used as for I-ITIC, but now 3-I (89.3 mg, 0.28 mmol) and IDTT-CHO (100 mg, 0.09 mmol) were used as the precursors. Yield: 94 mg (60%). 1H NMR δ (ppm): 9.00 (s, 0.52H), 8.85 (s, 1.92H), 8.35–8.38 (d, 1.40H), 8.21 (s, 3.20H), 8.01–8.07 (m, 1.98H), 7.65–7.66 (d, 1.95H), 7.58–7.60 (d, 0.60H), 7.13–7.23 (m, 16H), 2.55–2.60 (t, 8H), 1.57–1.62 (t, 7H), 1.28–1.30 (d, 25H), 0.84–0.88 (t, 12H). 13C NMR δ (ppm): 186.72, 159.62, 158.74, 155.88, 153.51, 147.82, 147.59, 143.98, 143.88, 143.55, 142.67, 141.22, 139.84, 139.79, 139.11, 138.92, 138.76, 137.86, 137.27, 137.13, 136.01, 134.18, 133.10, 128.98, 127.95, 126.37, 124.69, 121.89, 118.74, 114.63, 114.56, 114.39, 103.00, 102.21, 69.56, 63.34, 35.69, 31.78, 31.34, 29.27, 22.67, 14.17. MS (MALDI): calculated: 1679.74, found: 1679.2 (M+).
Conclusions
In conclusion, a series of F-, Cl-, Br- and I-containing conjugated molecules were simply prepared via Knoevenagel condensation and applied in FET devices and non-fullerene solar cells. Halogenated compounds were found to provide deep frontier energy levels and high crystal properties due to the electronegativity and heavy atom effect. As a consequence, halogenated conjugated molecules show high electron mobility up to 1.3 cm2 V−1 s−1 in FETs. Solar cells based on halogenated materials as electron acceptors also have PCEs around 9%, while non-halogenated analogues only provided a PCE of 6.4%. These results show the feasibility of halogenated conjugated molecules as new n-type materials in field-effect transistors and organic solar cells owing to the tailored energy levels and crystal properties.
Acknowledgements
This work was supported by the Recruitment Program of Global Youth Experts of China. The work was further supported by the National Natural Science Foundation of China (21574138 and 51603209) and the Strategic Priority Research Program (XDB12030200) of the Chinese Academy of Sciences.
Notes and references
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047–9050 CrossRef CAS PubMed.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- M.-H. Yoon, S. A. DiBenedetto, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 1348–1349 CrossRef CAS PubMed.
- Z. Bao, A. J. Lovinger and J. Brown, J. Am. Chem. Soc., 1998, 120, 207–208 CrossRef CAS.
- Y. Sakamoto, S. Komatsu and T. Suzuki, J. Am. Chem. Soc., 2001, 123, 4643–4644 CrossRef CAS PubMed.
- J. H. Schön, C. Kloc, Z. Bao and B. Batlogg, Adv. Mater., 2000, 12, 1539–1542 CrossRef.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- N. Leclerc, P. Chávez, O. Ibraikulov, T. Heiser and P. Lévêque, Polymers, 2016, 8, 11 CrossRef.
- Y. Y. Liang, D. Q. Feng, Y. Wu, S. T. Tsai, G. Li, C. Ray and L. P. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- D. J. Crouch, P. J. Skabara, M. Heeney, I. McCulloch, S. J. Coles and M. B. Hursthouse, Chem. Commun., 2005, 1465–1467 RSC.
- F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC.
- T. V. Rybalova and I. Y. Bagryanskaya, J. Struct. Chem., 2009, 50, 741–753 CrossRef CAS.
- W. Jiang, Y. Li and Z. Wang, Acc. Chem. Res., 2014, 47, 3135–3147 CrossRef CAS PubMed.
- M. L. Tang, J. H. Oh, A. D. Reichardt and Z. Bao, J. Am. Chem. Soc., 2009, 131, 3733–3740 CrossRef CAS PubMed.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed.
- Y.-Q. Zheng, Z. Wang, J.-H. Dou, S.-D. Zhang, X.-Y. Luo, Z.-F. Yao, J.-Y. Wang and J. Pei, Macromolecules, 2015, 48, 5570–5577 CrossRef CAS.
- A. M. Hiszpanski, J. D. Saathoff, L. Shaw, H. Wang, L. Kraya, F. Lüttich, M. A. Brady, M. L. Chabinyc, A. Kahn, P. Clancy and Y.-L. Loo, Chem. Mater., 2015, 27, 1892–1900 CrossRef CAS.
- S.-X. Sun, Y. Huo, M.-M. Li, X. Hu, H.-J. Zhang, Y.-W. Zhang, Y.-D. Zhang, X.-L. Chen, Z.-F. Shi, X. Gong, Y. Chen and H.-L. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 19914–19922 CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-J. Liu, J.-Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447–2452 RSC.
