
Palladium supported on an amphiphilic porous organic polymer: a highly efficient catalyst for aminocarbonylation reactions in water†
Yizhu
Lei
*a,
Yali
Wan
a,
Guangxing
Li
*b,
Xiao-Yu
Zhou
a,
Yanlong
Gu
b,
Jing
Feng
a and
Renshu
Wang
a
aDepartment of Chemistry and Chemical Engineering, Liupanshui Normal University, Liupanshui, Guizhou 553004, P. R. China. E-mail: yzleiabc@126.com
bSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Luoyu Road 1037, Wuhan, Hubei 430074, P. R. China. E-mail: ligxabc@163.com
First published on 16th March 2017
Abstract
Exploring environmentally friendly, efficient and recyclable heterogeneous catalysts for organic reactions in aqueous media is important for the development of green and sustainable processes. In this work, an amphiphilic porous organic polymer supported palladium catalyst (Pd@UPOP) was synthesized via a facile urea-forming condensation of commercially available 3,3′-diaminobenzidine and 1,4-phenylene diisocyanate, followed by immobilizing palladium acetate at room temperature. Physico-chemical characterization suggested that the obtained material possessed a good porous structure and amphiphilic properties. Aminocarbonylation of aryl iodides with amines in water showed that Pd@UPOP is more active than previously reported heterogeneous palladium catalysts. Under balloon pressure of carbon monoxide, a palladium catalyst loading as low as 0.5 mol% was sufficient for a 98% yield of N,N-diethylbenzamide in the model reaction, corresponding to a high turnover frequency of 98 h−1. The catalyst could be used for at least five consecutive runs with the catalytic activity being recovered easily after simple manipulations.
Introduction
Water is an ideal solvent for chemical transformations, since it offers the advantages of nontoxicity, nonflammability and low cost.1–3 Meanwhile, separation and recycling of homogeneous catalysts is considered a key green chemistry research area.4–9 From the standpoint of environmentally benign organic synthesis, the development of highly efficient immobilized catalysts and the use of water instead of organic compounds as a solvent are of great interest to chemists.10–15In recent years, palladium catalysts have been receiving tremendous research interest due to their excellent catalytic performance in various coupling reactions.16–21 Thus far, a considerable number of highly active palladium complexes have been developed for various coupling reactions of organic halides,22–26 as well as these reactions performed in water.27–31 However, separation, recovery and recycling of the expensive metal and ligands from the final reaction mixture is the problem to be addressed. In this regard, a number of heterogeneous catalysts were prepared via the method of immobilizing palladium complexes on different supports32–36 despite the fact that a substantial decrease of catalytic activity is frequently observed in the heterogeneous systems.
Among various catalytic supports, porous organic polymers (POPs), which emerged just a few years ago, have attracted great attention due to their unique properties such as large surface area, designable pore walls, high chemical stability and controllable chemical composition.37–40 One particular feature of POPs is their excellent chemical tunability that allows the precise incorporation of a wide range of target functionalities into their porous frameworks. POPs containing various functional units such as phthalocyanine, bipyridine, porphyrins, phosphine ligands, and triazine rings have been successfully prepared, and their performances in heterogeneous catalysis have also been investigated.41–43 The results showed that POPs offer a good opportunity to develop highly active heterogeneous catalysts, and some of them even exhibited higher catalytic activities than the corresponding homogeneous catalysts. However, the majority of POPs mainly consist of hydrophobic aromatic frameworks, which prevents their effective dispersion in water and results in poor contact between the substrates and the active sites.44,45 Recently, via the strategy of introducing hydrophilic urea linkages into the hydrophobic aromatic frameworks, Wang et al. reported an amphiphilic porous organic framework (UOF) supported palladium as a highly efficient catalyst for Suzuki–Miyaura cross-coupling reactions and selective reduction of nitroarenes in water.46 However, 1,3,5-benzenetriisocyanate, the important material for the synthesis of UOF, is commercially unavailable, and its preparation needs tedious reaction procedures and the use of hypertoxic chemicals.
On the other hand, an amide group is one of the most important building blocks in organic synthesis, and occurs frequently in nature.47–49 However, the traditional synthesis method of amides using carboxylic acid derivatives and amines has suffered from poor atom efficiency. Considering the importance of amides in chemistry and biology, the ACS GCI Pharmaceutical Roundtable listed “amide formation avoiding poor atom efficiency” as the top research priority for reactions currently.50 This has led to the rapid development of new and improved methods for amide synthesis in recent years.51–54 Among the various improved methods of amide synthesis, palladium-catalyzed aminocarbonylation of aryl halides represents a powerful and alternative technique for amide synthesis, since it has a number of advantages over traditional synthesis methods such as high atom efficiency and easily available feedstocks.55–59 However, most recent efforts were devoted to the development of active palladium complexes.60–66 Although homogeneous-based systems generally exhibit the advantages of high activity and selectivity, they still suffer from practical problems such as catalyst separation, recovery and recycling. To address these problems, palladium on various supports, such as silica,67,68 MCM-41,69 MOF-5,70 ZIF-8,71 organic polymers72,73 and porous organic polymers74,75 have been investigated for the aminocarbonylation reactions. However, to the best of our knowledge, studies on the applications of amphiphilic porous organic polymers in aqueous carbonylation have rarely been explored.
In continuation of our efforts to develop highly active heterogeneous catalysts for the carbonylation reactions,76–78 in this paper, a urea linked porous organic polymer supported palladium catalyst (Pd@UPOP) was prepared for aminocarbonylation reactions. The catalyst showed excellent catalytic activity for aminocarbonylation of aryl iodides in water. In addition, the catalyst could be used for up to five consecutive runs with negligible catalyst leaching during the reactions. Mechanism studies showed that the excellent performance of the catalyst should be attributed to its good amphiphilicity, porous structure and rich coordination sites.
