
Robust porous polymers enabled by a fast trifluoroacetic acid etch with improved selectivity for polylactide†
Amrita
Sarkar
and
Morgan
Stefik
 *
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina 29208, USA. E-mail: morgan@stefikgroup.com
First published on 20th February 2017
Abstract
Polylactide is a widely used sacrificial block for the preparation of porous polymers from ordered block copolymers. Although numerous etching strategies were developed in the past decade, demonstrations to date are limited by slow etch rates that require as long as a week for the etching of few-mm thick films. Recent studies have also shown that NaOH etching of thin-films can degrade the morphology, highlighting the need for more selective processes. Here we report an aqueous trifluoroacetic acid etchant that results in an enhanced etch rate of 14 nm s−1 with greatly improved selectivity for poly(styrene-block-lactide). The high etch rate enables the complete removal of polylactide from 2 mm thick block copolymer films in 19 h. Furthermore, the improved etch selectivity enables the macroscopic preservation of morphologies as confirmed by both SAXS and SEM and yields pristine porous PS as confirmed by NMR and GPC.
Introduction
Mesoporous materials derived from block copolymers are crucial for numerous applications including size selective separations such as ultrafiltration for water treatment,1–3 controlled delivery of drugs,4–6 templating of surface patterns,7,8 and production of functional inorganic nanostructures.9–13 The self-assembly of block copolymers into ordered morphologies provides a scalable approach for the synthesis of ordered materials with uniform, nanoscale pores14–19 and high specific surface areas that may be chemically functionalized.20,21 The selective etching of a sacrificial block is the most widespread method of producing such porous polymers, with polylactide emerging as one of the most widely used sacrificial blocks.20–25 Despite numerous etching chemistries for poly(styrene-b-lactide) (PS-b-PLA) type polymers reported in the literature,23,24,26,27 there remain challenges in balancing the extent of an etch process against the preservation of the nanostructure. For example, a widely used alkaline etch route23,24 has been noted to damage the quality of thin-film pattern transfer after 15 minutes of mild 0.05 M NaOH contact, limiting thin film pattern retention to films less than 50 nm.27,28 The etching of thick polymer films is also challenging where high selectivity of the etchant is crucial to limit the degradation of the retained PS block while providing enough exposure time to etch through the PLA and across multiple grain boundaries. Etching through <1 mm thick PS-b-PLA with 0.5 M NaOH typically requires about 3–5 days, corresponding to an average etch rate of 1–1.5 nm s−1.12,13,23,24 For example, complete removal of PLA from <0.32 mm thick films required shear alignment of the morphology to reduce the grain boundary concentration and a 44 h etch with 0.5 M NaOH.23The preservation of chemical integrity of the PS matrix is crucial for nanostructure retention through a complete etch process. Here, we note that the ideal evidence for nanostructure retention over macroscopic regions is an ensemble measurement such as SAXS that samples a few mm3 to quantitatively confirm the preserved lattice parameters over ∼1013 unit cells, rather than localized microscopy data.12,13,23,24,29–33 There are examples of PS-b-PLA12 and related poly(norbornenylethylstyrene-styrene-b-lactide) (PNS-b-PLA)34 films that exhibited a shift in the SAXS pattern after NaOH etching. Such shifts of SAXS peak positions demonstrate a change in the scattering structure factor and are indicative of morphology shrinkage. Reports of PLA etching to date have yet to demonstrate the complete removal of PLA from ordered films thicker than 0.32 mm while preserving the morphology as confirmed by an ensemble measurement such as SAXS.
An ideal etchant would (1) quickly hydrolyze the PLA backbone while (2) maintaining orthogonality towards other blocks such as PS. Trifluoroacetic acid (TFA) is a widely used agent for hydrolytic ester cleavage in preparative organic chemistry owing to its high acidity (low pKa) and concomitant miscibility with both water and organic solvents. The latter criterion is subtly important where surface wetting of the resulting hydrophobic PS pores is a crucial factor for etching beyond the perimeter.35,36 Despite the remaining challenges for etching PS-b-PLA, TFA has not yet been investigated as an etchant for porous polymers. We note that TFA was previously used for hydrolysis of homogeneous solutions of PLA-based block copolymers.37 Here, we report a significantly higher etch rate of 14 nm s−1 using TFA to enable the complete etching of 2 mm thick films in less than 1 day while quantitatively preserving the starting morphology and without observable degradation of the remaining PS.
Experimental
Materials
Styrene (99%, Acros Organics) was used after passage through a basic alumina column. 3,6-Dimethy-1,4-dioxane-2,5-dione (99%, Acros Organics) or D,L-lactide was dried overnight in a vacuum oven at 40 °C just before use. 2-Hydroxyethyl-2-bromoisobutyrate (95%, Aldrich), tris-[2-(dimethylamino)ethyl]amine (97%, Aldrich), tin(II) ethylhexanoate (92%, Aldrich), benzoic acid (99.5%, Aldrich), trifluoroacetic acid (99.5%, Acros Organics) and sodium hydroxide (97%, pellet, Sigma Aldrich) were used as received. Copper(I) bromide (99.99%, Aldrich), cyclohexylamine (99%, Alfa Aesar), 3,5-bis(trifluoromethylphenyl isothiocyanate) (98%, Aldrich), and anhydrous chloroform (stabilized with amylene, 99.9%, Acros Organics) were stored inside a glove box and used as received. 1,8-Diazabicyclo[5.4.0]undec-7-ene (98%, Aldrich) was dried over calcium hydride and then filtered in a glovebox using a nylon syringe filter (0.45 μm). Tetrahydrofuran (99.5%, Macron), methanol (99.8%, BDH), and toluene (99.5%, Macron) were used as received.Synthesis of hydroxyl terminated polystyrene (PS-OH) macroinitiator
The PS-OH macroinitiator was synthesized via ARGET-ATRP using a reagent ratio of [styrene]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[2-hydroxyethylbromoisobutyrate]:[Me6TREN]:[Cu(I)]:[Sn(II)] = 300:1:0.105:0.005:0.1. To a Schlenk flask, 66 mL of inhibitor free styrene (576 mmol) and 278 μL of 2-hydroxy-2-ethylbromoisobutyrate (1.92 mmol) were added. The flask was degassed by three freeze–pump–thaw (FPT) cycles and backfilled with nitrogen (N2) gas. A catalyst stock solution of 1 mL of toluene containing 1.4 mg of Cu(I)Br (0.0096 mmol), 53.8 μL (0.2 mmol) of Me6TREN ligand and 62.2 μL of Sn(II) ethylhexanoate (0.192 mmol) was added to the reaction flask under flowing N2 gas. This reaction mixture was then placed into a pre-heated oil bath with constant stirring. The polymerization was continued for 10 h and at the end the reaction mixture was cooled with ice water before exposing the solution to air. The crude polymerization solution was diluted with THF and precipitated two times into 10 fold excess of cold methanol. The product was filtered and vacuum dried at 50 °C for 24 h. The number average molecular weight (Mn) of PS-OH of 12.9 kg mol−1 and molar mass dispersity Đ = 1.21 were determined using a PS calibrated GPC.
