DOI:
10.1039/C6QM00274A
(Research Article)
Mater. Chem. Front., 2017,
1, 1093-1106
N-doped ZnO–MoS2 binary heterojunctions: the dual role of 2D MoS2 in the enhancement of photostability and photocatalytic activity under visible light irradiation for tetracycline degradation†
Received
21st October 2016
, Accepted 18th December 2016
First published on 19th December 2016
Abstract
In this work, we report the fabrication of binary semiconductor heterojunctions comprising N-doped ZnO nanorods loaded with two-dimensional MoS2 nanoflowers in varying amounts, using a facile hydrothermal synthesis method. These semiconductor heterojunctions have been demonstrated to be highly efficient photocatalysts with enhanced performance under visible light irradiation for the degradation of a pharmaceutical pollutant, tetracycline. The superior photocatalytic activity of the heterojunctions can be attributed to the synergistic effect of N-doping of ZnO and loading of MoS2 leading to higher absorption of visible light, efficient separation of photogenerated charge carriers and rapid charge transfer to reaction sites, as per the conduction band potentials of both N-doped ZnO and MoS2. In addition, the two-dimensional nanoflower morphology of MoS2 provides more reaction sites for the adsorption of pollutants, due to its large surface area. Furthermore, the transfer of holes from the valence band of N-doped ZnO to the valence band of MoS2 prevents the photocorrosion of N-doped ZnO resulting in enhanced photostability of the catalyst during the reaction.
1. Introduction
Recently, pharmaceutical waste materials, mainly antibiotics, have generated a serious threat to the ecosystem and human life.1,2 These pharmaceutical residues, mainly the antibiotic tetracycline (TC), are widely used in human medicine and are hence discharged into the environment by pharmaceutical industries and as hospital effluents.3 They threaten human health by inducing the proliferation of multiresistant bacterial strains. Moreover, these antibiotic residues, mainly TC, have the potential to induce adverse environmental effects in aquatic life even at low concentrations and hence perturb the ecosystem through the drinking water and food chain.4,5 Excessive accumulation of these antibiotics can seriously intimidate human health by inducing central nervous system alterations, spermatogenesis anomalies, nephropathy, mutagenicity and many more lethal diseases.6,7 Hence there is an urgent need to develop the technology to effectively remove these antibiotic residues from water. Several techniques such as activated carbon adsorption, microbial degradation, UV/H2O2 processes, biodegradation and electrolysis have been applied in the past to tackle this problem.8,9 Recently, semiconductor based photocatalytic degradation has attracted much attention as a green, highly efficient technology for the removal of antibiotics and other pollutants from water.10–13
Among various semiconductors, TiO2 and ZnO are the most explored materials for various environmental remediation applications.14,15 ZnO is a very interesting and a better photocatalytic material than TiO2 because of its high electron mobility and significantly longer electron lifetime, which can effectively suppress the electron–hole recombination.15 ZnO has a direct band gap (3.37 eV), is photosensitive and low cost, and has a non-toxic nature and high exciton binding energy (60 meV). The large band gap value of ZnO restricts its use only to ultraviolet (UV) excitation (which constitutes only about 5% of the solar spectrum), and the fast recombination of photogenerated charge carriers limits its practical applications.16 Over the past few years, unremitting efforts have been made to improve the photocatalytic efficiency of ZnO based semiconductor materials and to enhance the absorption range towards the visible region by adopting various strategies, such as doping with noble metals to form ZnO–Au,17 ZnO–Ag,18 and ZnO–Pt19 nanocomposites, coupling with other semiconductor materials, composite formation with carbon based materials, such as graphene, etc.20–23 Hence, it is always desirable to develop a photocatalyst with enhanced response from the UV to visible spectral range, which constitutes about 43% of the solar spectrum. Semiconductor materials can be fabricated as visible light responsive by non-metal doping as a cost effective method in comparison to noble metal doping. Nitrogen has been widely used as a dopant to substitute oxygen because of its closest atomic size, electronegativity and small formation energy required for the substitution; hence it has attracted remarkable attention and successfully applied to ZnO for various photocatalytic applications.14 Furthermore, there could be a significant overlapping between the p orbital of the N-dopant with the valence band O 2p orbital which results in the suppression of charge recombination and enhancement in the photocatalytic performance.24 Although it has been reported that N-doped ZnO did not show much enhanced photocatalytic performance, such efforts have been perused to make ZnO a visible light active material.25
There are several recent studies which have successfully investigated the fabrication of binary heterojunctions of various interesting semiconductor materials (Ag2O, Ag2S, Ag2CO3, Bi2O3, Bi2O2CO3 and TiO2) as an effective strategy for enhancing their photocatalytic activity and stability.26–30 These binary heterojunctions show high efficiency for the removal of various pollutants by harvesting visible light efficiently. In addition to this, binary heterojunction formation of ZnO with other semiconductors, such as ZnO–In2O3,31 ZnO–Bi2O3,32 ZnO–Fe2O3,33 ZnO–SnO2,34 ZnO–BiVO4,11 ZnO–CdS,35 ZnO–FeWO4,36 ZnO–Ag2O,37 have been recently reported for better photocatalytic activity and to overcome the shortcoming of low visible light utilization as well as fast recombination of charge carriers. These heterojunctions showed improved photocatalytic performance due to strong interfacial interaction between ZnO and the semiconductor interface, which eventually promotes the generation of more and highly reactive hydroxyl radicals (OH*) leading to a superior photocatalytic performance of nanocomposites for the degradation of various pollutants. In recent times, two-dimensional (2D) graphene-like materials have attracted great attention because of their excellent properties such as a high specific surface area as compared to 0D and 1D materials.38 MoS2 is one of such potential materials, which serves as an excellent cocatalyst in various photocatalytic applications,39–41 which could effectively replace the noble metals, mainly Pt, without compromising the activity. MoS2 is a visible light responsive semiconductor material with a narrow band gap (∼1.8 eV).39 MoS2 has layered 2D graphene-like structures, wherein the Mo atom is sandwiched between two layers of S atoms that are held together by van der Waals forces. However, the fast recombination of photogenerated charge carriers and the poor conductivity of MoS2 limit its application as a cocatalyst.42
In one of our previous works, we have investigated the photocatalytic performance of ZnO nanoparticles,43 thus in view of further improvement herein we report the N-doped ZnO nanorod (N-ZnO NR) and 2D MoS2 nanoflower (MNF) heterojunction (NZM heterojunction). Furthermore, 1D nanostructures such as ZnO NRs exhibit superior photocatalytic activity as compared to the spherical nanoparticles due to their dimensional anisotropy and higher aspect ratio which lead to a large number of electron–hole pairs on its surface that can flow in the direction of crystal length and significantly decrease the charge recombination.44 A N-doped ZnO NR offers a large surface area and an intimate contact with a 2D MNF to form a heterojunction with enhanced absorption in the visible range. Moreover the MNFs could effectively replace the use of rare, expensive noble metals, like Pt, as a cocatalyst and thus leads to a highly desirable cost-effective, environmentally benign catalyst material. In this work, various compositions with varying amounts of MNFs were prepared by employing a hydrothermal method. All novel NZM heterojunctions were characterized thoroughly by different microscopic and spectroscopic techniques to confirm their successful formation. The photocatalytic performance of all samples has been studied in detail by investigating the photocatalytic degradation of TC antibiotic under visible light (λ ≥ 420 nm) illumination. The enhanced photocatalytic performance of these binary heterojunctions along with a plausible mechanism has been investigated and discussed in detail.
2. Experimental section
2.1. Chemicals
For the synthesis of MNFs, sodium molybdate dihydrate (Na2MoO4·2H2O), thiourea (NH2CSNH2), and oxalic acid (H2C2O4) were purchased from Merck. Zinc chloride (ZnCl2), sodium hydroxide (NaOH), and aq. ammonia solution (NH3) for the ZnO NR and N-ZnO NR synthesis were also supplied by Merck. Triethanolamine (TEA) and benzoquinone (BQ) were supplied by Merck, and isopropanol (IPA) was supplied by Fisher Scientific. All chemicals were used as received without any further purification. Deionized water (18.2 MΩ cm) used in synthesis was obtained from a double stage water purifier (ELGA PURELAB Option-R7).
2.2. Synthesis of MoS2 nanoflowers
For the synthesis of pristine MoS2 nanoflowers (MNFs), a reported hydrothermal method was adopted.45 In short, 1.0 g of sodium molybdate dihydrate (Na2MoO4·2H2O) and 1.4 g of thiourea (NH2CSNH2) were dissolved in 80 mL of deionized water. Herein, Na2MoO4·2H2O and NH2CSNH2 served as Mo and S sources, respectively. This was followed by the addition of 0.4 g of oxalic acid (H2C2O4) to adjust the pH value to an acidic environment. The mixture was magnetically stirred for about 30 min and then transferred to a 100 mL Teflon-lined stainless-steel autoclave, and sealed tightly. Finally, the autoclave was heated in a drying oven at 180 °C for 24 h. After the completion of reaction, the autoclave was allowed to cool down to room temperature naturally. The obtained black colored products were washed by ultrasonic cleaning with ethanol and deionized water alternately several times to remove impurities and then dried in a vacuum oven at 60 °C for 12 h to obtain MNF powder.
2.3. Synthesis of ZnO and N-ZnO nanorods
ZnO nanorods (NRs) were synthesized by a reported hydrothermal method.46 In brief, 10 mL of 0.2 M zinc chloride (ZnCl2) solution in ethanol was added into 70 mL of 0.5 M sodium hydroxide (NaOH) solution dropwise with vigorous stirring. This was followed by ultrasonic treatment of the solution for 30 min for homogenization. Then, this homogenous solution was transferred to a 100 mL Teflon-lined stainless steel autoclave, sealed tightly and maintained at 180 °C for 12 h. White precipitates of ZnO NRs were collected by centrifugation and washed several times with deionized water and ethanol and finally dried at 60 °C for 5 h. Nitrogen doping was done as per a reported hydrothermal method.47 In short, about 0.2 g of as-prepared ZnO NRs was dispersed in 40 mL of deionized water with the help of ultrasonication. This was followed by the addition of 4 mL of aqueous ammonia solution under continuous stirring to form a clear solution. Now the resulting suspension was treated in a 50 mL Teflon-lined stainless steel autoclave, sealed tightly and maintained at 150 °C for 24 h. The final product was obtained after washing thrice with deionized water and ethanol and dried at 60 °C.
2.4. Synthesis of N-ZnO–MNF heterojunctions
N-doped ZnO–MNF heterojunctions were also synthesized by adopting a hydrothermal strategy. In brief, the as-prepared MNFs (0.05 g) were dispersed in 40 mL of DI water by subjecting to ultra-sonication for about 15 min. To this solution, 0.2 M ZnCl2 solution and 0.5 M NaOH solution were added as a source of Zn and O respectively. This suspension was kept under magnetic stirring for 30 min and 4 mL of aqueous ammonia solution was added, then the suspension was transferred into a 50 mL Teflon-lined stainless steel autoclave, sealed tightly and maintained at 180 °C for 12 h. After the reaction completion, the resultant solution was washed with deionized water and ethanol thrice. The final product was obtained by drying the precipitate at 60 °C for 6 h. By varying the amount of MNFs, different compositions were prepared having different wt% of MNFs, and the samples were labelled as NZM0.2, NZM0.5, NZM1, NZM2, and NZM3 indicating 0.2 wt%, 0.5 wt%, 1 wt%, 2 wt% and 3 wt% of MNFs loaded onto N-ZnO NRs, respectively.