Experimental
Materials
3,3′-Diaminobenzidine (DAB, 99%) and 1,4-phenylene diisocyanate (PPDI, 98%) were purchased from Meryer (Shanghai) Chemical Technology Co. Ltd, China. Palladium acetate, aryl iodides, amines, N,N-diethylbenzamide, K2CO3, and dichloromethane were of analytical reagent grade and used without further purification. Tetrahydrofuran (THF) was an analytical reagent and dehydrated by refluxing over sodium for 24 h. 5 wt% Pd/C catalyst was purchased from Shaanxi Rock New Materials Co. Ltd (China). CO with a purity of 99.99% was purchased from the local manufacturer.Synthesis of UPOP
Urea based amphiphilic porous organic polymer (UPOP) was prepared as follows. Generally, a solution of 3,3′-diaminobenzidine (0.85 g, 4.0 mmol) in 40 ml THF was added dropwise to a solution of 1,4-phenylene diisocyanate (1.28 g, 8.0 mmol) in 100 ml THF at 70 °C. The mixture was stirred and heated at 70 °C for 4 days. After the reaction, the mixture was filtered, and a yellow powder was obtained. Then, the resulting powder was washed with methanol, THF, and diethyl ether. After drying in vacuo at 60 °C for 12 h, UPOP was obtained as a yellow powder (1.94 g, 91.1% yield). Elemental analysis found: C 56.92, H 4.22, N 19.01.Preparation of catalysts
After drying in vacuo at 80 °C for 2 h, UPOP (1.0 g) was added to a dichloromethane solution (30 ml) containing palladium acetate (0.0443 g), and the mixture was stirred at room temperature for 12 h. Then, the solid was centrifugally separated, washed repeatedly with acetone, and purified by Soxhlet-extraction with acetone to remove palladium(II) species in the form of physical absorption. After drying in vacuo at 60 °C for 12 h, the final catalyst was obtained and denoted as Pd@UPOP. Atomic absorption spectroscopy analysis showed that the Pd content in Pd@UPOP was 2.0 wt%, this result suggested that nearly 97% Pd was successfully immobilized. For comparison, PdCl2(phen)@Y76 and Pd@KAPs(Ph-PPh3)75 were prepared as we previously reported.Aminocarbonylation reaction
The aminocarbonylation reaction was conducted in a 20 ml reaction flask and fitted with a condenser and a carbon monoxide balloon. A mixture of aryl iodide (1 mmol), amine (2 mmol), Na2CO3 (1.2 mmol), Pd@UPOP (0.5 mol%) and water (4 ml) was stirred at 100 °C under balloon pressure of CO. At the end of the reaction, the flask was cooled to room temperature and the catalyst was separated by simple centrifugation. For the model reaction, the filtrate was quantitatively analysed with GC by using N,N-dimethylacetamide as an internal standard. For the study of substrate scope, the filtrate was extracted with diethyl ether. The organic layer thus collected was dried over anhydrous Na2SO4, filtered, and then concentrated. Then, the residue was purified by preparative thin-layer chromatography (PTLC) using petroleum ether and ethyl acetate (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as eluting solvents. The product was analyzed by NMR and yields were based on aryl iodides. All the synthesized amides are known products that we have reported recently.60,75,76 For the recycling experiments, the solid catalyst was separated by centrifugation, washed three times with deionized water, and then engaged in a new catalytic cycle.
:1, v/v) as eluting solvents. The product was analyzed by NMR and yields were based on aryl iodides. All the synthesized amides are known products that we have reported recently.60,75,76 For the recycling experiments, the solid catalyst was separated by centrifugation, washed three times with deionized water, and then engaged in a new catalytic cycle.
Adsorption of chlorobenzene in water
The adsorption capacity of Pd@UPOP for chlorobenzene was determined as follows. Generally, in a 50 ml Teflon-lined steel autoclave, Pd@UPOP (0.3 g) was added to 2.0 g of chlorobenzene in 20 ml of deionized water under magnetic stirring. Then, the mixture was heated at 100 °C for 30 min. After cooling the reactor to room temperature, the mixture was centrifuged at 5000 rpm for 10 min. The obtained solid was extracted with 5 ml ethanol 4 times, and the collected ethanol was then analyzed with GC. Moreover, the solubility of chlorobenzene in water was analysed under the same conditions.Instruments and characterization