:[2-hydroxyethylbromoisobutyrate]:[Me6TREN]:[Cu(I)]:[Sn(II)] = 300:1:0.105:0.005:0.1. To a Schlenk flask, 66 mL of inhibitor free styrene (576 mmol) and 278 μL of 2-hydroxy-2-ethylbromoisobutyrate (1.92 mmol) were added. The flask was degassed by three freeze–pump–thaw (FPT) cycles and backfilled with nitrogen (N2) gas. A catalyst stock solution of 1 mL of toluene containing 1.4 mg of Cu(I)Br (0.0096 mmol), 53.8 μL (0.2 mmol) of Me6TREN ligand and 62.2 μL of Sn(II) ethylhexanoate (0.192 mmol) was added to the reaction flask under flowing N2 gas. This reaction mixture was then placed into a pre-heated oil bath with constant stirring. The polymerization was continued for 10 h and at the end the reaction mixture was cooled with ice water before exposing the solution to air. The crude polymerization solution was diluted with THF and precipitated two times into 10 fold excess of cold methanol. The product was filtered and vacuum dried at 50 °C for 24 h. The number average molecular weight (Mn) of PS-OH of 12.9 kg mol−1 and molar mass dispersity Đ = 1.21 were determined using a PS calibrated GPC.
Synthesis of co-catalyst bis(3,5-trifluoromethyl)phenylcyclohexylthiourea
The ROP co-catalyst was synthesized following a reported procedure.38 The synthesis was performed in a glovebox where 3,5-bis(trifluoromethyl)phenylisothiocyanate (3.37 mL, 18.5 mmol) and anhydrous THF (20 mL) were combined in a round bottom flask that was previously dried using a heat-gun and contained a magnetic stir bar. In the glovebox, cyclohexylamine (2.11 mL, 18.5 mmol) was added to the stirring solution dropwise via a syringe. The reaction continued at room temperature for 24 h and then THF was removed using a rotary evaporator. The white residue was recrystallized twice from chloroform and stored in the glove box after drying under vacuum at 50 °C for 48 h.Synthesis of PS-b-PLA
The PS-OH macroinitiator was chain extended from the terminal hydroxyl group via organocatalytic ROP using a reagent ratio of [PS-OH]:[lactide]:[DBU]:[thiourea] = 1:250:1.34:1.34. Before starting the reaction, the PS-OH macroinitiator and D,L-lactide monomer were each dried separately overnight at 40 °C under high vacuum before transfer to a glove box via a sealed Schlenk flask. The PS-OH (1 g, 77.1 μmol) and lactide monomer (2.8 g, 19.3 mmol) were combined with thiourea co-catalyst (37 mg, 103 μmol). All the reagents were dissolved in 5 mL of anhydrous chloroform. After dissolution, the DBU catalyst (41 μL, 103 μmol) was added to the polymerization mixture dropwise and the reaction was stirred at RT for 60 min. The reaction was terminated by adding benzoic acid (12.6 mg, 103 μmol). The polymer solution was precipitated twice into 10 fold excess of cold methanol. The white solid was collected by filtration and dried under vacuum at 40 °C for 24 h. The molar mass was determined to be Mn = 41.6 kg mol−1 with PLA Mn = 28.6 kg mol−1 (79% monomer conversion), as calculated from 1H NMR results. GPC confirmed a narrow molar mass dispersity Đ = 1.16.
Sample preparation
PS-b-PLA polymer films were prepared by solution casting from toluene (10 wt% of polymer, filtered through a 0.2 μm filter) into a Teflon dish placed on a hot plate set to 50 °C. The film was left to dry for 8 h and then transferred to a vacuum oven set to 40 °C for another 2 h to remove trace solvent. The dried sample was annealed at 110 °C for 1 h in a vacuum oven. The oven was vented to atmospheric pressure and the sample quenched to room temperature. The resulting “as-made” sample was used for subsequent etching experiments.PLA etching by TFA immersion
A portion of the as-made film 2 mm in thickness and >1 cm2 in area was immersed in a glass vial containing 10 mL of TFA solution. The sample etch rates were compared by maintaining a constant minimum sample dimension of 2 mm in thickness (see section PLA etch kinetics). TFA etchant solutions were prepared using a range of molarities from 0.01 to 8 M, as prepared in a 70:30 mixture by volume of methanol:water. The vial containing etchant and sample was placed in an oven pre-heated to 40 °C for the desired amount of time. After this etch treatment, the film was rinsed thoroughly with water and immersed in methanol for 10 minutes before drying in a vacuum oven set to 30 °C for 4 h. The procedure steps are shown in Fig. S6, ESI.†
PLA etching by TFA vapor
A portion of the as-made film 2 mm in thickness and ∼>1 cm2 in area was placed in an aluminum dish. The same range of TFA etchant solutions were examined for vapor etching. The resulting TFA solution (10 mL) was placed in a glass vial. The aluminum dish and glass vial were both placed within a plastic jar and the entire assembly was placed within an oven pre-heated to 40 °C for a predetermined amount of time. After this etch treatment, the film was rinsed thoroughly with water and immersed in methanol for 10 minutes before drying in a vacuum oven at 30 °C for 4 h. The procedure steps are shown in Fig. S7, ESI.†PLA etching by 0.5 M NaOH
A portion of the as-made film was immersed in 0.5 M NaOH in a 40:60 mixture by volume of methanol:water and etching was conducted for 3 days at 60 °C.12,13,23,24 After this etch treatment, the film was rinsed thoroughly with water and methanol. The extent of PLA etching and the morphology were monitored by 1H NMR and SAXS, respectively.