2.5. Materials characterization
X-ray diffraction (XRD) measurements were performed using a Rigaku Smart Lab 9 kW rotating anode X-ray diffractometer with Ni-filtered Cu Kα irradiation (λ = 0.1542 nm) at 45 kV and 100 mA in 2θ ranging from 10°–80° at a scan rate of 2° per minute and a step size of 0.02°. Raman spectroscopic measurements were done using a Horiba LabRAM high resolution UV-Vis-NIR instrument using 633 nm laser excitation. Morphology of the samples was investigated by using a scanning electron microscope (SEM), FEI Nova Nano SEM-450, and a transmission electron microscope (TEM), FEI Tecnai G2 20 S-twin microscope operated at 200 kV. Energy dispersive X-ray (EDAX) spectra were obtained using the same SEM and TEM instruments, and elemental mapping was performed using a TEM instrument. Further, the presence of nitrogen in N-ZnO and NZM1 heterojunctions has been confirmed by CHNSO analysis by using a Thermo Scientific (FLASH 2000) Organic Elemental Analyzer (OEA). The total organic carbon (TOC) estimation was done using a TOC-VCPH, Shimadzu ASI-V instrument. X-ray photoelectron spectroscopic (XPS) measurements were performed using a high resolution PREVAC photoemission spectrometer having an Al Kα (1486.6 eV) dual anode as the source operating at 12 kV anode voltage and 23 mA filament current. The XPS data were collected with a pass energy of 50 eV at 6.1 × 10−10 m bar vacuum using a Scienta R3000 electron energy analyzer. As an internal reference for the absolute binding energy, the C-1s peak (284.5 eV) was used. Initially the XPS unit was calibrated using the Fermi edge of Ag (KE 1482.544). The data obtained from the instrument were processed using CASA software; XPS software was developed by Fairley. The survey peak of the specimens was initially corrected for the C-1s peak and after that the quantitative estimate for the elemental analysis was performed. The high resolution XPS spectra as are also initially calibrated against the C-1s peak (284.5 eV). After that they are deconvoluted using CASA software to understand different electronic states that are present in the particular element. The baseline correction for each spectrum was initially carried out using a Shirley background correction. The deconvolution was then carried out using a combination of Gaussian and Lorentzian curves [GL(30)-meaning the Gaussian percentage is 30] in order to find out the chemical state and the compound of main elements present in the passive layer. The optical properties were analyzed by UV-vis diffuse reflectance spectroscopy (DRS) using a Perkin Elmer UV-/Vis-NIR Lambda 750 spectrophotometer, in which the polytetrafluoroethylene (PTFE) polymer was employed as an internal reflectance standard. The photocatalytic studies were performed using a homemade photoreactor setup consisting of two 45 W white light emitting CFL lamps, which produced an intensity of approximately 55000 lux, measured with a LX-101A digital luxmeter. The time-dependent UV-visible absorption spectra of the samples were recorded using a Shimadzu UV-2450 spectrophotometer in the wavelength range 200 nm to 800 nm. All the photoluminescence (PL) spectra were recorded on an Agilent Technologies Cary Eclipse fluorescence spectrometer.
2.6. Evaluation of photocatalytic activity
The photocatalytic activity of all prepared NZM heterojunctions and pristine samples was monitored by studying the photodegradation of TC antibiotic under visible light irradiation. In brief, 25 mg of photocatalyst was dispersed in 50 mL of aqueous solution of TC (0.01 g L−1). Before the suspension was irradiated, it was stirred magnetically for about 30 min in dark to establish the adsorption–desorption equilibrium between the photocatalyst and TC. Control experiments (without photocatalyst) were also performed under the same conditions. The TC concentration after adsorption–desorption equilibrium was regarded as the initial concentration (C0). After this equilibrium in the dark between TC and the photocatalyst, the suspension was illuminated with visible light. The reaction was analyzed after every 15 min and 1 mL of the suspension was taken out and centrifuged to precipitate the catalyst and the concentration of supernatant (containing the TC) was measured as a function of irradiation time using a UV-vis spectrophotometer. The photocatalytic degradation rate (%) of TC was calculated by applying the following formula:
where C0 is the initial absorbance of TC whereas Ct is the absorbance after sampling analysis.
3. Results and discussion
3.1. Crystal phase analysis
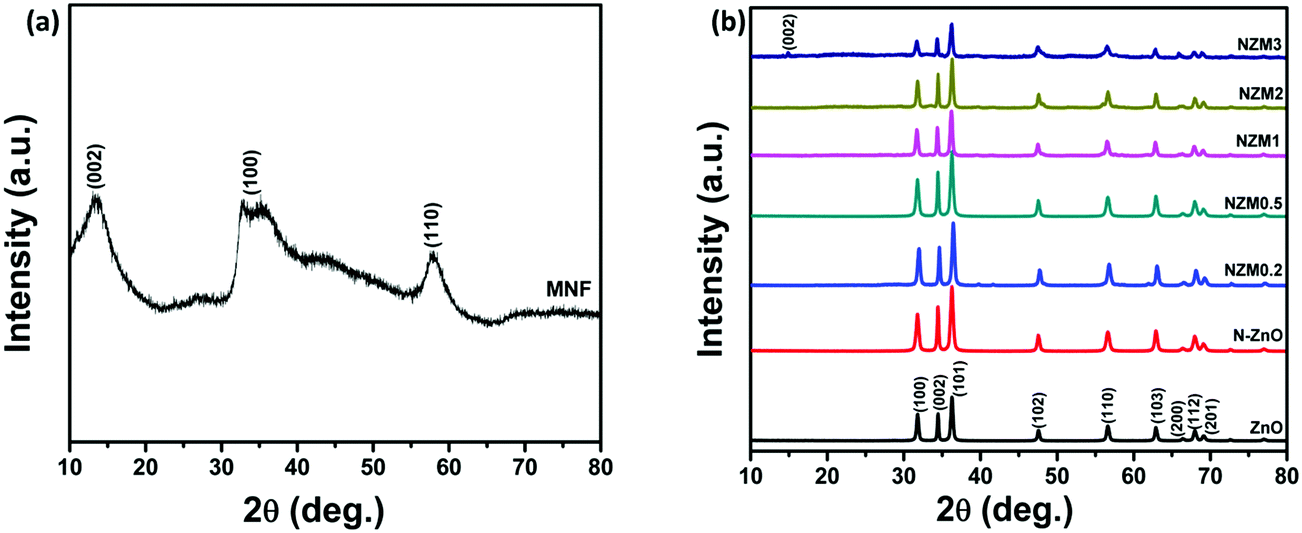
The crystal phase of ZnO, N-ZnO, MNFs and NZM heterojunctions was analyzed by X-ray diffraction (XRD), and the data are presented in Fig. 1. For pure MNFs, diffraction peaks at 2θ = 15.0°, 32.7° and 57.0° can be assigned to the (002), (100) and (110) reflection planes, respectively, of the hexagonal phase of MoS2 (a = b = 0.316, c = 1.230 nm, JCPDS no. 37-1492) (Fig. 1a).48 The most prominent diffraction peak at 2θ = 15.0° corresponds to the d-spacing of about 0.62 nm and the broader nature of this diffraction peak indicates the presence of a well stacked layered structure of MNFs in the material.49Fig. 1b presents the XRD patterns of pure ZnO NRs, N-ZnO NRs and all other heterojunctions (NZM0.2, NZM0.5, NZM1, NZM2 and NZM3). All the samples display diffraction peaks at 2θ = 31.7°, 34.3°, 36.1°, 47.4°, 56.5°, 62.7°, 66.3°, 67.9° and 69.0°, which could be attributed to (100), (002), (101), (102), (110), (103), (200), (112) and (201) lattice planes indicating a polycrystalline hexagonal wurtzite structure of ZnO (JCPDS no. 36-1451).50 It is noteworthy to mention here that the XRD patterns of N-ZnO display the same diffraction peaks as those of pristine ZnO, but are slightly broader. This broadening of peaks in doped samples indicates the decreased crystallinity due to N substitution. Moreover, the diffraction peaks appear at slightly higher 2θ angles, which reveal the small contraction of lattice parameters in N-ZnO.14 There are no characteristic diffraction peaks of MNFs in heterojunctions except in NZM3 in which a very small peak around 15.0° is observed corresponding to the (002) diffraction plane of MNFs. This could be attributed to the small amount of MoS2 present in the sample and its reduced stacking due to heterojunction formation with N-ZnO NRs.
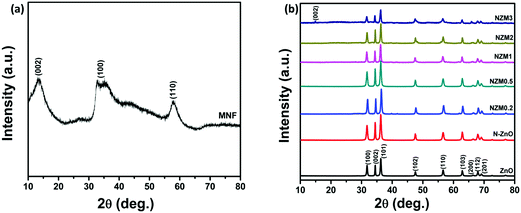 |
| | Fig. 1 (a) XRD patterns of MNFs. (b) XRD patterns of ZnO NRs, N-ZnO, NZM0.2, NZM0.5, NZM1, NZM2 and NZM3. | |
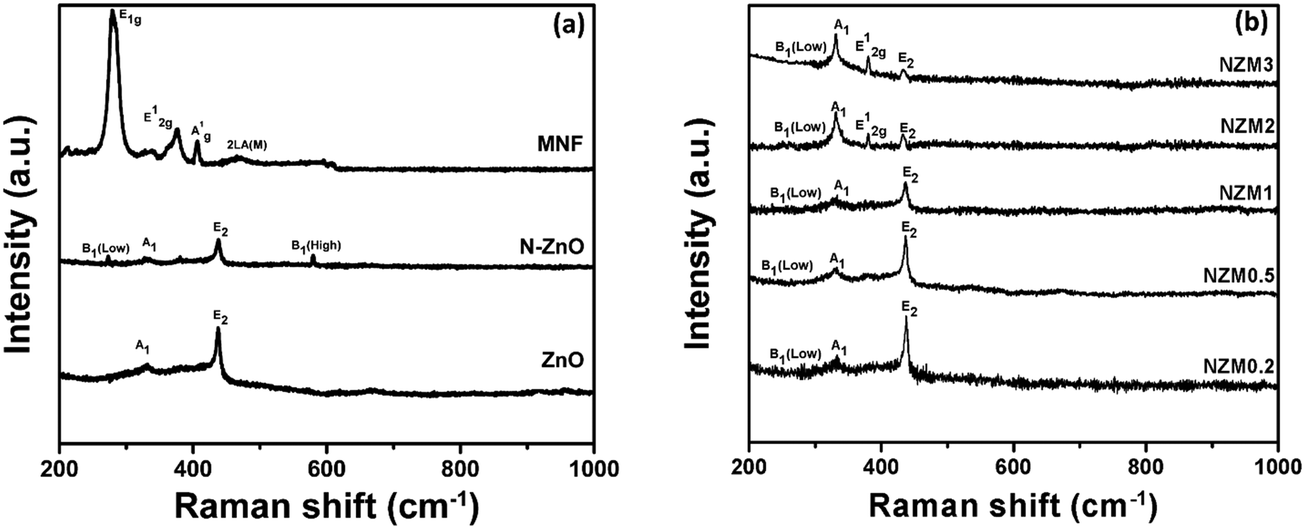
The crystal structure of the prepared doped materials was further investigated by Raman spectroscopy, which provides useful insights into the nature of defects and lattice disorder. Fig. 2(a) presents the Raman spectra of ZnO NRs, N-ZnO NRs, and MNFs. For pure ZnO NRs, Raman bands around 436 cm−1, 382 cm−1 and 331 cm−1 can be assigned to E2 (high), A1 (TO) (transverse optical) and A1 vibration modes, which are the characteristic bands of hexagonal wurtzite ZnO.43 The N-ZnO material also displayed the E2 (high), A1 (TO) (transverse optical) and A1 vibration modes revealing that the wurtzite structure of ZnO was preserved during nitrogen implantation. It is well reported in the literature51 that ion implantation introduces disorder activated Raman scattering (DARS), which is responsible for the emergence of the silent B1 (low) mode at 273 cm−1 and B1 (high) mode at 580 cm−1 in doped ZnO and confirms the successful N-doping in the crystal lattice of ZnO. In MNFs, Raman peaks around 287 cm−1 and 381 cm−1 can be assigned to E1g and E12g which correspond to the associated vibrations of two S atoms with respect to the Mo atom, and a peak around 407 cm−1 can be ascribed to A1g vibration mode and corresponds to the out-of-plane vibration of S atoms.42 The Raman peak around 454 cm−1 presents the typical peak of MoS2 and assigned as 2LA(M).42 Raman spectra of NZM0.2, NZM0.5, NZM1, NZM2 and NZM3 heterojunctions are presented in Fig. 2(b), and it can be seen clearly that the characteristic Raman bands which correspond to ZnO (A1 and E2 vibrational modes), N-ZnO (B1 mode) and MNFs remain intact in each sample indicating their successful combination to form binary heterojunctions.