Nitrogen sorption isotherms collected at 77 K were measured using a Micrometrics ASAP 2020 system, and the samples were pre-treated under vacuum at 100 °C for 5 h before the measurements. Fourier transform infrared spectra (FT-IR) were recorded in the region 4000–400 cm−1 on a Bruker Equinox 55 FT-IR spectrophotometer in KBr pellets. Simultaneous thermogravimetry-differential scanning calorimetry (TG-DSC) was performed on a NETZSCH STA 449 F5 instrument under a dynamic argon atmosphere, and the sample was heated from room temperature to 500 °C at a heating rate of 10 °C min−1. The elemental analysis was performed using an Elementar Vario MICRO Elemental analyzer. The structure of the polymer was characterized by X-ray diffraction (Bruker, D8 Advance instrument) with CuKα radiation. Atomic absorption spectroscopy (AAS) was performed on a PerkinElmer AA-300 spectrophotometer. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was carried out on a PerkinElmer Optima 8000. Scanning electron micrographs (SEM) were recorded on a SU8010 field-emission scanning electron microscope (FESEM, Hitachi, Japan). Transmission electron microscopy (TEM) and energy dispersive spectrometry (TEM-EDS) were measured on a Philips Tecnai G2 F30 instrument. X-ray photoelectron spectroscopy (XPS) measurements of the heterogeneous Pd catalysts were carried out on a VG multilab 2000 spectrometer using a Mg–AlKα X-ray source, and the C1s transition was adjusted to 284.8 eV. The contact angles were measured on a Dataphysics OCA20 contact angle system. 1H and 13C NMR spectra were recorded on a Bruker AVANCE III NMR spectrometer (400 and 100 MHz) at ambient temperature with CDCl3 as a solvent. Solid-state 13C NMR spectra were carried out on a Bruker AVANCE III 600 Bruker spectrometer equipped with a 4 mm double resonance MAS probe. The GC analysis was performed with an Agilent 1790 gas chromatograph equipped with a HP-5 capillary column and a flame ionization detector.Results and discussion
Characterization
The catalyst preparation steps are summarized in Scheme 1. Briefly, UPOP was prepared via a facile urea-forming condensation of DAB and PPDI. Then palladium acetate was immobilized on the obtained UPOP to produce Pd@UPOP.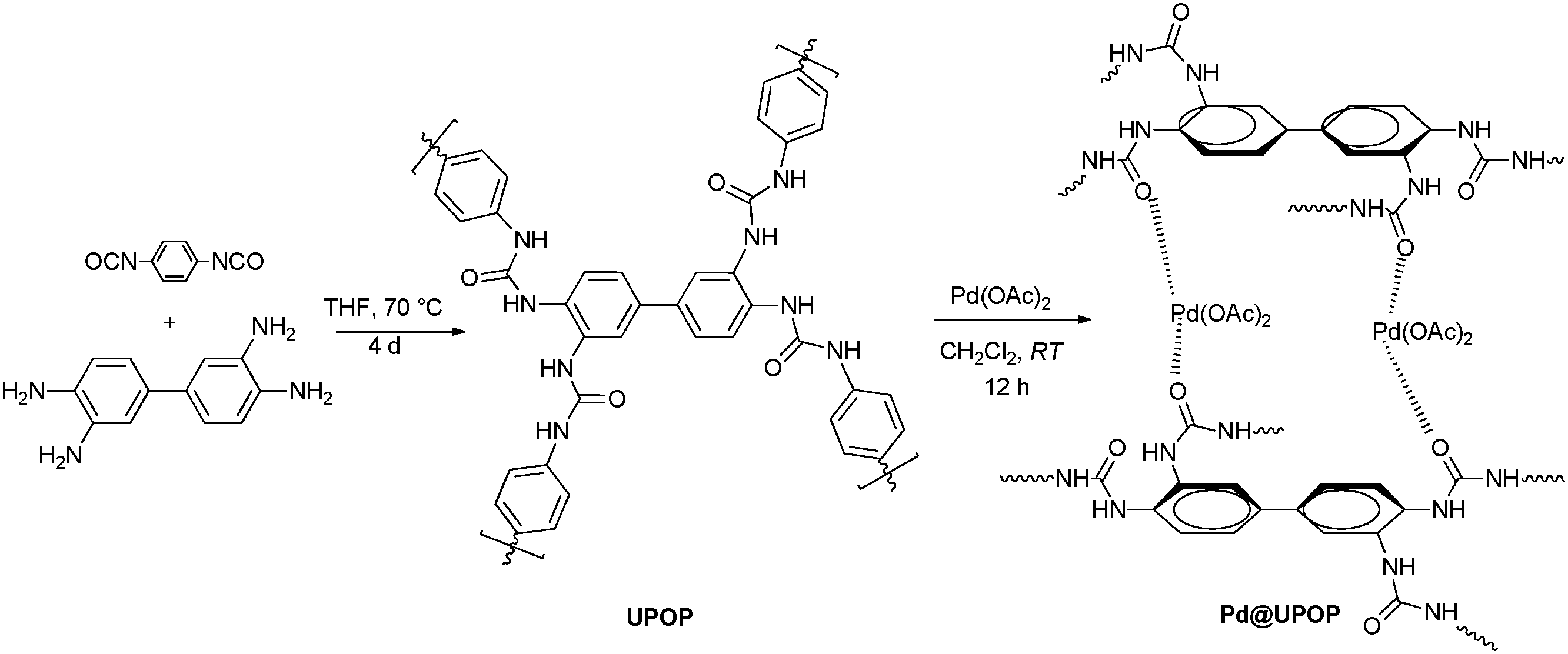 | ||
| Scheme 1 Synthesis of UPOP and Pd@UPOP. | ||
UPOP was thoroughly characterized by means of FT-IR, solid-state 13C NMR spectra, XRD, SEM, N2 adsorption–desorption, TG-DSC, XPS and contact angle measurements. Moreover, XPS measurement of the Pd@UPOP catalyst was carried out to understand the nature and oxidation state of Pd species. As depicted in Fig. 1, the FT-IR spectroscopy spectrum of UPOP showed that the stretching band of isocyanate (2277.8 cm−1) in PPDI disappeared, and a new stretching band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1666.4 cm−1 emerged; these results indicated that the condensation reaction between 3,3′-diaminobenzidine and 1,4-phenylene diisocyanate was finished. As depicted in Fig. 2, the solid-state 13C NMR spectra of UPOP and Pd@UPOP were almost identical. The resonance peak near 156 ppm should be attributed to the carbonyl carbon atom of the urea moiety, whereas the peaks near 135 and 125 ppm could be assigned to substituted aromatic carbon and non-substituted aromatic carbon respectively. The powder XRD study of UPOP indicated that it was an amorphous polymer (Fig. S1 in the ESI†).
O at 1666.4 cm−1 emerged; these results indicated that the condensation reaction between 3,3′-diaminobenzidine and 1,4-phenylene diisocyanate was finished. As depicted in Fig. 2, the solid-state 13C NMR spectra of UPOP and Pd@UPOP were almost identical. The resonance peak near 156 ppm should be attributed to the carbonyl carbon atom of the urea moiety, whereas the peaks near 135 and 125 ppm could be assigned to substituted aromatic carbon and non-substituted aromatic carbon respectively. The powder XRD study of UPOP indicated that it was an amorphous polymer (Fig. S1 in the ESI†).
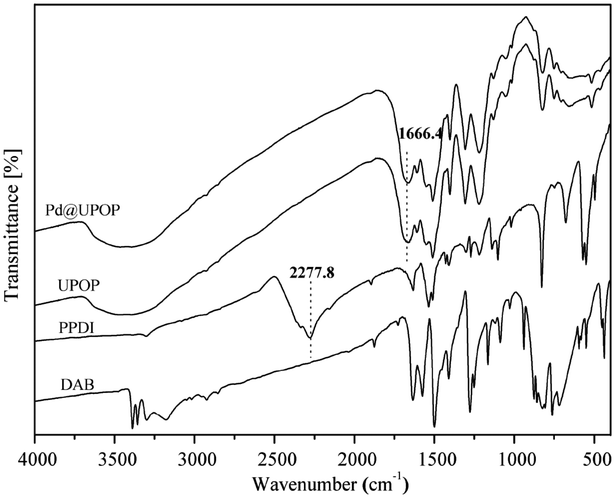 | ||
| Fig. 1 FT-IR spectra of DAB, PPDI, UPOP and Pd@UPOP. | ||
 | ||
| Fig. 2 Solid-state 13C NMR spectra of UPOP and Pd@UPOP. | ||
The elemental composition of the prepared UPOP was measured with elemental analysis, and the results showed that both the C and N contents were slightly lower than the corresponding theoretical values, which is a very common phenomenon in porous materials, and should be ascribed to the presence of trapped guest molecules in their porous structure.46 Further proof of the presence of guest molecules was obtained through TG-DSC analysis in argon. As depicted in Fig. S2 in the ESI,† the initial weight loss (3.5%) occurred at 25–100 °C, mainly ascribed to the removal of guest molecules. Whereas the rapid weight loss that takes place at 230–360 °C should be ascribed to the decomposition of UPOP, which suggested that UPOP could be stable up to 230 °C. Fig. 3 shows the SEM image of UPOP and TEM image of Pd@UPOP; the results suggested that UPOP exhibited 3D network morphology with abundant macroporous structures.