PLA etch kinetics
The etch rate was determined by monitoring the etching of planar films. Here the minimum film dimension was maintained at 2 mm in thickness. For such an anisotropic shape with an isotropic etchant, the film thickness will largely determine the etch time and allow the estimation of the etch rate by minimizing edge effects. The extent of PLA removal was monitored using 1H NMR on portions of film as a function of etch time. The disappearance of the non-convolved PLA proton (Fig. 4 proton-g) was compared to the non-convolved PS protons (Fig. 4 protons-a) to quantify the amount of PLA remaining relative to the starting diblock copolymer. The spectra were normalized to have the a-integral correspond to 5 × DPPS where the PLA remaining was quantified as DPPLA = g-integral. The etch rate was calculated using rate (nm s−1) = (film thickness (nm) × %PLA degraded)/(2 × etching time (s)). This calculation assumes a constant velocity of the etch interface and the factor of 2 accounts for etching from both of the exposed film surfaces. The assumption of linear etch rates was supported by time resolved measurements.Molecular characterization
All proton nuclear magnetic resonance (1H NMR) and heteronuclear single quantum coherence (HSQC) spectra were recorded on Bruker Avance III HD 300. NMR samples were prepared in deuterated chloroform (CDCl3) at a concentration of ∼1 wt%. Molecular weight (Mn) and molar mass dispersity (Đ) were determined using a Waters gel permeation chromatography (GPC) instrument equipped with a 515 HPLC pump, a 2410 refractive index detector, and three styragel columns (HR1, HR3, and HR4 in the effective molecular weight range of 0.1–5, 0.5–30, and 5–600 kg mol−1 respectively). Tetrahydrofuran (THF) was used as the eluent at 30 °C at a flow rate of 1 mL min−1. The GPC was calibrated with PS standards (2570, 1090, 579, 246, 130, 67.5, 34.8, 18.1, 10.4, 3.4, 1.6 kg mol−1) obtained from Polymer Laboratories. GPC samples were prepared by dissolving the sample in THF at a concentration of 2.0 mg mL−1 and were filtered (0.2 μm) just prior to injection.Small-angle X-ray scattering (SAXS)
X-ray scattering experiments were conducted using a SAXSLab Ganesha system at the South Carolina SAXS Collaborative. A Xenocs GeniX3D microfocus source was used with a Cu target to generate a monochromatic beam with 0.154 nm wavelength. The instrument was calibrated using National Institute of Standards and Technology (NIST) reference material, 640c silicon powder with the peak position at 2θ = 28.44° where 2θ is the total scattering angle. A Pilatus 300 K detector (Dectris) was used to collect the two-dimensional (2D) scattering patterns with the incident beam normal to the planar sample surface. 2D images were azimuthally integrated into one-dimensional (1D) data of intensity (I) versus q (momentum transfer). All data were acquired after one hour of measurement with an X-ray flux of ∼4.1 M photons per s incident upon the sample. SAXS data were fit using custom MATLAB software.Field emission scanning electron microscopy (FE-SEM)
After PLA etching, the samples were mounted on brass shims using carbon adhesive (Electron Microscopy Sciences) and imaged normal to the planar film surface. Samples were sputter coated with gold–palladium alloy using a Denton Desk II Sputter Coater with global rotation and tilt to avoid sample charging. Images were acquired using an acceleration voltage of 5 keV with an in-lens secondary electron detector. The working distance was fixed at 3.00 mm or less for the acquisition of the images.Results and discussion
A prototypical PS-b-PLA with a lamellar (LAM) morphology was used for development of etch conditions applied towards mm-thick films. The polymer was synthesized using sequential polymerization reactions from an initiator with both alkyl halide and alcohol functionalities. ARGET-ATRP39 was used to grow PS from the alkyl halide of the initiator. Subsequently, ROP38,40 was used to grow PLA from the alcohol group of the initiator. The PS-b-PLA had a molar mass of 41.6 kg mol−1 and contained 66 vol% of PLA with a molar mass dispersity (Đ) of 1.16 (Table 1).| Sample | M n,PS (kg mol−1) | M n,PLA (kg mol−1) | Total Mnc (kg mol−1) | Đ | f vPS | f vPLA | Morphologye |
|---|---|---|---|---|---|---|---|
| a Obtained from GPC analysis. b Obtained from 1H NMR analysis. c Obtained from a + b. d Volume fractions (fv) calculated using densities24,41 at 110 °C, PS = 1.02 g cm−3, PLA = 1.18 g cm−3. e Morphology determined by SAXS. | |||||||
| PS-b-PLA | 12.9 | 28.6 | 41.6 | 1.16 | 0.34 | 0.66 | LAM |
Ordered 2 mm thick films of PS-b-PLA were prepared by solvent casting, followed by an annealing treatment to yield the “as-made” film. The 2D SAXS pattern of the as-made film consisted of isotropic rings (Fig. S1, ESI†), indicating a homogeneous distribution of grain orientations with abundant grain boundaries as a worst-case scenario for etching. The scattering profile was consistent with LAM symmetry, having peaks observed at q/q* = 1, 2, 3, and 4 with a d-spacing of 33.9 nm. A 2 mm thick PS-b-PLA film was etched with TFA solution by direct immersion. The 1 M TFA etch kinetics were monitored by 1H NMR by the disappearance of PLA at δ 5.19 ppm. The TFA immersion etch kinetics were linear (r2 = 0.98) with a best-fit etch rate of 14 nm s−1 (Fig. 1).