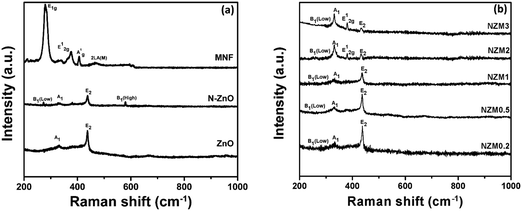 |
| | Fig. 2 (a) Raman spectra of ZnO, N-ZnO, and MNFs and (b) Raman spectra of NZM0.2, NZM0.5, NZM1, NZM2 and NZM3. | |
3.2. Morphology and compositional analysis
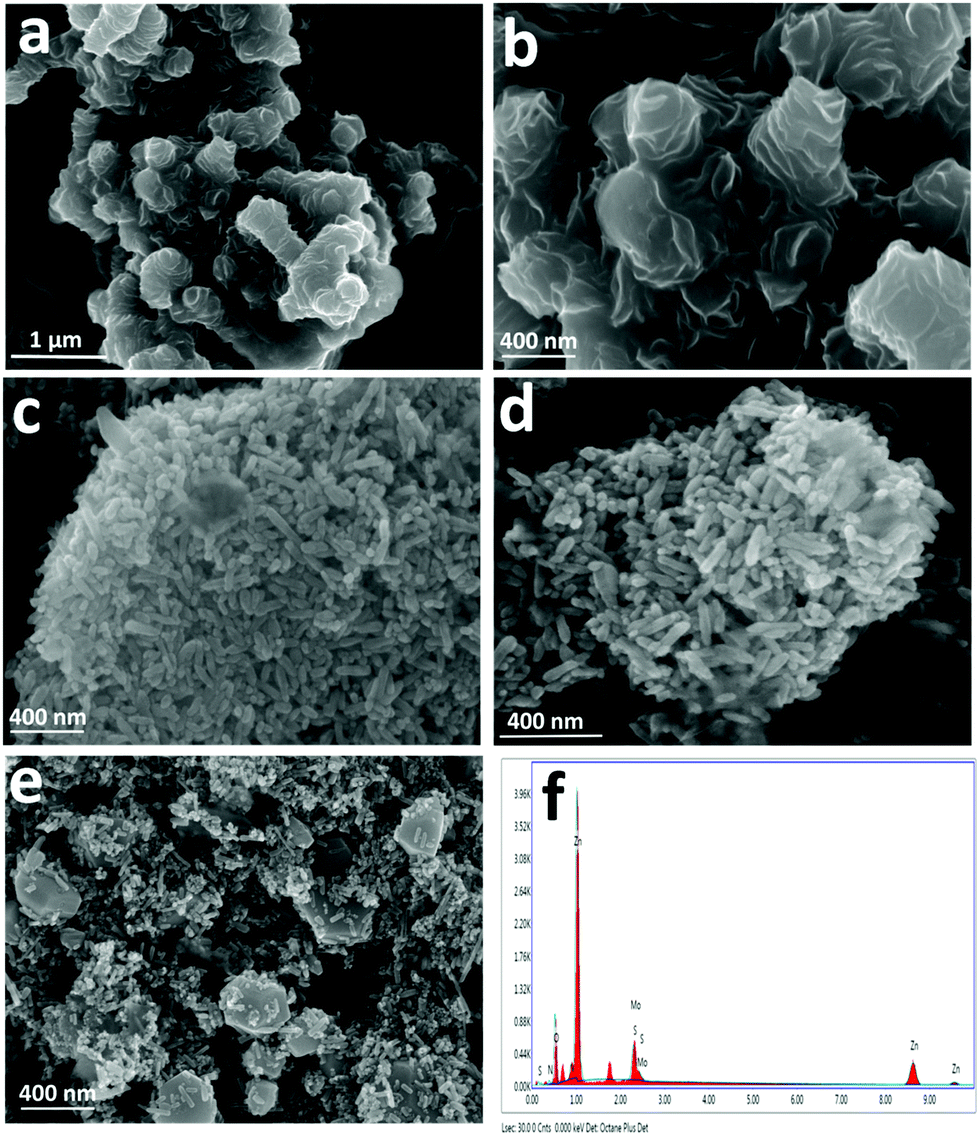
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses were performed to investigate the morphology and microstructures of pristine and NZM heterojunction samples. Fig. 3a and b show the SEM images of MNFs having a massive petal-like structure, which aggregated over each other to form a flower-like morphology with many grooves on its surface. Bare ZnO NRs and N-ZnO NRs possess a rod-like morphology having a length in the range of 200 nm (Fig. 3c and d). Fig. 3e presents the SEM images of the representative NZM1 heterojunction, which clearly reveals that N-ZnO NRs adhered well to the surface of MNFs. The MNF grooves, which are visible in Fig. 3a and b, cannot be seen in the NZM1 composite due to N-ZnO NRs, which are inserted into the surface of MNFs, confirming the formation of N-ZnO–MNF heterojunctions. The SEM images of other prepared heterojunctions (NZM0.2, NZM0.5, NZM2 and NZM3) are presented in Fig. S1 (refer the ESI†). EDAX patterns of NZM1 confirm the presence of all constituent elements (N, Zn, O, Mo and S) in the heterojunction; the corresponding spectrum is presented in Fig. 3f. The EDAX patterns and the corresponding atomic compositions of ZnO, N-ZnO, MNF, NZM0.2, NZM0.5, NZM1, NZM2 and NZM3 are presented in Fig. S2 (refer the ESI†). In addition, the CHNSO analysis of a representative sample, NZM1, and the control sample, N-ZnO, has also been performed (refer Fig. S3 in the ESI†). The results indicated the successful doping of nitrogen in the sample, and its atomic percentage was found to be 4.55%, which is in agreement with the values determined from EDAX analysis.
 |
| | Fig. 3 SEM images of (a) MNFs (low mag.), (b) MNFs (high mag.), (c) ZnO NRs, (d) N-ZnO NRs, and (e) NZM1 and (f) EDAX pattern of NZM1. | |
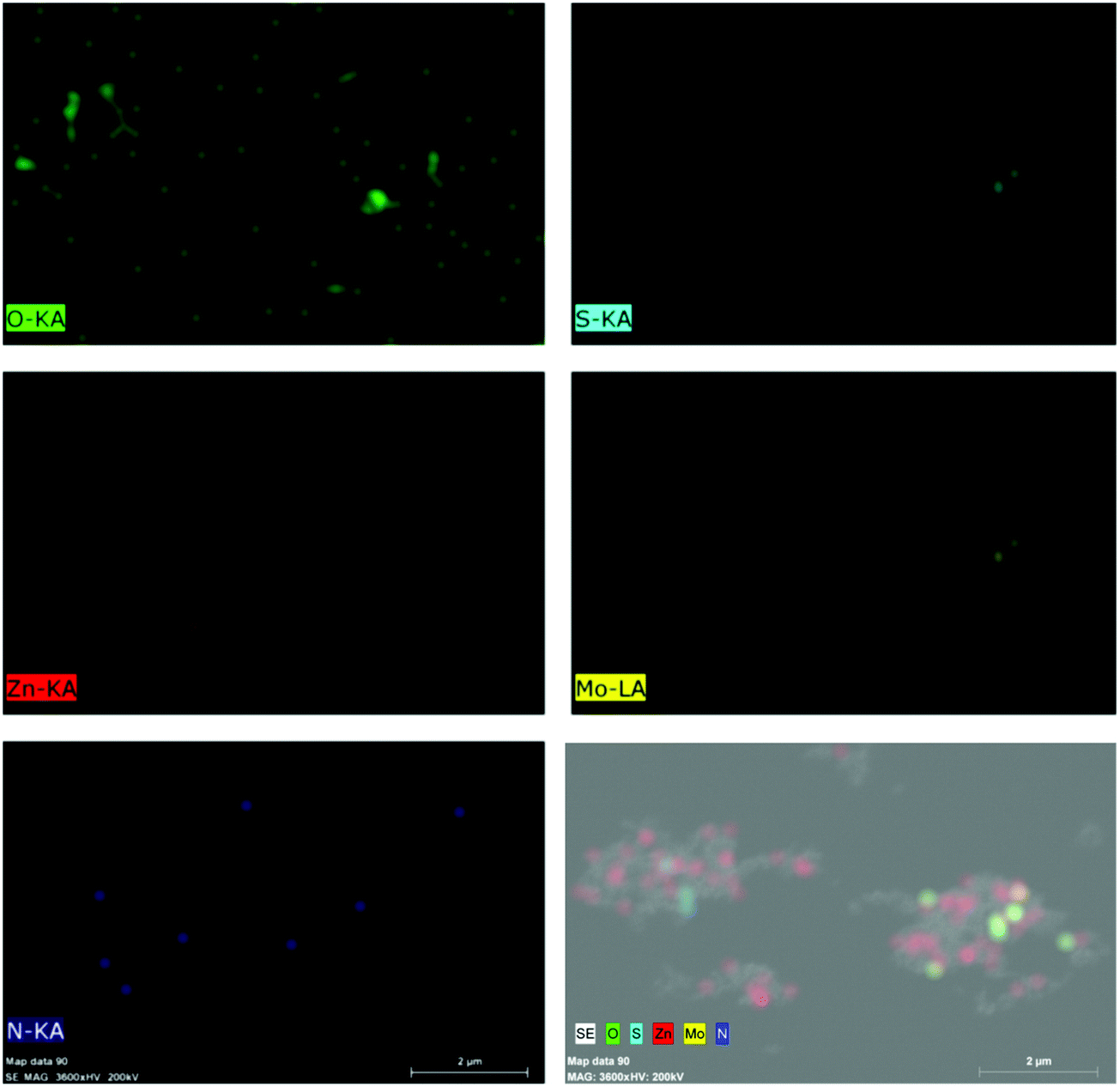
Further, Fig. 4 presents the TEM and HRTEM images of MNFs, N-ZnO NRs and the NZM1 heterojunction along with the EDAX data and Fig. S4 (refer ESI†) presents the TEM images of NZM2 and NZM3 heterojunctions. The HRTEM image of MNFs (Fig. 4b) confirms its hexagonal crystal structure as evidenced by XRD data because the lattice fringes with a d-spacing of 0.62 nm corresponds to the (002) diffraction plane which is the characteristic plane of MNFs. For N-ZnO NRs, the TEM image (Fig. 4c) also confirms its rod-like morphology and the length of around 200 nm, while the HRTEM image of N-ZnO (Fig. 4d) confirms the formation of a hexagonal wurtzite structure with a characteristic diffraction plane (100) with d-spacing corresponding to 0.28 nm. Moreover, N-ZnO NRs are inserted well over MNF petals to form a heterojunction which is clearly evidenced in the TEM images of NZM1 (Fig. 4e and f) where lattice fringes can be seen at the N-ZnO–MNF heterojunction. In the NZM1 heterojunction, N-ZnO NRs are deposited over the petal-like morphology of MNFs, which significantly decreases the groove path of adjacent petals in MNFs, due to which it appears as a sheet-like morphology (Fig. 4e), as evidenced in the literature.45 The presence of all the constituent elements in N-ZnO (N, Zn, O) and NZM1 (N, Zn, O, Mo and S) has been confirmed by EDAX spectra (Fig. 4g and h). Furthermore, the elemental mapping has also been performed on the NZM1 heterojunction, which also confirms the presence of all elements in the heterojunction (Fig. 5).