 | ||
| Fig. 3 (a) SEM image of UPOP; (b) TEM image of Pd@UPOP; (c) TEM image of Pd@UPOP after the 2nd recycle. | ||
The porous properties of the sample were analysed by nitrogen sorption analysis. As shown in Fig. 4(a), the results indicated that the physisorption isotherm is of type IV isotherm patterns according to IUPAC classification.79 A sharp increase in the adsorption amount of nitrogen at a high relative pressure showed the presence of large pores, which could be attributed to interparticle porosity. The appearance of hysteresis might be an indicator of the softness of UPOP, thus demonstrating the deformation and swelling of the network.80 The surface area of UPOP was 103 m2 g−1, which was calculated using the Brunauer–Emmet–Teller (BET) method. This low surface area could be ascribed to framework interpenetration, and the hydrogen bonds between urea groups are probably responsible for interpenetration during framework formation.46 The pore size distribution of UPOP, obtained from appropriate fitting of the density functional theory (DFT) model to the isotherm, is shown in Fig. 4(b). A wide distribution was obtained and centered at 1.4 nm.
 | ||
| Fig. 4 (a) Nitrogen adsorption (solid) and desorption (open) isotherms of UPOP; (b) pore size distribution of UPOP as determined by non-linear density functional theory. | ||
Given that urea groups could impart hydrophilic character of UPOP, UPOP was dispersed in H2O and hexane to investigate its hydrophilicity.46,81 As shown in Fig. S3 in the ESI,† UPOP could be well dispersed in water and retained in the aqueous phase in a water/hexane biphasic system. For comparison, the hydrophobic KAPs(Ph-PPh3) was also investigated. KAPs(Ph-PPh3) was suspended at the water surface, and it was well distributed in the hexane phase in a water/hexane biphasic system. The amphipathicity was further proved by examining the contact angle measurements (Fig. 5). The contact angle between water and UPOP was 45°, however, the contact angle between salad oil and UPOP was only 7°. These results indicated that the prepared UPOP had good amphiphilic properties. The nice hydrophilicity could be ascribed to the hydrophilic urea moieties, and the strong oleophilicity was ascribed to the presence of the hydrophobic composition of phenyl rings. Thus, a porous and amphiphilic catalytic support for heterogeneous catalysts was achieved.
 | ||
| Fig. 5 Images of contact angle measurements of UPOP: (a) photograph of a water droplet; (b) photograph of a salad oil droplet. | ||
To obtain information on the elemental compositions of the surface and coordination states of Pd species, XPS studies have been performed on the UPOP support as well as on the Pd@UPOP catalyst. In Fig. 6(a), an XPS study of UPOP revealed that C, N, and O elements were present in the porous polymer. The XPS spectrum of Pd@UPOP in Fig. 6(b) revealed that Pd species in the Pd@UPOP were present in the +2 oxidation state, corresponding to the binding energy (B.E.) of 337.6 eV in the Pd 3d5/2 level. In comparison with that of free Pd(OAc)2 (338.4 eV),82 the PdII binding energy in the Pd@UPOP shifted negatively by 0.8 eV, which should be attributed to the interaction of [Pd(OAc)2] with the UPOP support. These results demonstrated that PdII was successfully immobilized on the UPOP by coordination to urea groups rather than by physical adsorption of Pd(OAc)2 on the surface. The Pd content in Pd@UPOP was determined by using AAS analyses and found to be 2.0 wt%.
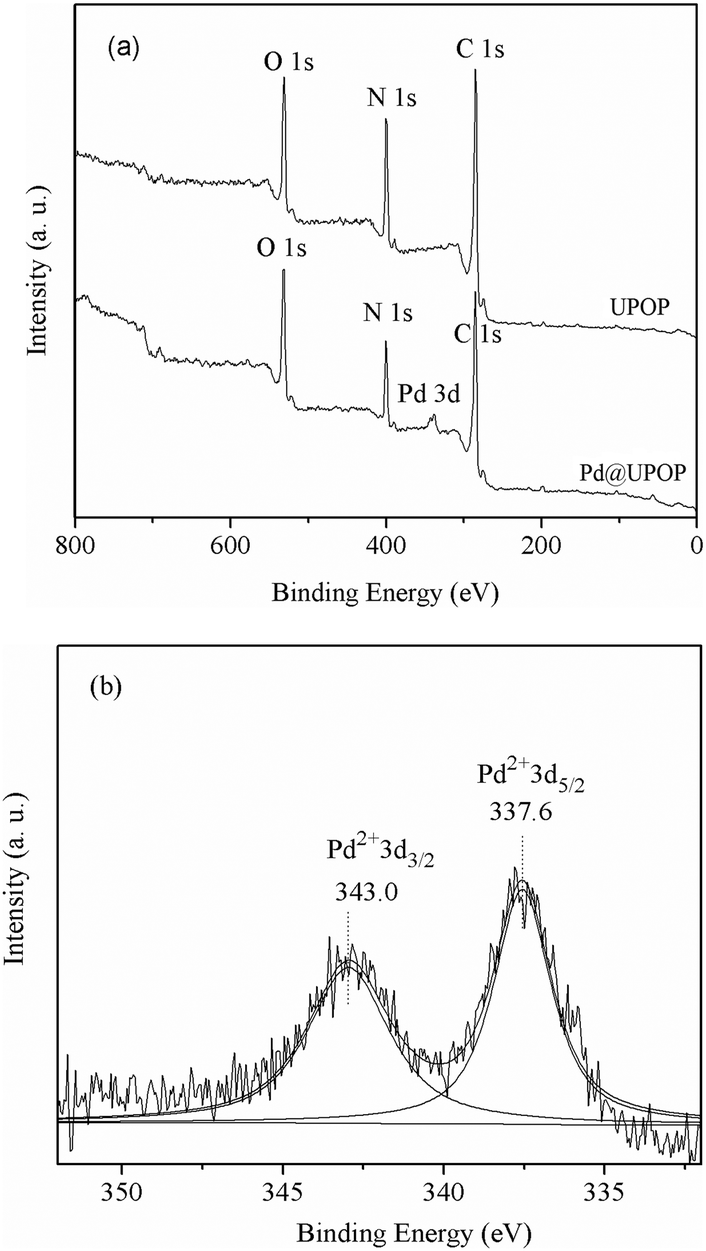 | ||
| Fig. 6 XPS spectra: (a) XPS spectra of UPOP and Pd@UPOP (elemental scan); (b) XPS spectrum of Pd on Pd@UPOP. | ||
Catalytic activity