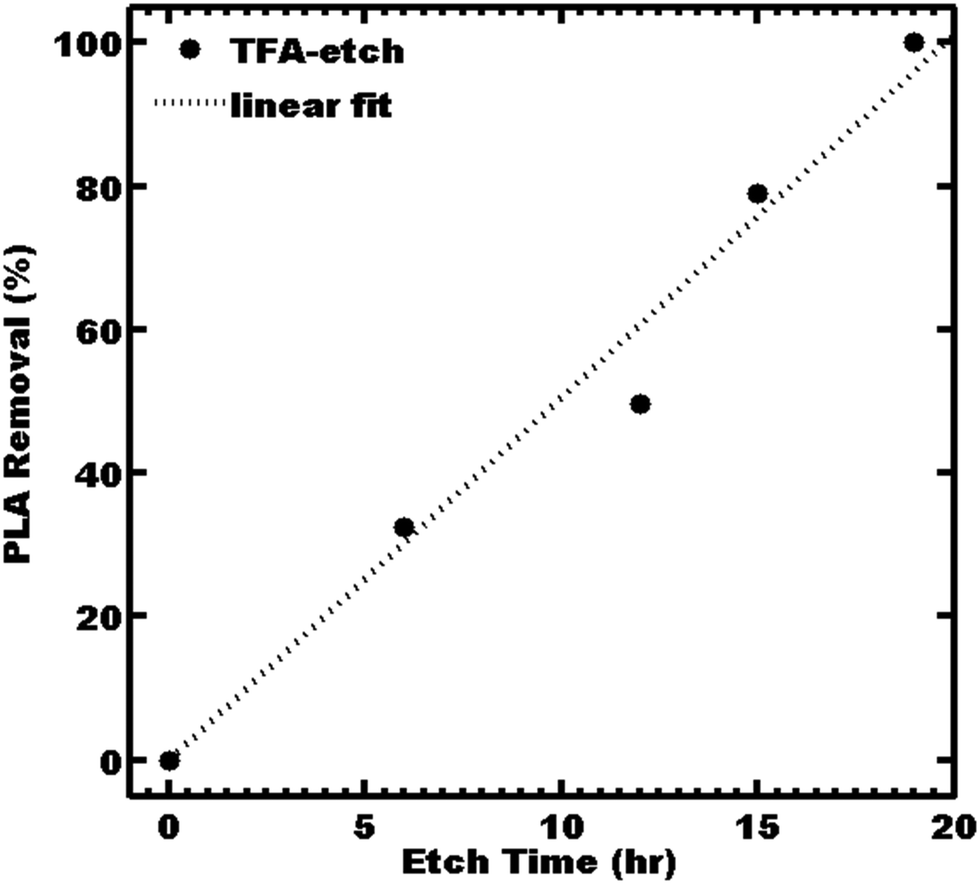 | ||
| Fig. 1 PLA etch kinetics from PS-b-PLA immersed 1 M TFA solution. | ||
The resulting fully etched sample was termed “TFA-etch” (Table 2). Sample TFA-etch was stable under ambient conditions and was mechanically rather brittle due to the high Tg of PS. For comparison, 0.5 M NaOH was reported to have an etch rate of 1–1.5 nm s−1 at 65 °C for <0.5 mm thick films.23,24 Similarly, a dilute 0.05 M NaOH was reported to yield initial etch rates of 0.02 nm s−19,27,42 and an average etch rate of 0.6–0.9 nm s−143 for thin films. Likewise an etch rate of 0.17 nm s−1 was found with yet more dilute 0.01 M NaOH.28 Broadly, NaOH etch rates are reported to increase with temperature.23,24 For comparison, an as-made film was etched with a popular 0.5 M NaOH etchant at 60 °C and was termed “NaOH-etch”.23,24 The PLA was completely removed after 3 days of etching, corresponding to an etch rate here of 3.8 nm s−1 (Table 2). This etch rate is considerably slower than the 14 nm s−1 found with immersion in 1 M TFA at 40 °C, despite the slightly reduced temperature.
| Sample name | Etch rate (nm s−1) | LAM morphology | d-Spacing (nm) (obtained by SAXS) | Observed SAXS reflections (q/q*) | Etch time (h) | Etch conditions |
|---|---|---|---|---|---|---|
| As-made | — | — | 33.8 | 1, 2 | 0 | — |
| TFA-etch | 14 | Preserved | 33.9 | 1, 2, 4 | 19 | Immersed in 1 M TFA |
| TFA-etch-vapor | 9.5 | Preserved | 33.1 | 1, 2, 4 | 32 | In contact with 8 M TFA vapor |
| NaOH-etch | 3.8 | Degraded | — | — | 72 | Immersed in 0.5 M NaOH |
The morphology before and after etching was compared by SAXS. Measurements of sample TFA-etch (Fig. 2a) exhibited the same series of highly ordered SAXS reflections, indicating that both the unit cell symmetry and lattice parameters were preserved through the TFA etch. The presence of mesopores is evidenced by mesostructure preservation (SAXS and SEM) combined with 1H NMR demonstrating complete removal of the PLA phase. Please note that the effects of different unit cell dimension and symmetry have been looked at extensively elsewhere.9,11,43 None the less, we also demonstrated 100% PLA removal and complete morphology retention for a hexagonal morphology using the same 1 M TFA etching procedure (Table S1 and Fig. S9, ESI†). SEM measurements of sample TFA-etch confirmed the preservation of the ordered morphology with a d-spacing of 31.8 ± 4.0 nm (Fig. 2b), consistent with the SAXS findings. The complete removal of PLA by 0.5 M NaOH required a significantly longer 3 days exposure to etchant and resulted in morphology degradation evidenced by a few weak SAXS shoulders (Fig. S2, ESI†). Although NaOH has been widely used successfully to etch thinner <1 mm films, these data suggest that modified conditions may be more efficacious for few-mm thick films. Highly selective etchants are needed to enable the etching of macroscopic films that support large-volume production of nanostructured materials, which can be beneficial in e.g. catalytic applications. The use of a fast TFA etch implies a shortened exposure to etchant and was demonstrated to preserve the PS nanostructure through the complete removal of PLA from 2 mm thick films (Table 2).