 |
| | Fig. 4 TEM images of (a) MNFs, (b) MNFs (HR), (c) N-ZnO NRs, (d) N-ZnO NRs (HR), (e) NZM1, and (f) NZM1 (HR), and (g and h) EDAX patterns of N-ZnO and NZM1. | |
 |
| | Fig. 5 Elemental mapping of the NZM1 heterojunction showing the presence of constituent elements (N, Zn, O, Mo and S). | |
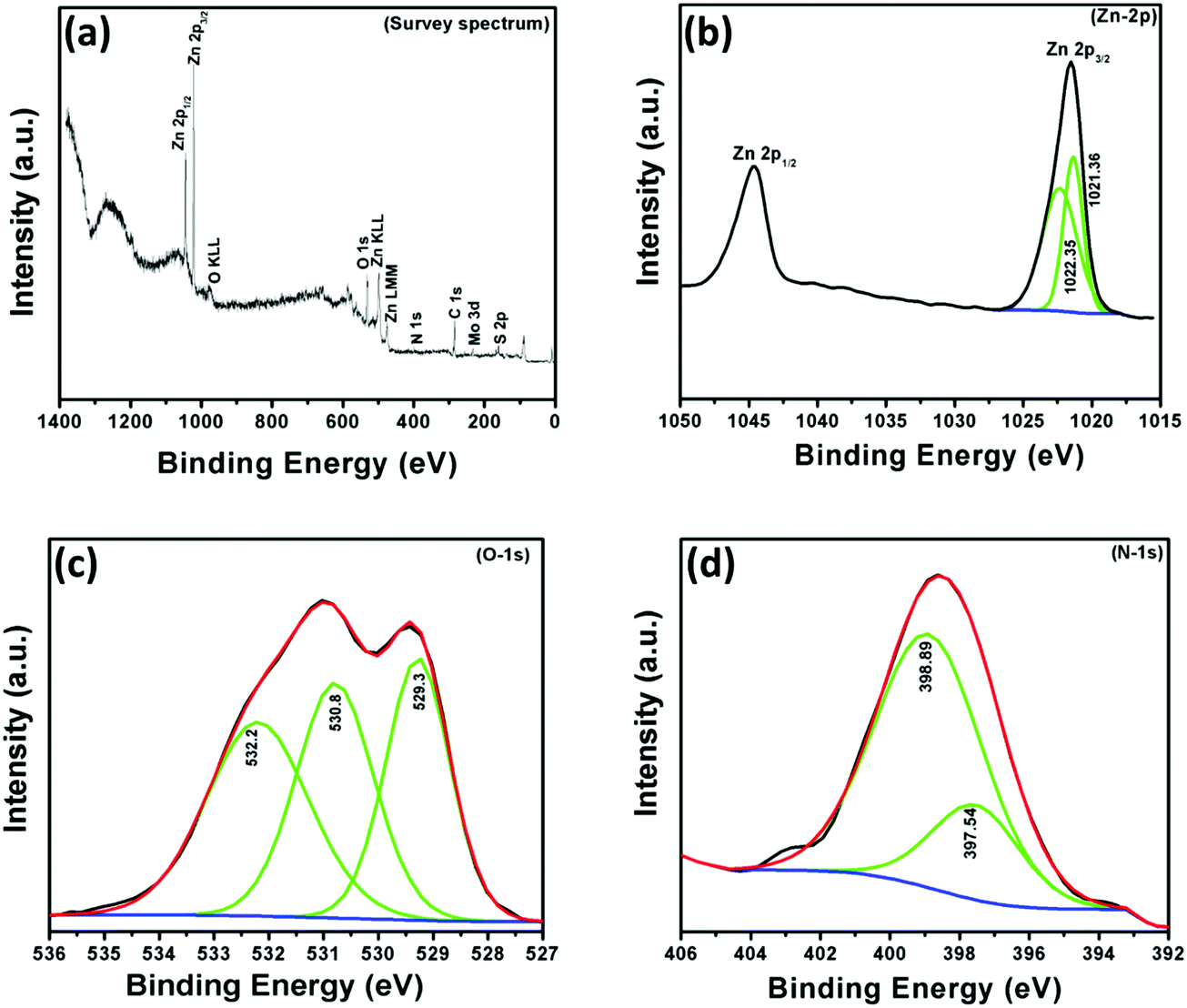
In order to understand the chemical composition, oxidation state and N-doping of ZnO, X-ray photoelectron (XPS) spectroscopic measurements were performed in the region of 0–1400 eV. The survey scan spectrum of the NZM1 heterojunction reveals the presence of Zn, O, N, Mo, and S elements on the sample surface (Fig. 6). It is noteworthy to mention that the elemental analysis made on the basis of XPS shows more of the elements that are present on the surface and does not exactly account for the bulk composition. The elemental analysis shows the presence of both MoS2 and N-doped ZnO, wherein the atomic percentage of nitrogen was determined to be 4.728% in the sample. The binding energy peak at 284.5 eV is the adventitious carbon peak from the instrument and is used as the internal standard. The XPS data for the NZM1 heterojunction can be seen in Fig. 6, the XPS peaks at binding energies 1021.4 eV and 1044.6 eV could be assigned to Zn 2p3/2 and Zn 2p1/2 orbitals of Zn 2p respectively. The Zn 2p3/2 peak could be deconvoluted into two peaks at 1021.36 eV and 1022.35 eV. This shows that Zn is mostly present in two electronic environments. The peak at 1022.35 eV possessing more binding energy should be representative of Zn2+ being bonded to a more electronegative moiety i.e., O of the ZnO lattice;14 however the other binding energy should reflect Zn2+ being bound to N lower in electronegativity than O. The prominent presence of the N-1s peak in the XPS data and the Zn2+ being present at two states definitely hints towards the N being doped in the ZnO lattice in the anionic sites. In addition, O 1s shows three binding energy peaks at 529.3 eV, 530.8 eV and 532.2 eV, which indicates the presence of three different types of oxygen species in the sample. The initial O-peak could be assigned to lattice O contribution of ZnO, while the one at 530.8 eV could be related to the presence of the O (–OH) molecule on the surface of the photocatalyst.14 The doping of N at the anionic sites should result in the formation of several O-vacancies in order to maintain the lattice charge neutrality. The third peak of O 1s at 532.2 eV would be representative of the O-vacancies that will be created in the lattice of ZnO.52 The N 1s XPS peak shows an interesting story. The very presence of the N 1s peak confirms the N-doping at the anionic sites of ZnO. The N 1s band could be deconvoluted into two peaks with binding energies at 398.9 eV and 397.5 eV, respectively. The two peaks represent the N in two different chemical environments, respectively. The higher binding energy peak should represent the O–Zn–N moiety in the ZnO lattice, whereas the lower energy peak would represent that of the Zn–N bond in the N-doped ZnO lattice.53,54 Furthermore, the Mo 3d XPS spectrum (not shown here) has peaks at 232.4 eV and 226.8 eV attributed to Mo 3d3/2 and Mo 3d5/2. The peak at 226.8 eV confirms the presence of the Mo4+ state representing MoS2.45 The presence of N 1s by XPS analysis in two different electronic environments confirms that it has been successfully doped in the crystal lattice of ZnO to replace O. The other XPS data are also conclusive of the above fact only. Usually the peak of Mo4+ is obtained at 228 eV. However in the present heterojunction structure Mo4+ is obtained at 226.8 eV, which is at least suggestive of the fact that definitely the electronic cloud of the Mo is to certain extent perturbed by the N-doped ZnO's Fermi edge, probably causing a Schottky-barrier type between them.
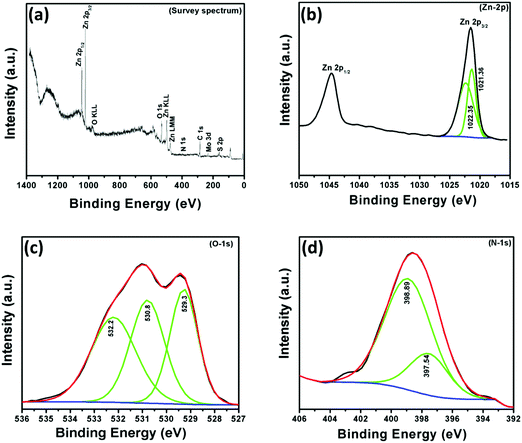 |
| | Fig. 6 X-ray photoelectron (XPS) spectra of NZM1: (a) survey spectrum, (b) Zn-2p, (c) O-1s and (d) N-1s. | |
3.3. Optical property studies
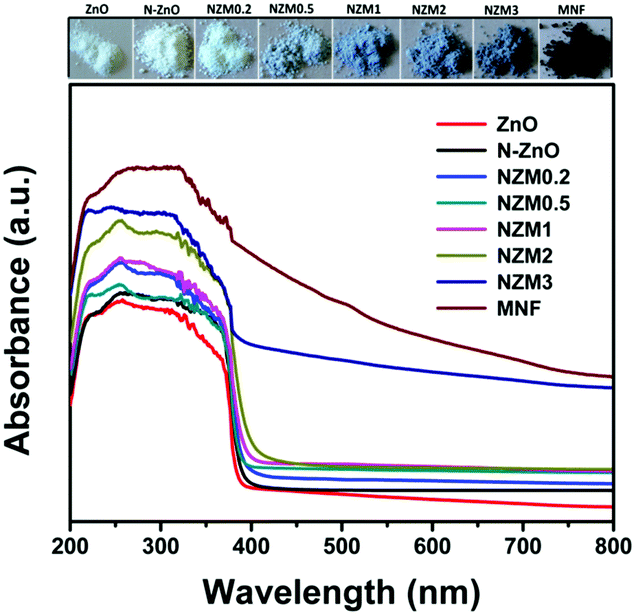
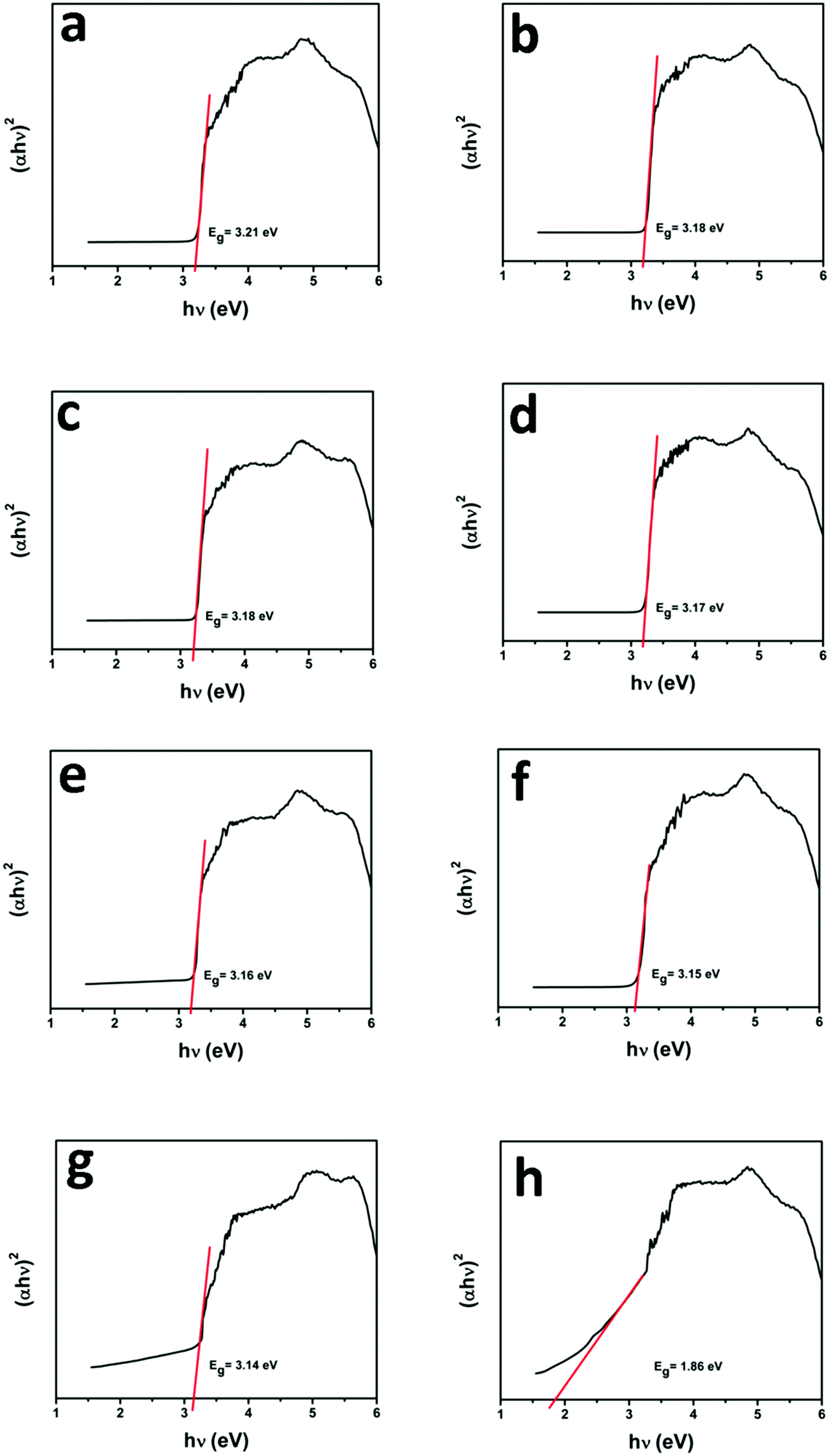
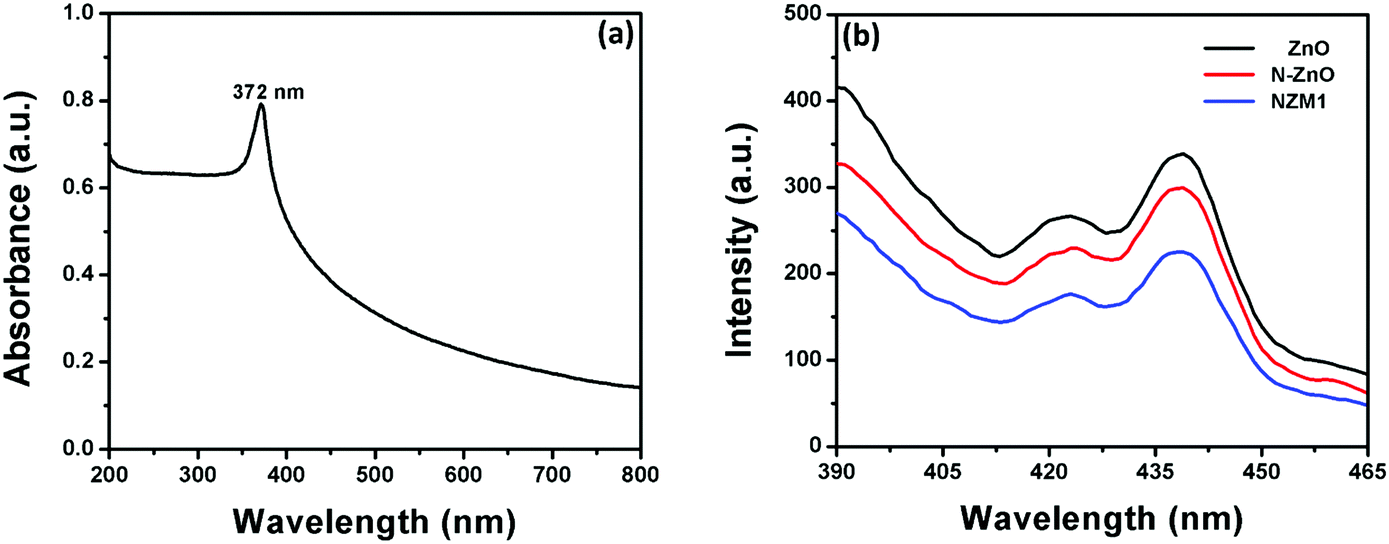
UV-vis diffuse reflectance spectroscopy (DRS) was employed in the range of 200–800 nm to characterize the optical properties of all prepared samples. The optical absorption of all semiconductor materials is directly affected by their electronic band gap structure, which determines the photocatalytic activity of materials. It is clear from Fig. 7 that bare ZnO NRs exhibit the characteristic fundamental absorption edge around 385 nm, which corresponds to the band gap energy of 3.21 eV. It is clear from spectra that ZnO NRs show no absorption in the visible region (λ > 400 nm). N substitution significantly affects the electronic structure of ZnO as its absorption edge shifts to around 390 nm corresponding to the band gap energy of 3.18 eV. Thus after N-doping, the band gap of ZnO is reduced from 3.21 eV to 3.18 eV, which leads to the creation of additional energy levels induced by N-doping on top of the valence band (VB) of ZnO shifting its absorption towards the visible region. MNFs show absorption in the whole range of 200–800 nm. An increase in the content of MNF in N-ZnO influences the light absorption ability of the heterojunction resulting in decreased reflectivity and enhancement of the visible light absorption ability, which is evident from Fig. 8, wherein plots of the transformed Kubelka–Munk function vs. the energy of light (3.18 eV for NZM0.2 and 3.14 eV for NZM3 composition) are shown. Thus DRS measurements clearly reveal that the synergetic effect of N-doping and MNF loading onto ZnO NRs is responsible for the enhanced absorption of the photocatalyst in the visible region and makes better utilization of the solar spectrum which influences its photocatalytic performance.