The catalytic performance of the thereby obtained Pd@UPOP catalyst was evaluated in an aminocarbonylation reaction in water, and the aminocarbonylation of iodobenzene with diethylamine was chosen as a model reaction. When the model reaction was performed with 0.5 mol% Pd usage and in the presence of K2CO3 as a base, a 99% yield of N,N-diethylbenzamide was observed within 4 h (Table 1, entry 1). For comparison, some effective heterogeneous catalytic systems that have previously been reported were also tested under the same conditions. Pd/C,83 one of the most commonly used heterogeneous catalysts for aminocarbonylation, gave N,N-diethylbenzamide in a 35% yield (Table 1, entry 2). PdCl2(phen)@Y, a highly effective heterogeneous catalyst that we have previously reported,76 showed low catalytic activity (Table 1, entry 3). These results suggested that Pd@UPOP presented obvious advantages over Pd/C and PdCl2(phen)@Y, although all of them were well dispersed in water. We suspected that the excellent catalytic performance of Pd@UPOP should be attributed to its special amphiphilic porous structure. The good hydrophilicity made it effectively disperse in water and have nice contact with the organic substrates; meanwhile, the porous structure and strong oleophilicity provided a microenvironment with a high concentration of organic substrates inside the frameworks. To demonstrate our theory, Pd@KAPs(Ph-PPh3), an oleophilic heterogeneous catalyst75 that exhibited high aminocarbonylation activity in DMF, was tested in water. As we expected, no target amide was detected and the catalyst was suspended at the solution surface during the reaction (Table 1, entry 4). As proof of the high concentration of organic substrates inside the frameworks, the adsorption experiment showed that the adsorption capacity of Pd@UPOP for chlorobenzene was 3653 mg g−1. While the solubility of chlorobenzene in water was only 0.4 mg g−1 under the same conditions. With the above results in hand, the effect of reaction time was studied. When reducing the reaction time to 2 h, the reaction also proceeded well, and a 98% yield of N,N-diethylbenzamide was achieved (Table 1, entry 5). Thus, even under milder reaction temperature, a higher turnover frequency (98 h−1) than that of the previously reported heterogeneous catalysts was obtained.66–75,83 Previous studies have shown that bases have a remarkable influence on the reactivity of the aminocarbonylation reaction. The widely used bases for aminocarbonylation, including K2CO3, Na2CO3 and Et3N, were examined and the results revealed that K2CO3 and Na2CO3 were suitable bases for this reaction, both giving 98% yields of the desired amide (Table 1, entries 5–7). Although K2CO3 and Na2CO3 performed equally well, considering the economy of sodium salt over potassium salt, Na2CO3 was chosen for further optimization.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Pd [wt%] | Base | Time (h) | Yieldb (%) |
| a Reaction conditions: catalyst (0.5 mol% Pd), iodobenzene (1 mmol), HNEt2 (2 mmol), deionized water (4 ml), base (1.2 mmol), 100 °C, CO (1 atm), stirring speed (450 rpm). b GC yield. | |||||
| 1 | Pd@UPOP | 2.0 | K2CO3 | 4 | 99 |
| 2 | Pd/C | 5 | K2CO3 | 4 | 35 |
| 3 | PdCl2(phen)@Y | 2.1 | K2CO3 | 4 | 14 |
| 4 | Pd@KAPs(Ph-PPh3) | 0.8 | K2CO3 | 4 | — |
| 5 | Pd@UPOP | 2.0 | K2CO3 | 2 | 98 |
| 6 | Pd@UPOP | 2.0 | Na2CO3 | 2 | 98 |
| 7 | Pd@UPOP | 2.0 | Et3N | 2 | 96 |

Substrate scope
With these optimal conditions in hand, we then explored the scope and generality of the catalytic system in water. Firstly, the effect of various amines was examined by using iodobenzene as a substrate. As shown in Table 2, linear and branched aliphatic amines, such as dimethylamine, dipropylamine, dibutylamine and diisopropylamine, gave the target amides in 94%, 95%, 96% and 90% yields, respectively (Table 2, entries 2–5). It should be mentioned that dimethylamine is a gas at room temperature and has a boiling point of 7 °C. This catalytic system in water benefited the addition of dimethylamine by using aqueous solution instead of gaseous dimethylamine. Cyclic aliphatic amines, such as morpholine, 4-methylpiperdine and pyrrolidine also performed well, affording the corresponding amides in 89%, 96% and 93% yields, respectively (Table 2, entries 6–8). As an example of an aryl amine, aniline was also tested and gave the corresponding amides in a 92% yield (Table 2, entry 9). Ammonia, a weaker nucleophile, was also tested under the optimal conditions. Unfortunately, the reaction did not run at all (Table 2, entry 10).|