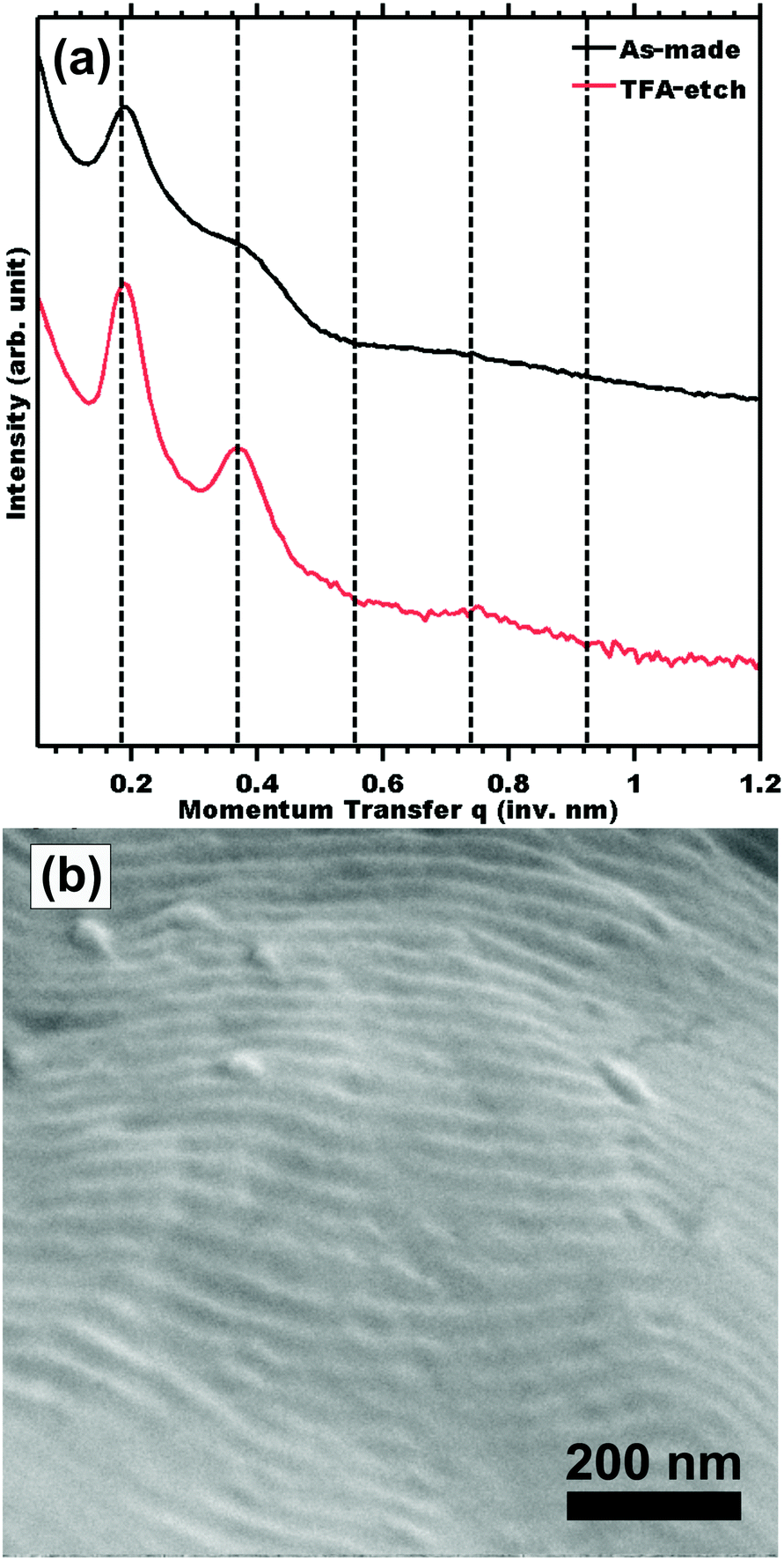 | ||
| Fig. 2 (a) SAXS of as-madePS-b-PLA compared to fully etched sample TFA-etch by immersion in 1 M TFA. The samples were indexed for LAM symmetry with peaks indicated at q/q* = 1, 2, 3, and 4 where q = 4πsin(θ)/λ. The lines in (a) were offset vertically for clarity. (b) Top-view SEM image of mesoporous PS in sample TFA-etch. | ||
The selectivity of TFA as an etchant was further examined in terms of PS degradation by both GPC and 1H NMR. During synthesis, GPC demonstrated chain extension from the PS-OH macroinitiator to the final PS-b-PLA diblock copolymer. After TFA etching, GPC elugrams exhibited a nearly identical molar mass distribution to that of the parent PS-OH macroinitiator (Table 3), consistent with 100% PLA removal without observable damage to PS (Fig. 3). Please note that PS does not contain ester groups and is not expected to degrade by hydrolysis during etching.