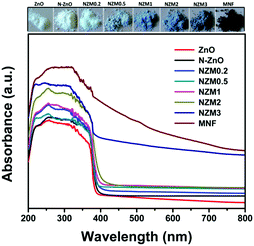 |
| | Fig. 7 UV-vis diffuse reflectance spectra (DRS) of ZnO NRs, N-ZnO, NZM0.2, NZM0.5, NZM1, NZM2, and NZM3. | |
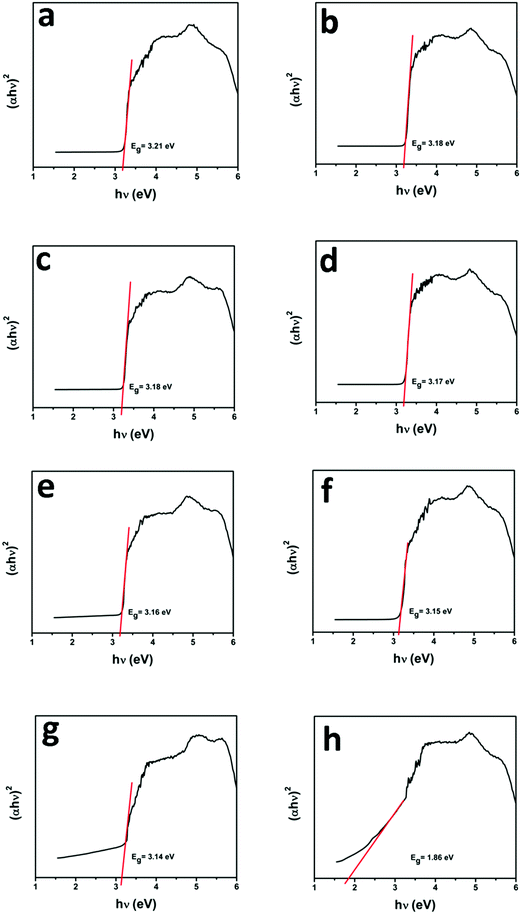 |
| | Fig. 8 Plot of the transformed Kubelka–Munk function vs. the energy of light: (a) ZnO NRs, (b) N-ZnO, (c) NZM0.2, (d) NZM0.5, (e) NZM1, (f) NZM2, (g) NZM3 and (h) MNFs. | |
3.4. Photocatalytic activity studies
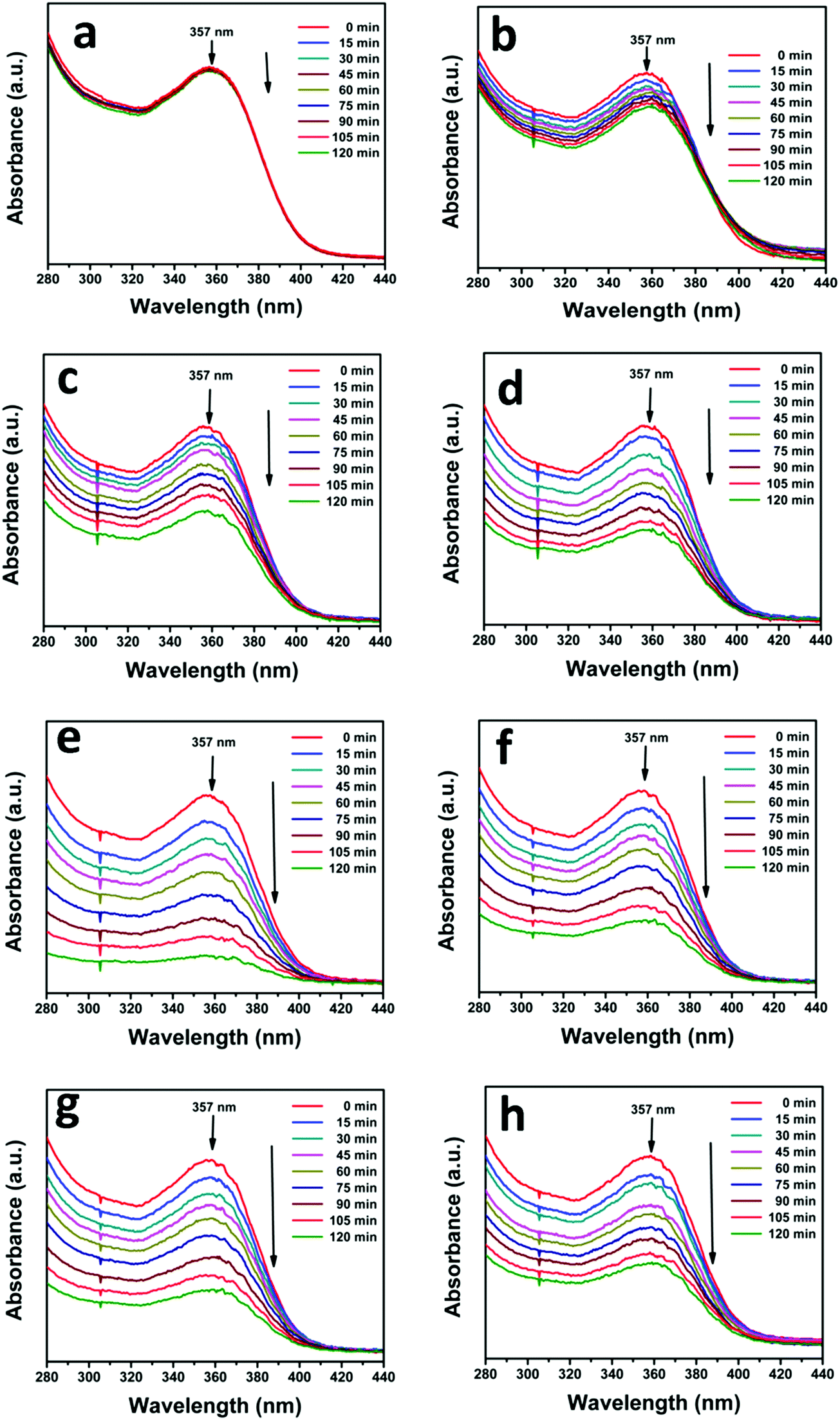
The photocatalytic activity of the as-prepared N-ZnO–MNF heterojunctions was evaluated by investigating the photodegradation of TC antibiotic under visible light irradiation. For this study, the characteristic absorption peak of TC at 357 nm was monitored using a UV-vis spectrophotometer at regular intervals of time, and the corresponding absorption spectra are shown in Fig. 9 for all heterojunctions. Initially the photodegradation of a blank TC solution was examined without a catalyst as a control experiment under visible light irradiation and only negligible degradation was observed.