- Y. Kunugi, K. Takimiya, Y. Toyoshima, K. Yamashita, Y. Aso and T. Otsubo, J. Mater. Chem., 2004, 14, 1367–1369 RSC.
- T. Okamoto, M. L. Senatore, M. M. Ling, A. B. Mallik, M. L. Tang and Z. N. Bao, Adv. Mater., 2007, 19, 3381–3384 CrossRef CAS.
- N. Hergue, P. Leriche, P. Blanchard, M. Allain, N. Gallego-Planas, P. Frere and J. Roncali, New J. Chem., 2008, 32, 932–936 RSC.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- C. Z. Li, C. C. Chueh, F. Z. Ding, H. L. Yip, P. W. Liang, X. S. Li and A. K. Y. Jen, Adv. Mater., 2013, 25, 4425–4430 CrossRef CAS PubMed.
- M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS PubMed.
- J. S. Kim, Z. Fei, S. Wood, D. T. James, M. Sim, K. Cho, M. J. Heeney and J.-S. Kim, Adv. Energy Mater., 2014, 4, 1400527 CrossRef.
- M. Jeffries-El, B. M. Kobilka and B. J. Hale, Macromolecules, 2014, 47, 7253–7271 CrossRef CAS.
- C.-H. Tsai, A. Fortney, Y. Qiu, R. R. Gil, D. Yaron, T. Kowalewski and K. J. T. Noonan, J. Am. Chem. Soc., 2016, 138, 6798–6804 CrossRef CAS PubMed.
- W. C. Tsoi, D. T. James, E. B. Domingo, J. S. Kim, M. Al-Hashimi, C. E. Murphy, N. Stingelin, M. Heeney and J.-S. Kim, ACS Nano, 2012, 6, 9646–9656 CrossRef CAS PubMed.
- R. D. Pensack, Y. Song, T. M. McCormick, A. A. Jahnke, J. Hollinger, D. S. Seferos and G. D. Scholes, J. Phys. Chem. B, 2014, 118, 2589–2597 CrossRef CAS PubMed.
- A. A. Jahnke, B. Djukic, T. M. McCormick, E. Buchaca
Domingo, C. Hellmann, Y. Lee and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 951–954 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS PubMed.
- Z. Li, K. Jiang, G. Yang, J. Y. L. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 13094 CrossRef CAS PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- Y. Lin, F. Zhao, Y. Wu, K. Chen, Y. Xia, G. Li, S. K. K. Prasad, J. Zhu, L. Huo, H. Bin, Z.-G. Zhang, X. Guo, M. Zhang, Y. Sun, F. Gao, Z. Wei, W. Ma, C. Wang, J. Hodgkiss, Z. Bo, O. Inganäs, Y. Li and X. Zhan, Adv. Mater., 2017, 29, 1604155 CrossRef PubMed.
- Y. Yang, Z.-G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610–616 CAS.
- Y. Li, X. Liu, F.-P. Wu, Y. Zhou, Z.-Q. Jiang, B. Song, Y. Xia, Z.-G. Zhang, F. Gao, O. Inganas, Y. Li and L.-S. Liao, J. Mater. Chem. A, 2016, 4, 5890–5897 CAS.
- X. Gong, G. Li, S. Feng, L. Wu, Y. Liu, R. Hou, C. Li, X. Chen and Z. Bo, J. Mater. Chem. C, 2017, 5, 937–942 RSC.
- D. M. Stoltzfus, J. E. Donaghey, A. Armin, P. E. Shaw, P. L. Burn and P. Meredith, Chem. Rev., 2016, 116, 12920–12955 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 2231–2234 CrossRef CAS PubMed.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- N. A. Ran, J. A. Love, C. J. Takacs, A. Sadhanala, J. K. Beavers, S. D. Collins, Y. Huang, M. Wang, R. H. Friend, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2015, 28, 1482–1488 CrossRef PubMed.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- R. W. Higgins, C. L. Hilton and S. D. Deodhar, J. Org. Chem., 1951, 16, 1275–1277 CrossRef CAS.
- D. Leblois, S. Piessard, G. Le Baut, P. Kumar, J.-D. Brion, L. Sparfel, R.-Y. Sanchez, M. Juge, J.-Y. Petit and L. Welin, Eur. J. Med. Chem., 1987, 22, 229–238 CrossRef CAS.
- J. Wilbuer, G. Schnakenburg and B. Esser, Eur. J. Org. Chem., 2016, 2404–2412 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details, synthesis, characterization and device fabrication. See DOI: 10.1039/c7qm00025a |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Wenbin
Lai
a,
Andong
Zhang
ab,
Hui
Huang
*a,
Wenbin
Lai
a,
Andong
Zhang
ab,
Hui
Huang