| Samples | M n (kg mol−1) | Đ |
|---|---|---|
| M n (kg mol−1) and Đ obtained by GPC. | ||
| PS-OH | 13.0 | 1.21 |
| PS-b-PLA | 57.6 | 1.16 |
| TFA-etch | 13.0 | 1.20 |
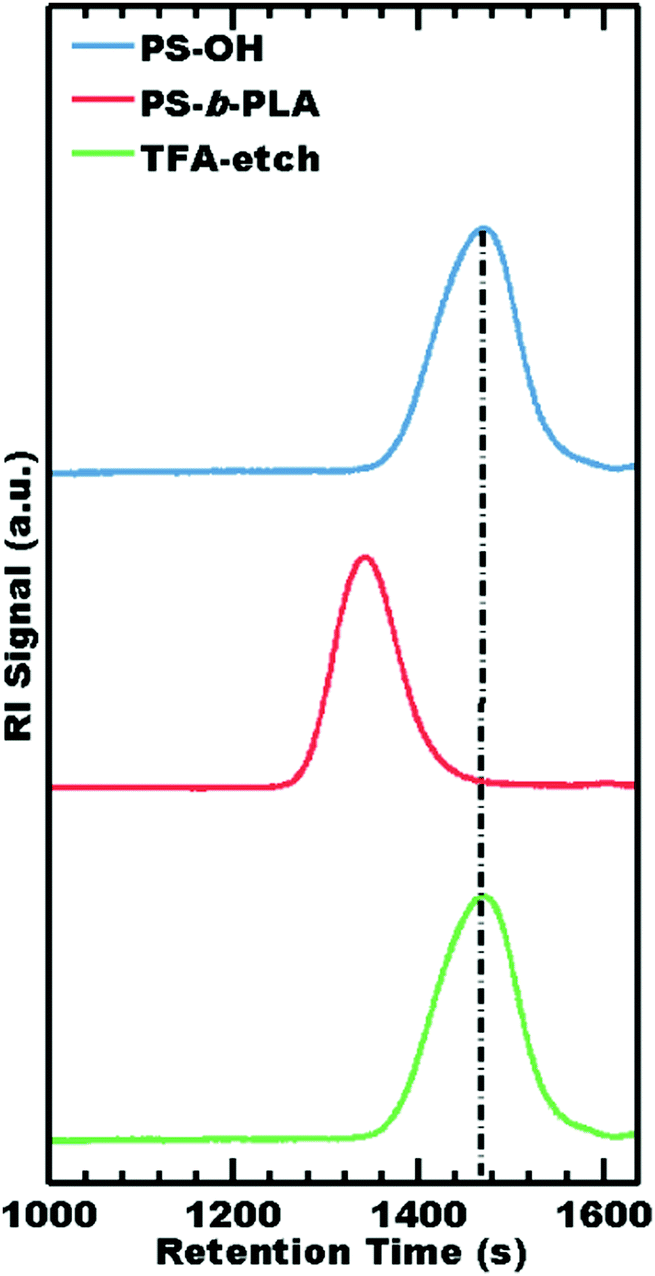 | ||
| Fig. 3 GPC traces of the PS-OH used to make PS-b-PLA as well as the fully etched sample TFA-etch. | ||
Complementary 1H NMR data also showed complete removal of PLA, proton g at 5.19 ppm, without noticeable changes to the PS spectra (Fig. 4d). Please note that the new peak at 1.5 ppm was attributed to water (Fig. 4d) and was confirmed to be not associated with the PS carbon by heteronuclear single quantum coherence (HSQC) spectroscopy (Fig. S3, ESI†). In contrast, the NMR spectra of the NaOH-etch sample exhibited spectral changes in the 1H δ1.0–2.0 ppm range with two new correlated carbon peaks (δ40–44 ppm) by HSQC (Fig. S5, shown by an arrow, ESI†) that may correspond to damage to the PS backbone after the extended etch process needed for 2 mm thick films. We note that the initiator group (proton-d at 3.5 ppm) was removed by both NaOH and TFA etching procedures (Fig. 4c and d).
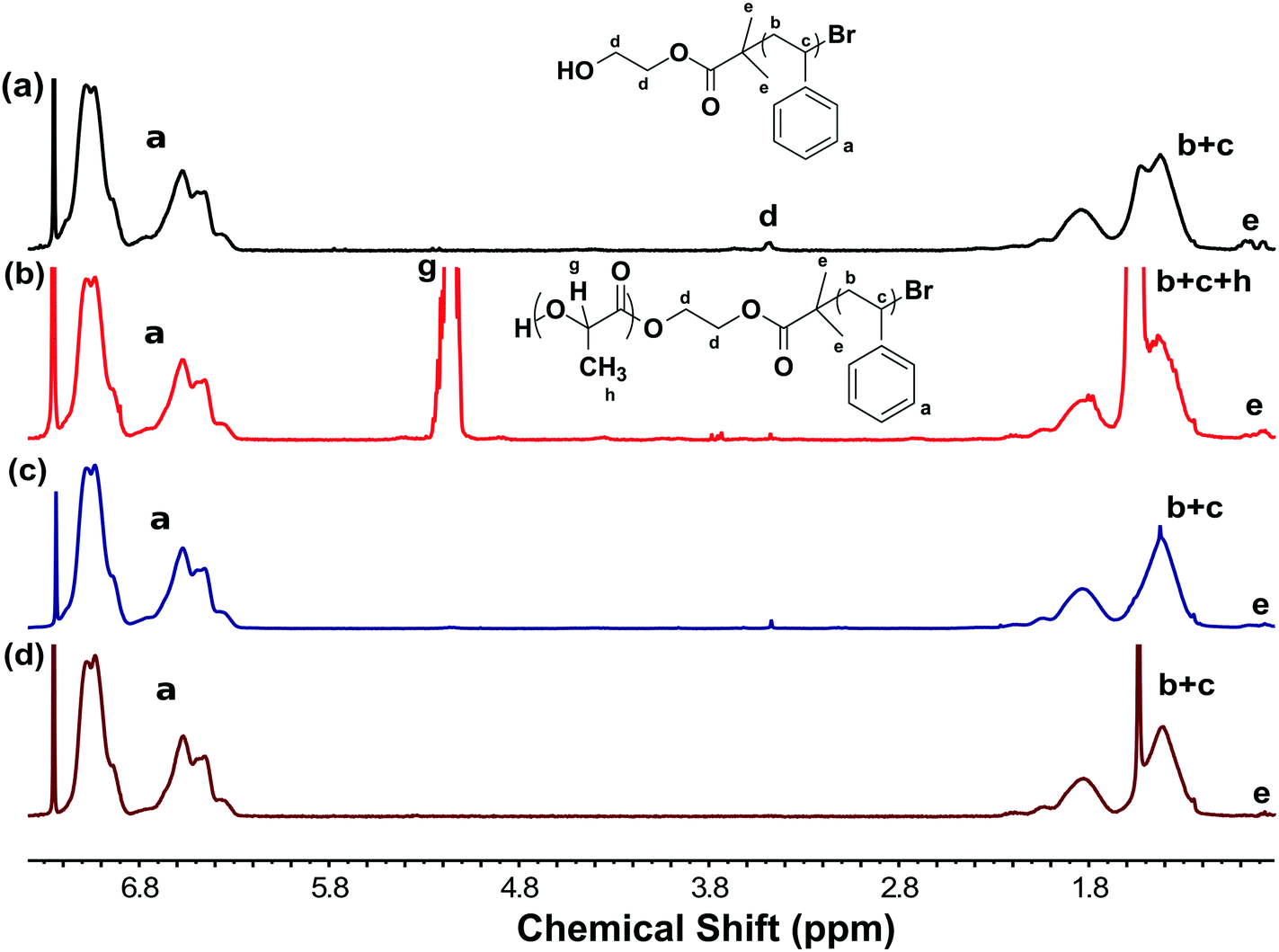 | ||
| Fig. 4 1H NMR spectra of (a) PS-OH (b) PS-b-PLA before etching and samples (c) NaOH-etch and (d) TFA-etch. | ||
Indeed, bases are industrially used for the deliberate degradation of PS with reported products of styrene, ethyl benzene, toluene, cumene and indane derivatives.44,45 However, these side products were not observed in the NaOH-etch solution or in rinsates as examined by 1H-NMR. In contrast, the HSQC spectra of TFA-etch and PS-OH samples exhibited the same peak correlations without apparent changes to the polymer (Fig. S3 and S4, ESI†). TFA was demonstrated to enable remarkable etch selectivity with fast PLA removal and without apparent damage to PS.