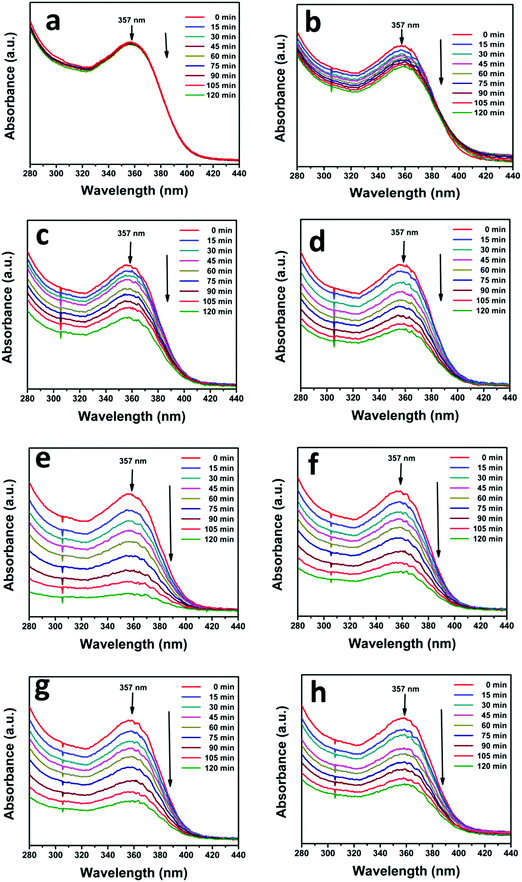 |
| | Fig. 9 Successive time dependent UV-vis spectra of photocatalytic degradation of TC under visible light irradiation (a) without a catalyst and with (b) ZnO NRs, (c) N-ZnO, (d) NZM0.2, (e) NZM0.5, (f) NZM1, (g) NZM2 and (h) NZM3. Experimental conditions: 25 mg of the photocatalyst sample was dispersed in 50 mL of aqueous solution of TC (0.01 g L−1). | |
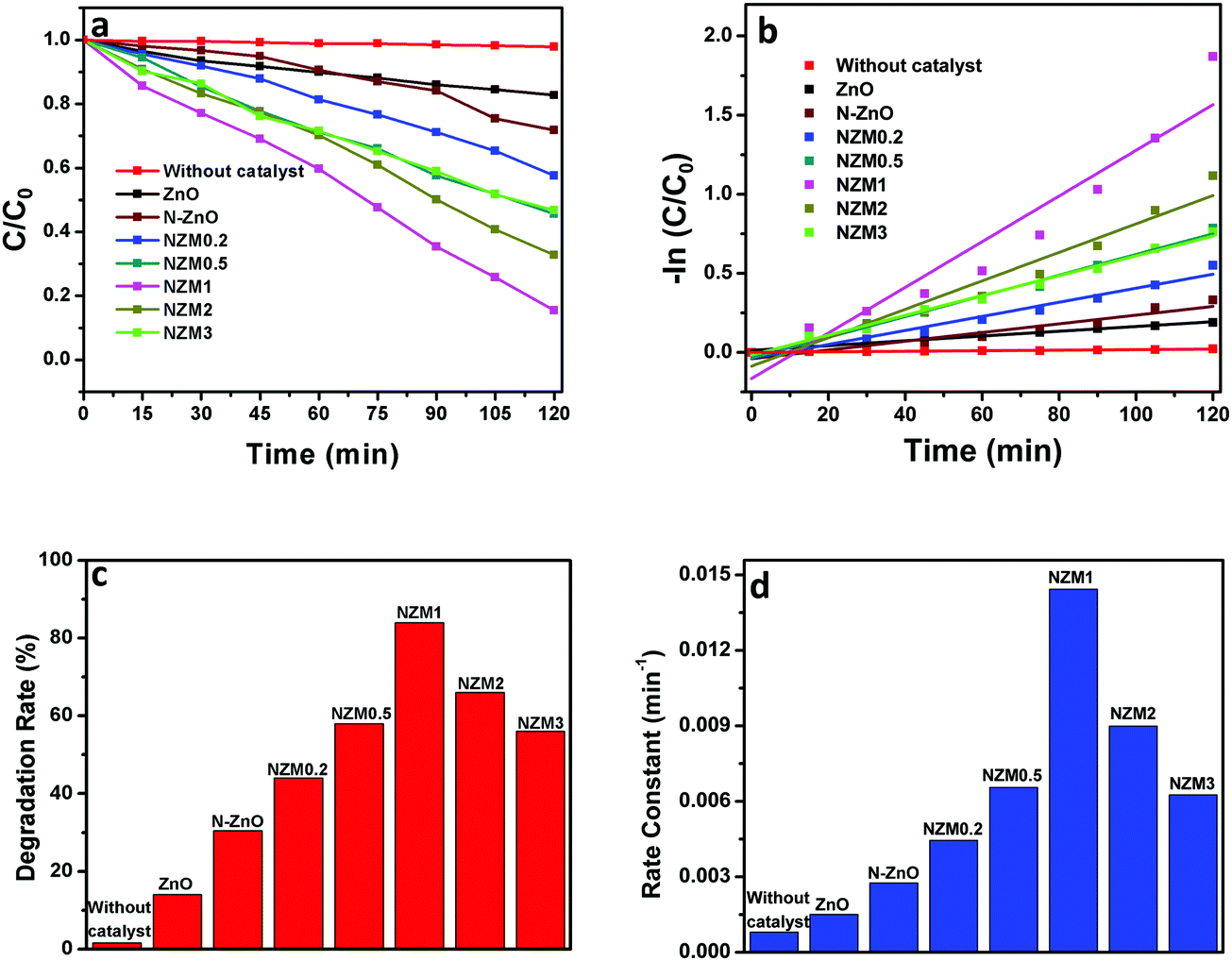
It can be very clearly seen from Fig. 9 that N-ZnO–MNF heterojunctions exhibit considerably higher photocatalytic activity as compared to bare ZnO NRs and N-ZnO NRs for which the 16% and 32% degradation rates were achieved respectively. The degradation rates (%) calculated for NZM0.2, NZM0.5, NZM1, NZM2 and NZM3 were 44%, 58% and 84%, 66% and 56% respectively, for 120 min of irradiation. Hence these results clearly reveal that the increasing MNF loading considerably increases the degradation rate (%) from 44% to 84% in NZM0.2 and NZM1 heterojunctions, respectively, indicating that MNF loading can effectively promote the charge transfer on the interface and improve the photocatalytic performance significantly. The maximum photocatalytic activity was achieved with a heterojunction having 1 wt% of MNF loading onto N-ZnO NRs. The photocatalytic degradation of TC follows the pseudo first order kinetics as per the Langmuir–Hinshelwood model, ln(C/C0) = −kt, and the corresponding C/C0 plots as a function of time for all the samples are presented in Fig. 10(a and b). Furthermore, Fig. 10(c and d) presents the comparative degradation rates (%) of all prepared samples for TC in a histogram along with their rate constant values. The pseudo first order rate constant (k), half-life time of reaction (t1/2) and the linear regression coefficient (R2) for all the catalysts under visible light irradiation are presented in Table 1.
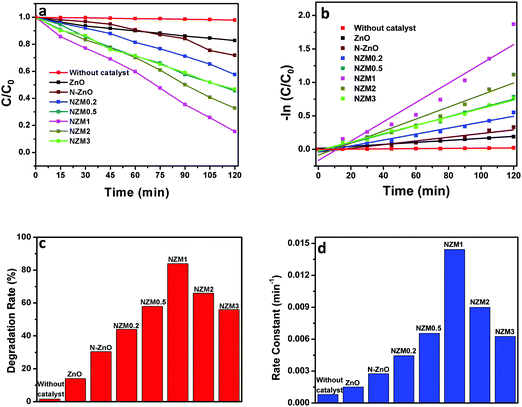 |
| | Fig. 10 (a) Kinetic curves for the degradation of TC, (b) a −ln(C/C0) vs. time curve for degradation of TC, (c) a histogram showing a comparative degradation rate (%) of TC under visible light illumination and (d) a bar graph showing the values of rate constants for all the photocatalysts. | |
Table 1 Summary of kinetic data for the photocatalytic degradation of TC using prepared photocatalysts under visible light irradiation
| Sr. no. |
Photocatalyst |
Rate constant k (min−1) |
t
1/2 (min−1) |
R
2
|
| 1 |
Without catalyst |
0.17 × 10−3 |
4024 |
0.9732 |
| 2 |
ZnO |
1.51 × 10−3 |
459 |
0.9884 |
| 3 |
N-ZnO |
2.76 × 10−3 |
251 |
0.9128 |
| 4 |
NZM0.2 |
4.45 × 10−3 |
157 |
0.9674 |
| 5 |
NZM0.5 |
6.55 × 10−3 |
105 |
0.9911 |
| 6 |
NZM1 |
14.43 × 10−3 |
48 |
0.9174 |
| 7 |
NZM2 |
8.99 × 10−3 |
77 |
0.9485 |
| 8 |
NZM3 |
6.26 × 10−3 |
110 |
0.9913 |
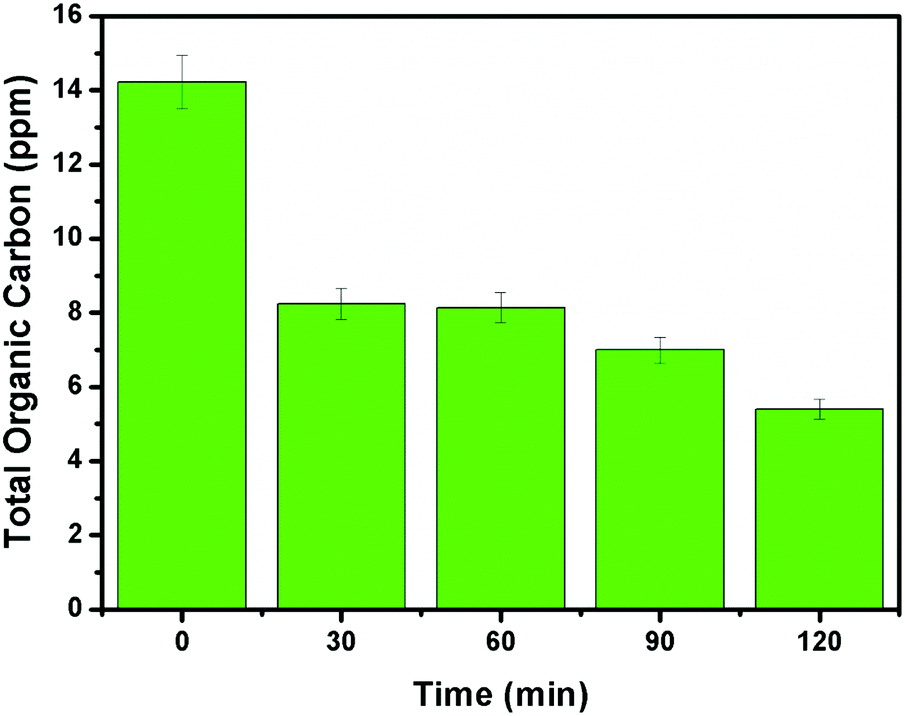
It can be evidenced from Fig. 10d that the rate constant of the NZM1 heterojunction (0.01443 min−1) is about 10 times higher than bare ZnO NRs (0.00151 min−1). In addition, it can be seen clearly from histograms that the photocatalytic activity of N-ZnO for TC degradation increases initially with MNF loading until it reaches 1 wt% and decreases upon further increase in the amount of MNFs. At higher concentrations, MoS2 acts as a recombination center for photogenerated charge carriers and increases the sample opacity, which results in the reduced photocatalytic activity.48 This explains the reason for the superior photocatalytic activity of NZM1 heterojunctions towards antibiotic degradation as compared to other prepared samples under visible light irradiation. Additionally, due to the anisotropic conductivity of MoS2,55 thin layered MoS2 can lessen the poor charge transport from layer to layer and shorten the electron transport time and distance, thus improving the efficiency in the utilization of the photogenerated electrons for TC degradation. Moreover, MoS2 is active in the visible region and the synergetic effect of N-doping and MNF loading efficiently increases the light absorption in the visible region along with the rapid charge transfer across the N-ZnO–MNF heterojunction to accelerate the photocatalytic activity. Furthermore, the oxidative mineralization of TC solution was primarily examined by measuring the total organic carbon (TOC) content as a function of irradiation time in the presence of the photocatalyst. As a representative example, the TOC content determined in the case of the NZM1 heterojunction is presented in Fig. 11, which clearly shows that the TOC profile of TC solution diminishes with irradiation time revealing the fragmentation of the complex molecular structure of TC during photocatalytic reaction.
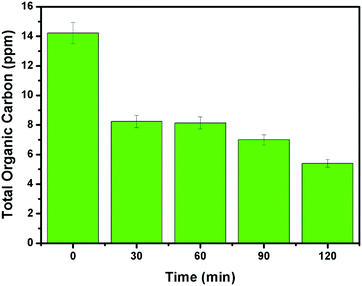 |
| | Fig. 11 Total organic carbon (TOC) content analysis of TC solution as a function of irradiation time. Experimental conditions: [TC]0 = 10 mg L−1; photocatalyst = NZM1 (0.5 g L−1). | |
3.5. Photoluminescence studies
Photoluminescence (PL) spectra of pure ZnO NRs, N-ZnO NRs and the NZM1 heterojunction were measured to determine the extended lifetime of photogenerated charge carriers during the photocatalytic process. It is well known that PL emission intensity results from the recombination of photogenerated charge carriers in semiconductor materials.53Fig. 12b presents the PL emission spectra measured at room temperature. The excitation wavelength was chosen to be 372 nm, as the UV-vis spectrum of ZnO NRs exhibits a strong absorption peak at this wavelength (Fig. 12a). It can be clearly seen from the PL spectra of ZnO NRs that they exhibit the highest emission intensity with a peak around 440 nm, which originates mainly because of the electron–hole recombination, oxygen vacancy and zinc interstitial defects.56 However, the N doping in ZnO NRs reduces the PL intensity in N-ZnO NRs, which clearly indicates the low recombination rate of photogenerated charge carriers and their transfer to reaction sites. The PL spectra of the NZM1 heterojunction shows a notably lower PL emission intensity band as compared to pure ZnO NRs in which the higher recombination rate of photogenerated charge carriers results in high emission intensity. With the loading of MNFs onto N-ZnO, the low PL intensity indicates the lower recombination rate, fast charge transfer across the heterojunction, in addition to enhancing the visible light absorption, which prevent N-ZnO from photocorrosion in long photocatalytic reactions. It is noteworthy to mention here that multilayered MoS2 has no photoluminescence and only the monolayer MoS2 can exhibit photoluminescence properties.45
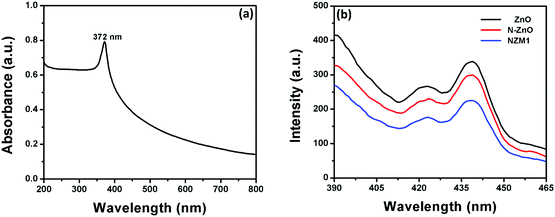 |
| | Fig. 12 (a) UV-visible absorption spectrum of ZnO NRs and (b) PL emission spectra of ZnO NRs, N-ZnO and NZM1. | |
3.6. Mechanism of photocatalytic activities
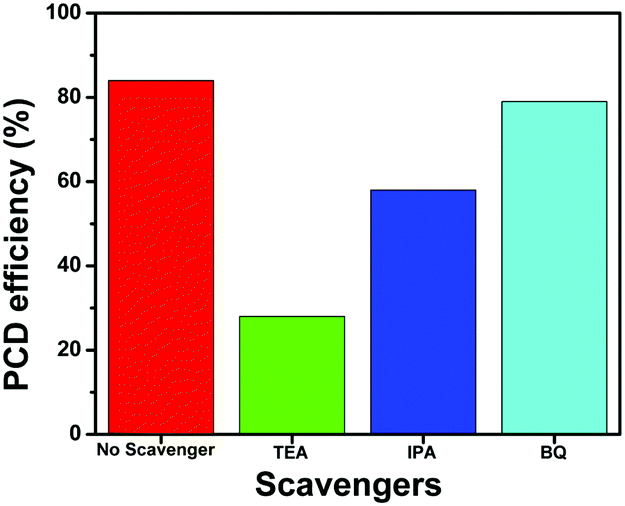
To further investigate the role of active species such as h+, *OH and O2−* in the photocatalytic degradation of TC, the active species trapping experiments were performed using the NZM1 catalyst, as it showed the best catalytic activity among all the heterojunctions. For these experiments, triethanolamine (TEA) was used as a h+ scavenger, isopropanol (IPA) as a *OH scavenger and benzoquinone (BQ) as an O2−* scavenger during the photocatalytic degradation reaction. From the results (Fig. 13), it is clear that with the addition of TEA, photocatalytic degradation drops to 28% indicating the major role played by h+ in TC degradation, while with IPA, the degradation rate drops to around 58% suggesting that *OH also plays an active role in TC degradation. Furthermore, in the presence of BQ, the degradation rate obtained was almost the same as that in the absence of the scavenger (around 80%) indicating that O2−* radicals are not generated during photocatalytic degradation. Thus, from these experiments, we conclude h+ and *OH as major active species in photocatalytic degradation of TC under visible light irradiation which is in good agreement with the obtained photocatalytic activity results described earlier.