|
|||
|---|---|---|---|
| Entry | Amine | Product | Yieldb (%) |
| a Reaction conditions: Pd@UPOP (0.5 mol% Pd), iodobenzene (1 mmol), amine (2 mmol), deionized water (4 ml), Na2CO3 (1.2 mmol), 100 °C, CO (1 atm), stirring speed (450 rpm), 2 h. b Isolated yields. | |||
| 1 | HNEt2 |
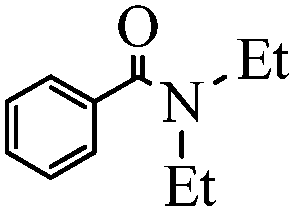
|
95 |
| 2 | HNMe2 |
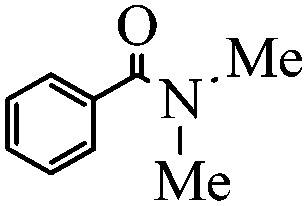
|
94 |
| 3 | HN(n-Pr)2 |
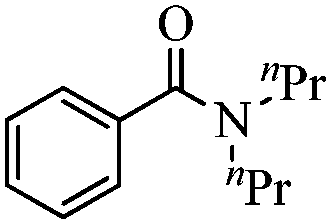
|
95 |
| 4 | HN(n-Bu)2 |
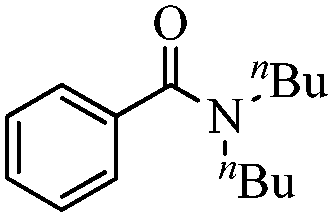
|
96 |
| 5 |

|
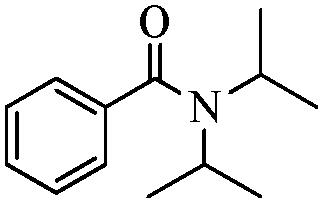
|
90 |
| 6 |
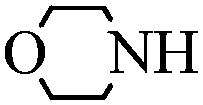
|

|
89 |
| 7 |
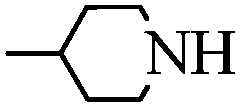
|
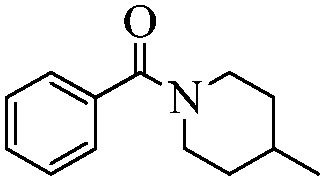
|
96 |
| 8 |
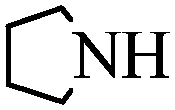
|
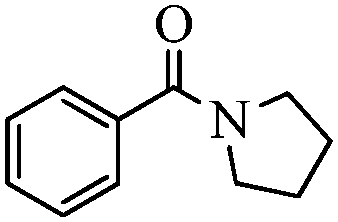
|
93 |
| 9 |

|
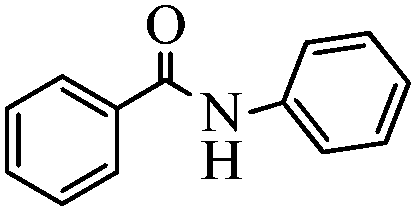
|
92 |
| 10 | NH3 |
|
— |

Next, the effect of varying aryl halides was examined by using diethylamine as a substrate (Table 3). In general, this aminocarbonylation reaction was also equally effective with both electron-rich, electron-deficient and bulky aromatic iodoarene derivatives (Table 3, entries 1–9). The various substituted aryl iodides were converted efficiently to the desired amides in good to excellent yields within 3 h. 3-Iodopyridine, as a representative example of (hetero)aryl iodide, afforded the target amide in a 95% yield (Table 3, entry 10). Bromobenzene was also tested. Unfortunately, no target amide was detected after the reaction (Table 3, entry 11). However, 2-bromopyridine gave the target amide in 82% yield (Table 3, entry 12).
|
|
|||
|---|---|---|---|
| Entry | Aryl halide | Reaction time (h) | Yieldb (%) |
| a Reaction conditions: Pd@UPOP (0.5 mol% Pd), aryl halide (1 mmol), diethylamine (2 mmol), deionized water (4 ml), Na2CO3 (1.2 mmol), 100 °C, CO (1 atm), stirring speed (450 rpm). b Isolated yields. | |||
| 1 |

|
2 | 96 |
| 2 |
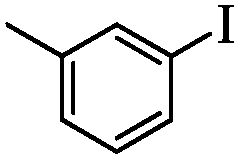
|
2 | 92 |
| 3 |
|
3 | 93 |
| 4 |
|
3 | 97 |
| 5 |
|
2 | 95 |
| 6 |
|
2 | 98 |
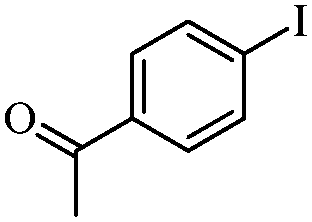
| 7 |
|
2 | 84 |
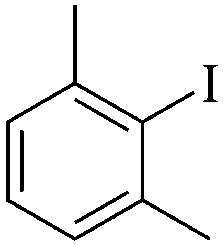
| 8 |
|
2 | 82 |
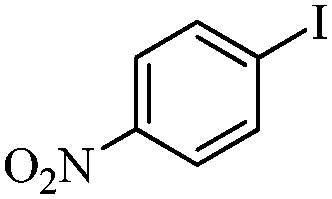
| 9 |
|
2 | 87 |
| 10 |

|
2 | 95 |
| 11 |
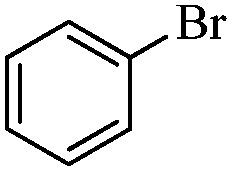
|
4 | — |
| 12 |

|
4 | 82 |
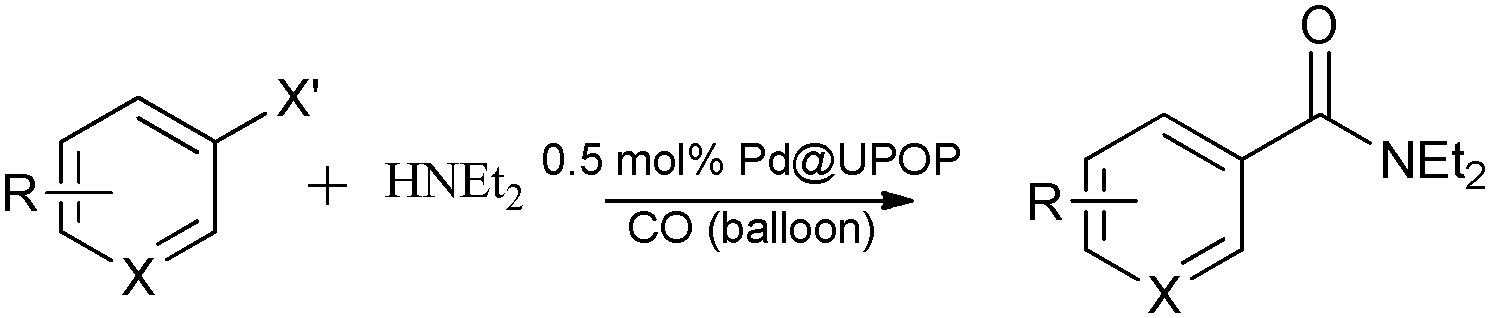
The recycling of the catalyst
In addition to catalytic activity, recovery, reuse of the catalyst and leaching of Pd metal in solution are also crucial for an outstanding heterogeneous catalyst. Recycling capacity was evaluated by examining the reaction between aminocarbonylation of iodobenzene with diethylamine in the presence of 0.5 mol% palladium. After every cycle, the catalyst was easily separated by simple centrifugation, and the yield of N,N-diethylbenzamide was analysed by GC. As shown in Fig. 7, Pd@UPOP exhibited good reusability and could be reused for five cycles with only a slight decrease of catalytic activity. Moreover, we tested the Pd content in the filtrate of the first cycle by means of ICP-AES, and the results showed that palladium leaching was very low, ca. 0.047 ppm, indicating that 99.9% of Pd species were still on the solid catalyst after the reaction. The excellent recycling ability and low palladium leaching should be attributed to the rich coordination sites. To test the coordination capacity of UPOP, UPOP (0.2 g) was added to a dichloromethane solution (30 ml) containing palladium acetate (0.0886 g). After stirring the mixture at room temperature for 12 h, the obtained solid was handled by the procedures described for the preparation of the catalysts, and the AAS results showed that the saturated Pd loading capacity of UPOP was 15.94 wt%. To assess whether Pd nanoparticles were responsible for the true catalytic species or not, TEM (Fig. 3(c)) and XPS studies were performed for the 2nd recycled Pd@UPOP (Fig. S5 in the ESI†). The TEM image showed that no palladium particles were formed, and the XPS spectra further confirmed that the Pd was still in the Pd2+ state after the 2nd recycle. Moreover, a mercury drop test was also carried out.84 When one drop of Hg0 was added to the reaction mixture before the aminocarbonylation of iodobenzene and diethylamine was performed, Pd@UPOP still gave the target amides in 79% GC yield. These results suggested that the catalytic species was Pd2+.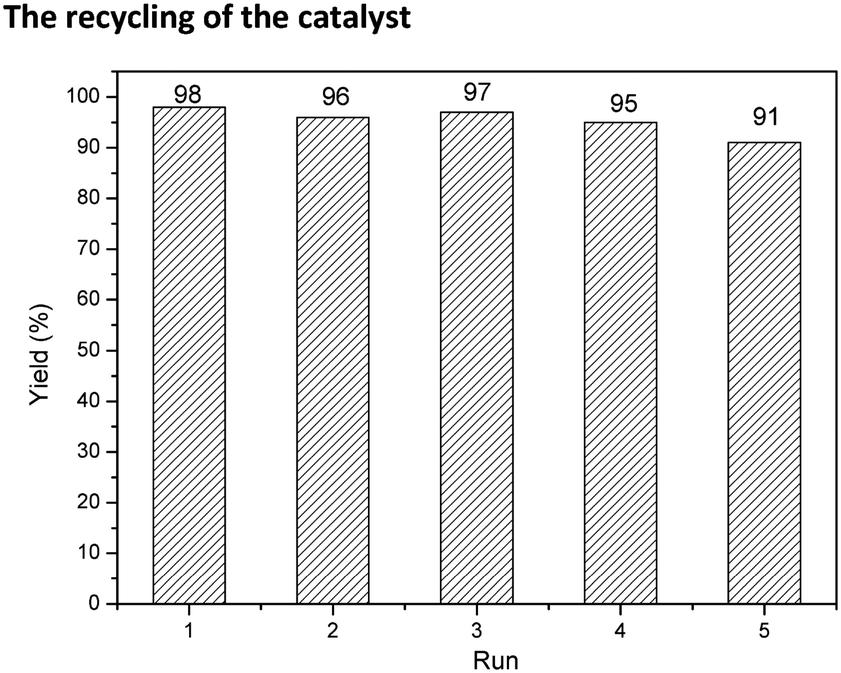 | ||