Wide ranges of TFA etching conditions were examined. Etching by both direct immersion and vapor exposure was investigated separately with the experimental setups shown in Fig. S6 and S7 (ESI†). In both cases, the PLA etch rate increased with TFA concentration where etching by direct immersion was faster than by vapor exposure for a particular TFA molarity (Fig. 5).
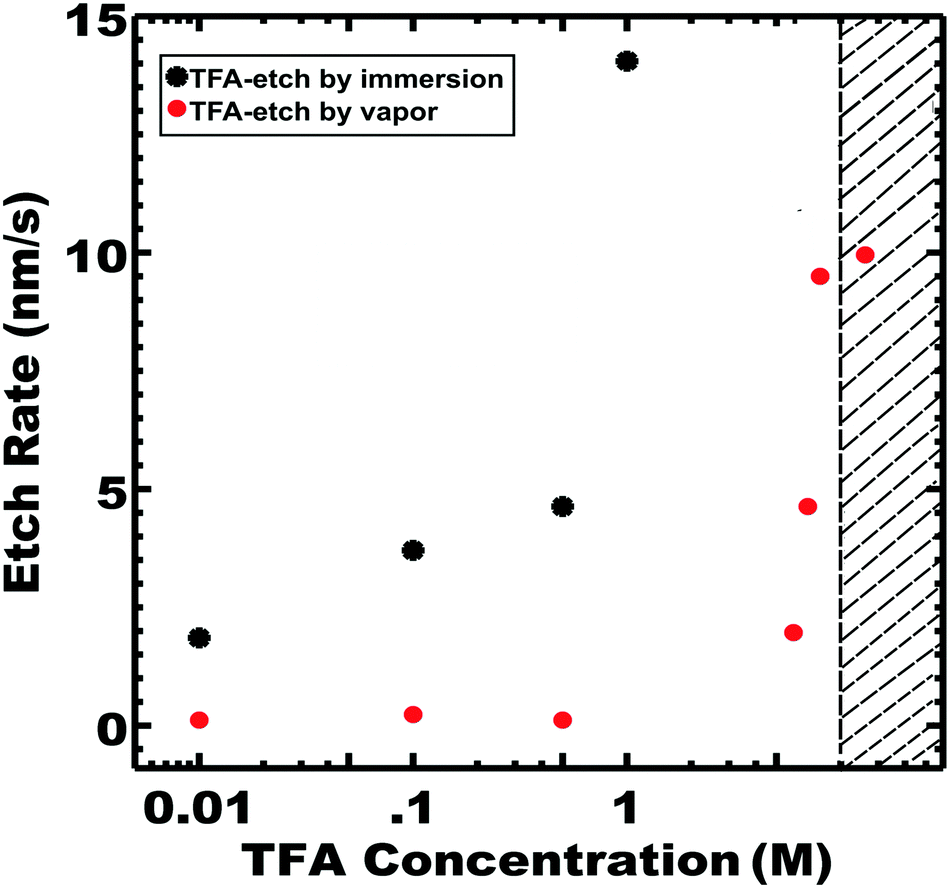 | ||
| Fig. 5 Etch rates with varying TFA concentration by either immersion or vapor exposure. The etch rates were calculated after a 24 h etch at 40 °C, except for 1 M TFA immersion and 8 M TFA vapor where PLA was completely removed in less time. SAXS was used to examine the morphology at the end of each process. The lamellar morphology was generally preserved, except for samples within the shaded region. | ||
Also in both cases, an upper limit of TFA concentration was observed where the ordered morphology was lost, presumably due to plasticization of the glassy PS. For example, immersion in 8 M TFA led to the fastest observed rate of 139 nm s−1, corresponding to 100% PLA removal in 2 h; however SAXS indicated collapse of the ordered morphology (Fig. 5 shown in the shaded region). Such a high TFA concentration may plasticize the PS and lead to the collapse of the porous morphology. Optimized conditions enabled the fastest etch rates that preserved the parent morphology. The optimum etch conditions were found to be 1 M TFA for immersion etching and 8 M TFA for vapor etching. The latter optimized 8 M TFA vapor etch was linear (r2 = 0.97) with a best fit etch rate of 9.5 nm s−1 (Fig. S8, ESI†). Considering the absence of liquid solvent, the observed linear kinetics are surprising since the resulting pores are expected to be filled with lactic acid until the later rinse step. SAXS and SEM measurements also confirmed morphology preservation of TFA-etch-vapor after complete PLA removal (Fig. 6).