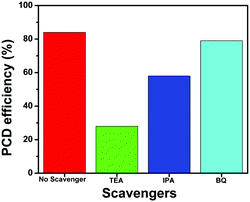 |
| | Fig. 13 Effect of scavengers on the photocatalytic degradation (PCD) efficiency of TC using the NZM1 heterojunction. | |
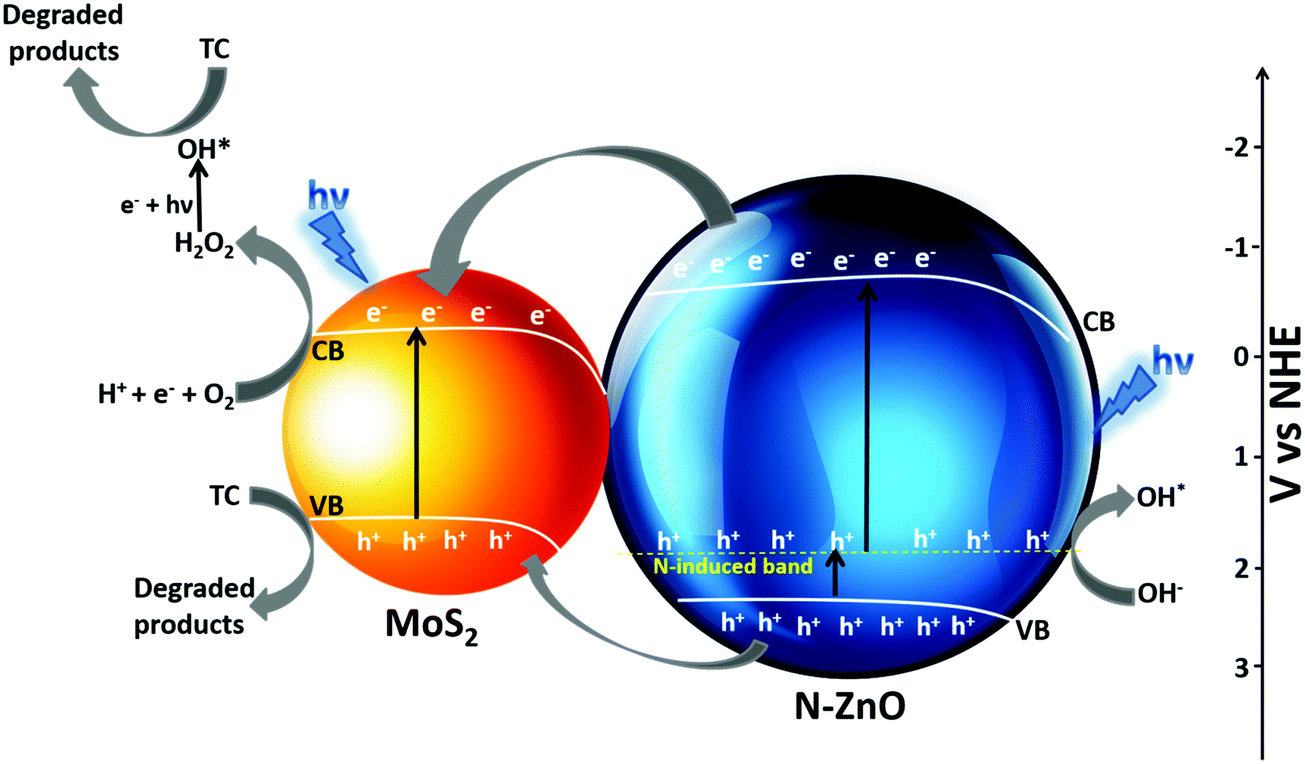
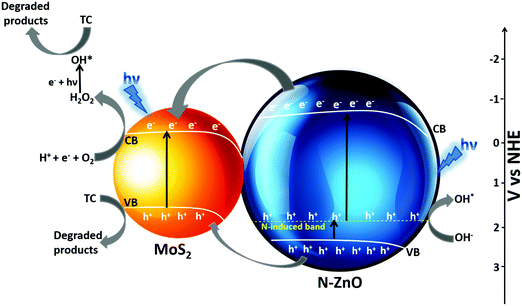
The possible mechanism for photocatalytic degradation of TC over NZM photocatalysts is proposed and presented in Scheme 1, which shows the charge separation and transfer activity at the N-ZnO–MNF heterojunction. The band gap positions (CB and VB) for N-ZnO and MoS2 were calculated by applying the following equations as reported in the literature:10
where
ECB and
EVB are the conduction band and valence band edge potentials,
X is the electronegativity of the semiconductor (ZnO is 5.79 eV and MoS
2 is 5.32 eV);
57Ee is the energy of free electrons on the hydrogen scale (4.5 eV); and
Eg is the band gap energy of the semiconductor (N-ZnO is 3.18 eV and MoS
2 is 1.86 eV) as calculated from DRS data. By applying the above-mentioned equation, the
ECB obtained for N-ZnO and MoS
2 were determined to be −0.3 eV and −0.11 eV, respectively.
 |
| | Scheme 1 Possible mechanism of the photocatalytic activity of the N-ZnO–MNF heterojunction for the degradation of TC under visible light irradiation. | |
The photoinduced charge transfer across the heterojunction is based on the conduction band (CB) edge potential of N-ZnO (−0.3 eV vs. NHE) and the CB edge potential of MoS2 (−0.11 eV vs. NHE). It is well reported in the literature that the loading of MoS2 acts as a photosensitizer molecule in the heterojunction because of which N-doped ZnO can utilize the visible region after sensitization.58 The synergetic effect of MoS2 loading and N doping causes N-ZnO to absorb visible light on illumination, the photon energy higher or equal to the band gap energy of N-ZnO and MoS2 excites the electrons to the CB band from the VB of both the materials, thus electrons are generated in the CB and holes are generated in the VB. The N-induced band near the VB in N-ZnO also contributes to the effective charge separation as holes are easily promoted to it, which are excited finally to the CB of N-ZnO by light absorption. Moreover, some of the holes from the N-induced band can generate the highly reactive OH* which directly degrade TC into degradation products.59 Due to the more negative CB potential of N-ZnO than the CB potential of MoS2, photogenerated electrons from the CB of ZnO are transferred to the CB of MoS2 at the N-ZnO–MNF interface. This is followed by the simultaneous hole transfer from the VB of N-ZnO to the VB of MoS2 as electrons and holes gain energy by moving down and up, respectively, as reported in a previous study.48 Thus MoS2 plays a very significant role in preventing ZnO from photocorrosion and makes it more photostable as holes can photocorrode an oxide catalyst, ZnO + 2h+ → Zn2+ + ½O2.60 The transferred electrons to the CB of MoS2 could not produce O2−* from dissolved O2 by photoreduction because of the more positive CB edge potential of MoS2 (−0.11 eV vs. NHE) than the standard potential of O2/O2−* (−0.33 eV vs. NHE).10 However, the reduction potential of O2/H2O2 was 0.695 eV vs. NHE, which means that electrons can react with O2 and H+ to produce H2O2 which eventually produces *OH. Meanwhile, the photoinduced holes of the VB of MoS2 cannot oxidize adsorbed H2O molecules to produce *OH as per the VB edge potential of MoS2 (1.75 eV vs. NHE), which is more negative than the standard redox potentials of *OH/−OH (2.38 eV vs. NHE) and *OH/H2O (2.72 eV vs. NHE).10 Holes along with OH* radicals, which are very reactive oxidative species degrade TC into degradation products. The whole degradation mechanism can be described as,
| N-ZnO–MoS2 + hν (visible) → N-ZnO–MoS2 (h+ VB + e− CB) |
| N-ZnO (e− CB) → MoS2 (e− CB) |
| N-ZnO (h+ VB) → MoS2 (h+ VB) |
| MoS2 (e− CB) + H+ + O2 → H2O2 |
| H2O2 + e− + hν → *OH + OH− |
| *OH + h+ + TC → degradation products |
Hence it can be concluded that doping ZnO with nitrogen and loading with MNFs result in an extended absorption towards the visible range, suppress the photogenerated charge recombination, and the large surface area of 2D MNFs helps in the fast electron transfer across the heterojunction, which results in the enhanced photocatalytic performance of the photocatalyst towards antibiotic (TC) degradation, in addition to preventing the photocorrosion of N-ZnO. Hence, it is hereby demonstrated that MoS
2 plays a dual role in enhancing the photocatalytic performance and increasing the photostability of the N-ZnO material.
3.7. Stability of the photocatalyst
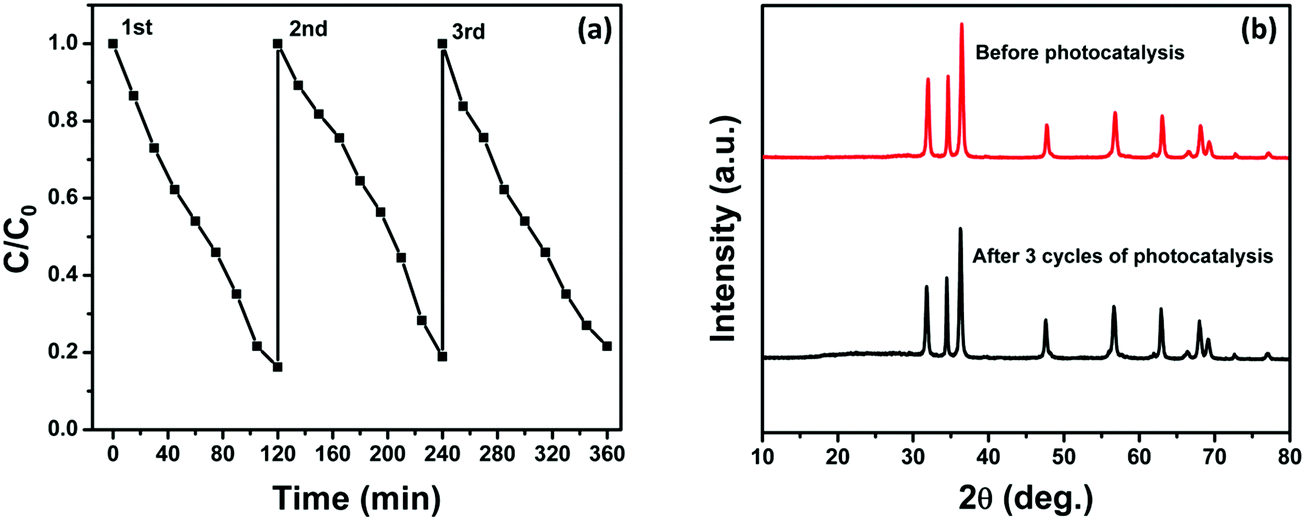
We have also investigated the stability and reusability of the NZM1 photocatalyst over three cycles for a period of 360 min irradiation. Fig. 14a clearly shows only a negligible loss in photocatalytic activity which might be due to the loss of the photocatalyst during each cycle of reusability, which involves the recovery of the photocatalyst by centrifugation followed by washing and then drying. Fig. 14b presents the XRD patterns of the reused photocatalyst after 360 min of irradiation which reveals that the structure of the catalyst remains intact and hence proves the better photostability of the catalyst. Furthermore, it demonstrates that the MoS2 loading makes ZnO resistant to photocorrosion and more stable in long photocatalytic reactions.