| Fig. 7 Reuse of the Pd@UPOP catalyst for the aminocarbonylation of iodobenzene. | ||
Conclusions
A highly efficient and recyclable heterogeneous palladium catalyst for aqueous aminocarbonylation was successfully prepared via a facile urea-forming condensation reaction. The obtained heterogeneous catalyst possessed good amphiphilic properties and exhibited higher catalytic activity than the previously reported heterogeneous palladium catalysts for aminocarbonylation. The catalyst can be reused for up to five consecutive cycles with negligible palladium leaching. Based on the results of the mechanism discussions, the excellent recycling ability and low palladium leaching should be attributed to the rich coordination sites of the prepared UPOP. While the superior catalytic performances of the prepared Pd@UPOP were mainly attributed to the amphiphilic porous structure. The good hydrophilicity made it effectively disperse in water and have nice contact with the substrate; the porous structure and the strong oleophilicity provided a microenvironment with a high concentration of organic substrates inside the frameworks. Thus, this protocol provides an efficient heterogeneous palladium catalyst for an eco-friendly aminocarbonylation process and some clues to prepare amphiphilic heterogeneous catalysts from urea-based materials.Acknowledgements
This work is partially supported by the Science and Technology Fund Project of Guizhou Province (qian ke he ji chu [2016]1133), the Youth Talent Growth Project of Educational Department of Guizhou Province (qian jiao he KY [2016]264), the Science and Technology Creative Team Project of Liupanshui Normal University (LPSSYKJTD201501, LPSSYKJTD201601), Guizhou Key Built Discipline Project (ZDXK[2016]24), the School-Level Key Discipline of Chemistry (LPSSYZDXK201602), Guizhou Solid Waste Recycling Laboratory of Coal Utilization (No. 2011-278), and Guizhou Scientific Research Experimental Platform of Coal Clean and Effective Use (No. [2011]4003).References
- C.-J. Li and T.-H. Chan, Organic reactions in aqueous media, Wiley, New York, 1997 Search PubMed.
- A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed.
- M.-O. Simona and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- Á. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600 CrossRef CAS PubMed.
- K. Natte, H. Neumann and X. F. Wu, Catal. Sci. Technol., 2015, 5, 4474–4480 CAS.
- J. Chen, K. Natte and X. F. Wu, Tetrahedron Lett., 2015, 56, 6413–6416 CrossRef CAS.
- F. Zhu, Y. Li, Z. Wang and X. F. Wu, Catal. Sci. Technol., 2016, 6, 2905–2909 CAS.
- C. L. Li, X. Qi and X. F. Wu, J. Mol. Catal. A: Chem., 2015, 406, 94–96 CrossRef CAS.
- P. Kleman, P. Barbaro and A. Pizzano, Green Chem., 2015, 17, 3826–3836 RSC.
- E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607–4692 CrossRef CAS PubMed.
- A. Hassine, S. Sebti, A. Solhy, M. Zahouily, C. Len, M. N. Hedhili and A. Fihri, Appl. Catal., A, 2013, 450, 13–18 CrossRef CAS.
- Y. M. A. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 3190–3198 CrossRef CAS PubMed.
- S. Minakata and M. Komatsu, Chem. Rev., 2009, 109, 711–724 CrossRef CAS PubMed.
- Y. Uozumi, Y. Matsuura, T. Arakawa and Y. M. A. Yamada, Angew. Chem., Int. Ed., 2009, 48, 2708–2710 CrossRef CAS PubMed.
- C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed.
- N. Kambe, T. Iwasakia and J. Teraob, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC.
- G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151–5169 RSC.