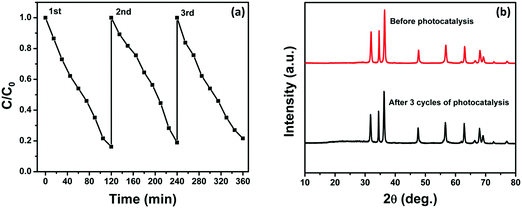 |
| | Fig. 14 (a) Three reaction cycles of photocatalytic degradation of TC by NZM1 and (b) XRD patterns of NZM1 before and after photocatalytic reaction. | |
4. Conclusions
In conclusion, we have reported the successful preparation of N-doped ZnO NRs loaded with MoS2 nanoflowers to form a visible light active N-ZnO–MNF heterojunction by adopting a hydrothermal method. The as-prepared heterojunctions have been thoroughly characterized using powder X-ray diffraction for crystal phase analysis, scanning electron microscopy and transmission electron microscopy for surface morphology, elemental analysis for compositional studies, ultraviolet-visible diffuse reflectance spectroscopy and photoluminescence for optical properties. The synergetic effect of both N-doping and MNF loading have been found to provide more reaction sites due to the high surface area of 2D MNFs, increased absorption in visible light and prevents ZnO from photocorrosion by rapid interfacial charge transfer across the N-ZnO–MNF heterojunction. The prepared heterojunction shows excellent activity towards the photocatalytic degradation of TC antibiotic under visible light irradiation and composition having 1 wt% of MNF (NZM1) showed the best TC degradation rate (84%) as compared to the bare ZnO NRs and other reported compositions (NZM0.2, NZM0.5, NZM2 and NZM3). Thus this study provides a detailed insight into the fabrication of visible light active doped ZnO based materials with enhanced photocatalytic performance and photostability.
Acknowledgements
We are thankful to the Advanced Materials Research Centre (AMRC), IIT Mandi for laboratory and the characterization facilities. VK acknowledges the financial support from the Department of Science and Technology, India under the INSPIRE Faculty Award. SK acknowledges the Research Fellowship from University Grants Commission (UGC), India.
References
-
M. E. Assessment, Ecosystems and human well-being, Island Press, Washington, DC, 2003 Search PubMed.
- C. Liu, G. Wu, J. Chen, K. Huang and W. Shi, New J. Chem., 2016, 40, 5198–5208 RSC.
- T. Jing, Y. Wang, Q. Dai, H. Xia, J. Niu, Q. Hao, S. Mei and Y. Zhou, Biosens. Bioelectron., 2010, 25, 2218–2224 CrossRef CAS PubMed.
- P. T. P. Hoa, S. Managaki, N. Nakada, H. Takada, A. Shimizu, D. H. Anh, P. H. Viet and S. Suzuki, Sci. Total Environ., 2011, 409, 2894–2901 CrossRef CAS PubMed.
- Y.-J. Lee, S.-E. Lee, D. S. Lee and Y.-H. Kim, Environ. Toxicol. Pharmacol., 2008, 26, 216–221 CrossRef CAS PubMed.
- R. Hao, X. Xiao, X. Zuo, J. Nan and W. Zhang, J. Hazard. Mater., 2012, 209, 137–145 CrossRef PubMed.
- X. Wu, Y. Wei, J. Zheng, X. Zhao and W. Zhong, Bioresour. Technol., 2011, 102, 5924–5931 CrossRef CAS PubMed.
- X. Liu, P. Lv, G. Yao, C. Ma, P. Huo and Y. Yan, Chem. Eng. J., 2013, 217, 398–406 CrossRef CAS.
- L. F. Da Silva, J.-C. M'Peko, J. Andres, A. Beltrán, L. Gracia, M. I. Bernardi, A. Mesquita, E. Antonelli, M. L. Moreira and V. R. Mastelaro, J. Phys. Chem. C, 2014, 118, 4930–4940 CAS.
- M. Yan, Y. Wu, F. Zhu, Y. Hua and W. Shi, Phys. Chem. Chem. Phys., 2016, 18, 3308–3315 RSC.
- S. Balachandran, N. Prakash, K. Thirumalai, M. Muruganandham, M. Sillanpää and M. Swaminathan, Ind. Eng. Chem. Res., 2014, 53, 8346–8356 CrossRef CAS.
- V. Sharma, S. Kumar and V. Krishnan, ChemistrySelect, 2016, 1, 2963–2970 CrossRef CAS.
- V. Sharma, S. Kumar and V. Krishnan, Mater. Chem. Phys., 2016, 179, 129–136 CrossRef CAS.
- S. Kumar, A. Baruah, S. Tonda, B. Kumar, V. Shanker and B. Sreedhar, Nanoscale, 2014, 6, 4830–4842 RSC.
- Y. Li, W. Xie, X. Hu, G. Shen, X. Zhou, Y. Xiang, X. Zhao and P. Fang, Langmuir, 2009, 26, 591–597 CrossRef PubMed.
- Y. Peng, S. Qin, W.-S. Wang and A.-W. Xu, CrystEngComm, 2013, 15, 6518–6525 RSC.
- Q. Wang, B. Geng and S. Wang, Environ. Sci. Technol., 2009, 43, 8968–8973 CrossRef CAS PubMed.
- R. Saravanan, N. Karthikeyan, V. Gupta, E. Thirumal, P. Thangadurai, V. Narayanan and A. Stephen, Mater. Sci. Eng., C, 2013, 33, 2235–2244 CrossRef CAS PubMed.
- C. Yu, K. Yang, Y. Xie, Q. Fan, C. Y. Jimmy, Q. Shu and C. Wang, Nanoscale, 2013, 5, 2142–2151 RSC.
- B. Li and H. Cao, J. Mater. Chem., 2011, 21, 3346–3349 RSC.
- B. Li, T. Liu, Y. Wang and Z. Wang, J. Colloid Interface Sci., 2012, 377, 114–121 CrossRef CAS PubMed.
- J. Li, L. Wei, C. Yu, W. Fang, Y. Xie, W. Zhou and L. Zhu, Appl. Surf. Sci., 2015, 358, 168–174 CrossRef CAS.
- S. Kumar, R. Sharma, V. Sharma, G. Harith, V. Sivakumar and V. Krishnan, Beilstein J. Nanotechnol., 2016, 7, 1684–1697 CrossRef.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. Hong, A. Manivannan, M. Zhi, M. Li and N. Wu, J. Am. Chem. Soc., 2009, 131, 12290–12297 CrossRef CAS PubMed.
- H. Fu, T. Xu, S. Zhu and Y. Zhu, Environ. Sci. Technol., 2008, 42, 8064–8069 CrossRef CAS PubMed.
- C. Yu, G. Li, S. Kumar, K. Yang and R. Jin, Adv. Mater., 2014, 26, 892–898 CrossRef CAS PubMed.
- C. Yu, W. Zhou, L. Zhu, G. Li, K. Yang and R. Jin, Appl. Catal., B, 2016, 184, 1–11 CrossRef CAS.
- C. Yu, W. Zhou, H. Liu, Y. Liu and D. D. Dionysiou, Chem. Eng. J., 2016, 287, 117–129 CrossRef CAS.
- C. Yu, L. Wei, W. Zhou, D. D. Dionysiou, L. Zhu, Q. Shu and H. Liu, Chemosphere, 2016, 157, 250–261 CrossRef CAS PubMed.
- C. Yu, L. Wei, J. Chen, Y. Xie, W. Zhou and Q. Fan, Ind. Eng. Chem. Res., 2014, 53, 5759–5766 CrossRef CAS.
- Z. Wang, B. Huang, Y. Dai, X. Qin, X. Zhang, P. Wang, H. Liu and J. Yu, J. Phys. Chem. C, 2009, 113, 4612–4617 CAS.
- S. Balachandran and M. Swaminathan, J. Phys. Chem. C, 2012, 116, 26306–26312 CAS.
- W. Wu, S. Zhang, X. Xiao, J. Zhou, F. Ren, L. Sun and C. Jiang, ACS Appl. Mater. Interfaces, 2012, 4, 3602–3609 CAS.
- M. T. Uddin, Y. Nicolas, C. L. Olivier, T. Toupance, L. Servant, M. M. Müller, H.-J. Kleebe, J. R. Ziegler and W. Jaegermann, Inorg. Chem., 2012, 51, 7764–7773 CrossRef CAS PubMed.
- S. Mukhopadhyay, I. Mondal, U. Pal and P. S. Devi, Phys. Chem. Chem. Phys., 2015, 17, 20407–20415 RSC.
- Y. Ma, Y. Guo, H. Jiang, D. Qu, J. Liu, W. Kang, Y. Yi, W. Zhang, J. Shi and Z. Han, New J. Chem., 2015, 39, 5612–5620 RSC.
- H. Abdullah, D.-H. Kuo, Y.-R. Kuo, F.-A. Yu and K.-B. Cheng, J. Phys. Chem. C, 2016, 120, 7144–7154 CAS.
- B. Radisavljevic, A. Radenovic, J. Brivio, I. V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- S. Kumar, V. Sharma, K. Bhattacharyya and V. Krishnan, New J. Chem., 2016, 40, 5185–5197 RSC.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4 Search PubMed.
- H. Li, K. Yu, X. Lei, B. Guo, H. Fu and Z. Zhu, J. Phys. Chem. C, 2015, 119, 22681–22689 CAS.
- K. Huang, Y. Li, S. Lin, C. Liang, H. Wang, C. Ye, Y. Wang, R. Zhang, D. Fan and H. Yang, Powder Technol., 2014, 257, 113–119 CrossRef CAS.
- T. Yi-hao, Z. Hang, W. Yin, D. Ming-hui, J. Guo and Z. Bin, Micro Nano Lett., 2015, 10, 432–434 Search PubMed.
- Y. Min, G. He, Q. Xu and Y. Chen, J. Mater. Chem. A, 2014, 2, 2578–2584 CAS.
- K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS PubMed.
- J. Zhang, L. Sun, J. Yin, H. Su, C. Liao and C. Yan, Chem. Mater., 2002, 14, 4172–4177 CrossRef CAS.
- F. Manjón, B. Mari, J. Serrano and A. Romero, J. Appl. Phys., 2005, 97, 053516 CrossRef.
- G. Wang, K. Bhattacharyya, J. Parrondo and V. Ramani, Chem. Eng. Sci., 2016, 154, 81–89 CrossRef CAS.
- A. Bhirud, S. Sathaye, R. Waichal, C.-J. Park and B. Kale, J. Mater. Chem. A, 2015, 3, 17050–17063 CAS.
- W. Zhou, K. Zhou, D. Hou, X. Liu, G. Li, Y. Sang, H. Liu, L. Li and S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 21534–21540 CAS.
- H. Tributsch and J. Bennett, J. Electroanal. Chem. Interfacial Electrochem., 1977, 81, 97–111 CrossRef CAS.
- H. Zeng, W. Cai, J. Hu, G. Duan, P. Liu and Y. Li, Appl. Phys. Lett., 2006, 88, 171910 CrossRef.
- Y. Xu and M. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- N. Tian, Z. Li, D. Xu, Y. Li, W. Peng, G. Zhang, F. Zhang and X. Fan, Ind. Eng. Chem. Res., 2016, 55, 8726–8732 CrossRef CAS.
- S. Kuriakose, B. Satpati and S. Mohapatra, Phys. Chem. Chem. Phys., 2015, 17, 25172–25181 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00274a |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here.