- C. Zhang, J. Liu and C. Xia, Catal. Sci. Technol., 2015, 5, 4750–4754 CAS.
- X. F. Wu, Chem. – Eur. J., 2015, 21, 12252–12265 CrossRef CAS PubMed.
- J. B. Peng, X. Qi and X. F. Wu, ChemSusChem, 2016, 9, 2279–2283 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555–1564 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- X. F. Wu, RSC Adv., 2016, 6, 83831–83837 RSC.
- S. Zheng, Y. Wang, C. Zhang, J. Liu and C. Xia, Appl. Organomet. Chem., 2014, 28, 48–53 CrossRef CAS.
- C. Zhang, J. Liu and C. Xia, Org. Biomol. Chem., 2014, 12, 9702–9706 CAS.
- S. Kumar, G. K. Rao, A. Kumar, M. P. Singh and A. K. Singh, Dalton Trans., 2013, 42, 16939–16948 RSC.
- G. Zhang, Y. Luan, X. Han, Y. Wang, X. Wen, C. Ding and J. Gao, Green Chem., 2013, 15, 2081–2085 RSC.
- D.-H. Lee, J.-Y. Jung and M.-J. Jin, Chem. Commun., 2010, 46, 9046–9048 RSC.
- V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS PubMed.
- K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173–6177 CrossRef CAS PubMed.
- P. Yang, R. Ma and F. Bian, ChemCatChem, 2016, 8, 3746–3754 CrossRef CAS.
- M. Gholinejad, A. Neshat, F. Zareh, C. Nájera, M. Razeghi and A. Khoshnood, Appl. Catal., A, 2016, 525, 31–40 CrossRef CAS.
- Z. Dong and Z. Ye, Appl. Catal., A, 2015, 489, 61–71 CrossRef CAS.
- M. I. Burrueco, M. Mora, C. Jiménez-Sanchidrián and J. R. Ruiz, Appl. Catal., A, 2014, 485, 196–201 CrossRef CAS.
- A. S. Roy, J. Mondal, B. Banerjee, P. Mondal, A. Bhaumik and S. M. Islam, Appl. Catal., A, 2014, 469, 320–327 CrossRef CAS.
- Q. Sun, Z. Dai, X. Meng, L. Wang and F.-S. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- M. Rose, ChemCatChem, 2014, 6, 1166–1182 CAS.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809–8816 CrossRef CAS.
- D. Mullangi, S. Nandi, S. Shalini, S. Sreedhala, C. P. Vinod and R. Vaidhyanathan, Sci. Rep., 2015, 5, 10876 CrossRef PubMed.
- L. Li, Z. Chen, H. Zhong and R. Wang, Chem. – Eur. J., 2014, 20, 3050–3060 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- D. W. Zhang, X. Zhao, J. L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zarks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- Y. Li, F. Zhu, Z. Wang and X.-F. Wu, ACS Catal., 2016, 6, 5561–5564 CrossRef CAS.
- W. Li and X.-F. Wu, Chem. – Eur. J., 2015, 21, 14943–14948 CrossRef CAS PubMed.
- Q. Wang, Y. Su, L. Li and H. Huang, Chem. Soc. Rev., 2016, 45, 1257–1272 RSC.
- P. Xie, C. Xia and H. Huang, Org. Lett., 2013, 15, 3370–3373 CrossRef CAS PubMed.
- S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367–10389 RSC.
- S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1–35 CrossRef CAS PubMed.
- I. Omae, Coord. Chem. Rev., 2011, 255, 139–160 CrossRef CAS.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC.
- Y. Lei, S. Xiao, G. Li, Y. Gu, H. Wu and K. Shi, Appl. Organomet. Chem. DOI:10.1002/aoc.3637.
- T. Kawamoto, A. Sato and I. Ryu, Chem. – Eur. J., 2015, 21, 14764–14767 CrossRef CAS PubMed.
- R. S. Mane and B. M. Bhanage, RSC Adv., 2015, 5, 76122–76127 RSC.
- S. T. Gadgea and B. M. Bhanage, Org. Biomol. Chem., 2014, 12, 5727–5732 Search PubMed.
- T. Fang, X.-H. Gao, R.-Y. Tang, X.-G. Zhang and C.-L. Deng, Chem. Commun., 2014, 50, 14775–14777 RSC.
- T. Xu and H. Alper, J. Am. Chem. Soc., 2014, 136, 16970–16973 CrossRef CAS PubMed.
- J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841–16844 CrossRef CAS PubMed.
- F. Tinnis, O. Verho, K. P. J. Gustafson, C.-W. Tai, J.-E. Bäckvall and H. Adolfsson, Chem. – Eur. J., 2014, 20, 5885–5889 CrossRef CAS PubMed.
- M. Cai, H. Zhao and Y. Huang, J. Mol. Catal. A: Chem., 2005, 238, 41–45 CrossRef CAS.
- W. Y. Hao, J. C. Sha, S. R. Sheng and M. Z. Cai, Catal. Commun., 2008, 10, 257–260 CrossRef CAS.
- T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1805–1807 RSC.
- T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406–1410 CrossRef CAS.
- Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148–153 CrossRef CAS.
- S. M. Islam, K. Ghosh, A. S. Roy and R. A. Molla, RSC Adv., 2014, 4, 38986–38999 RSC.
- R. A. Molla, M. A. Iqubal, K. Ghosh, A. S. Roy and S. M. Islam, RSC Adv., 2014, 4, 48177–48190 RSC.
- Y. Lei, X. Zhang, Y. Gu, J. Hu, G. Li and K. Shi, Transition Met. Chem., 2016, 41, 1–7 CrossRef CAS.
- H. Mei, J. Hu, S. Xiao, Y. Lei and G. Li, Appl. Catal., A, 2014, 475, 40–47 CrossRef CAS.
- Y. Lei, R. Zhang, L. Wu, Q. Wu, H. Mei and G. Li, Appl. Organomet. Chem., 2014, 28, 310–314 CrossRef CAS.
- J. Hu, Y. Gu, Z. Guan, J. Li, W. Mo, T. Li and G. Li, ChemSusChem, 2011, 4, 1767–1772 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- H. Ren, T. Ben, F. Sun, M. Guo, X. Jing, H. Ma, K. Cai, S. Qiu and G. Zhu, J. Mater. Chem., 2011, 21, 10348–10353 RSC.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2012, 24, 471–479 CrossRef CAS.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 2221–2224 CrossRef CAS.
- M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33–79 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00331a |
| This journal is © the Partner Organisations 2017 |