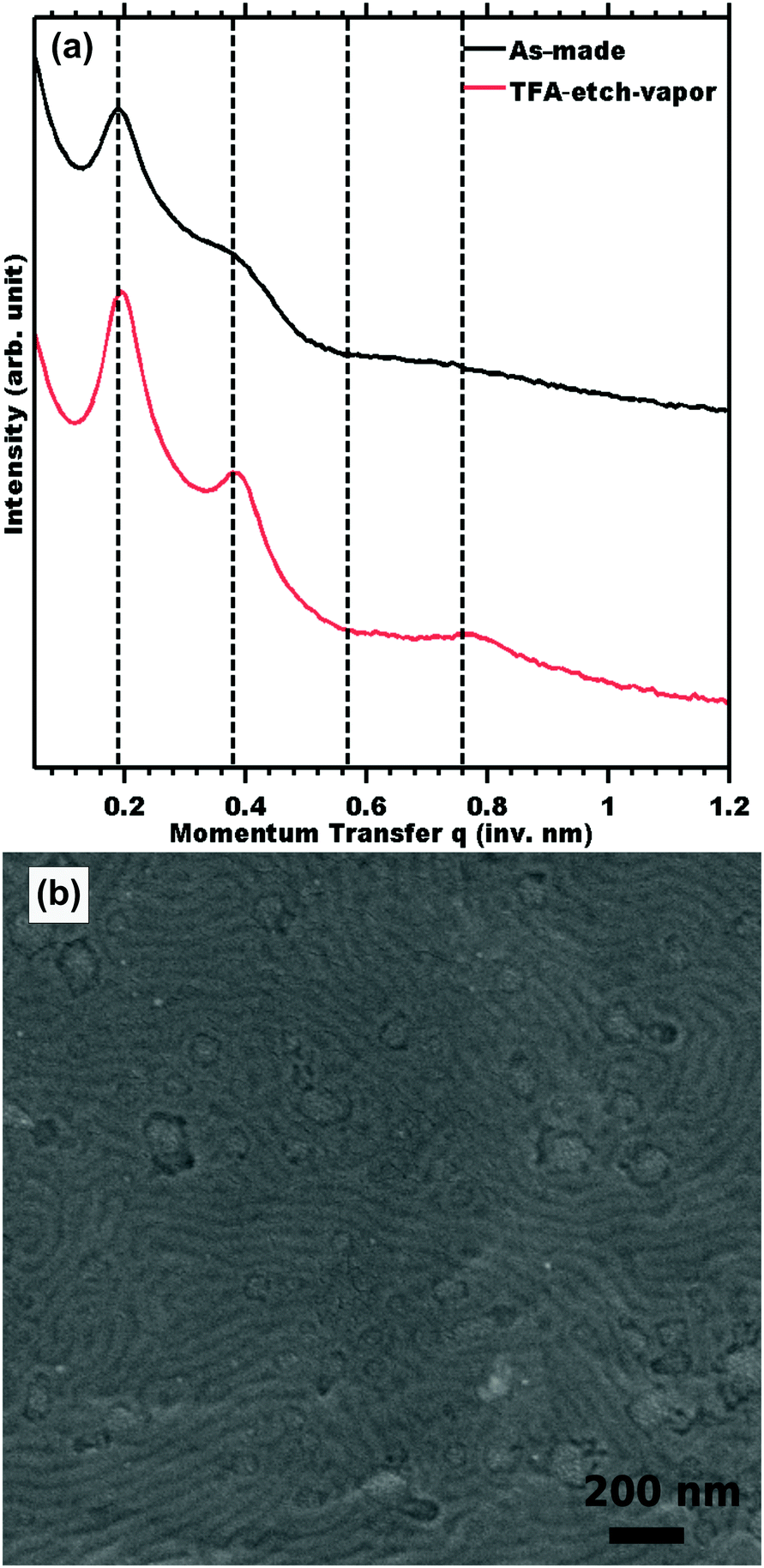 | ||
| Fig. 6 SAXS of as-made film and after subsequent etching by 8 M TFA vapor at 40 °C for 32 h (a). The samples were indexed for LAM symmetry with peaks indicated at q/q* = 1, 2, 3, and 4 where q = 4πsin(θ)/λ. The lines in (a) were offset vertically for clarity. (b) Top-view SEM images of mesoporous PS monolith resulting from TFA-vapor-etch. | ||
Conclusions
In summary, TFA was found to be an ideal etchant for PLA removal from thick block copolymer films. The high etch rate of up to 14 nm s−1 enabled the complete removal of PLA from 2 mm thick films in less than 1 day. The use of a short etch process decreases the extent of PS damage to below the detection limit. Etching with optimized TFA solutions also demonstrated preservation of the parent morphology as determined by SAXS and SEM. A combination of GPC and NMR confirmed that the only product was pristine porous PS with a molar mass distribution indistinguishable from that of the starting macroinitiator. The combination of 1H-NMR, HSQC, and GPC indicates that the TFA etch process did not induce detectable damage to the PS block, despite replacing the adjacent initiator group with a COOH. We anticipate that this significantly improved TFA etch process will impact numerous nanomaterial developments based upon access to porous polymers.Acknowledgements
M. S. thanks the University of South Carolina for start-up funds. This work made use of the South Carolina SAXS Collaborative, supported by the NSF Major Research Instrumentation program (award #DMR-1428620). We thank the NMR facility at USC and P. J. Pellechia for his help with HSQC analysis. We thank Dr Chuanbing Tang for helpful discussions and suggestions.References
- G. Liu, J. Ding, T. Hashimoto, K. Kimishima, F. M. Winnik and S. Nigam, Chem. Mater., 1999, 11, 2233–2240 CrossRef CAS.
- M. A. Hillmyer, Adv. Polym. Sci., 2005, 190, 137–181 CrossRef CAS.
- K.-V. Peinemann, V. Abetz and P. F. W. Simon, Nat. Mater., 2007, 6, 992–996 CrossRef CAS PubMed.
- J. Cleland, A. Daugherty and R. Mrsny, Curr. Opin. Biotechnol., 2001, 12, 212–219 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS PubMed.
- S. Y. Yang, J.-A. Yang, E. S. Kim, G. Jeon, E. J. Oh, K. Y. Choi, S. K. Hahn and J. K. Kim, ACS Nano, 2010, 4, 3817–3822 CrossRef CAS PubMed.
- Y. Lu, H. Fan, A. Stump, T. L. Ward, T. Rieker and C. J. Brinker, Nature, 1999, 398, 223–226 CrossRef CAS.
- M. Q. Li and C. K. Ober, Mater. Today, 2006, 9, 30–39 CrossRef CAS.
- E. J. W. Crossland, S. Ludwigs, M. A. Hillmyer and U. Steiner, Soft Matter, 2007, 3, 94–98 RSC.
- E. J. W. Crossland, M. Nedelcu, C. Dukati, S. Ludwigs, M. A. Hillmyer, U. Steiner and H. J. Snaith, Nano Lett., 2009, 9, 2813–2819 CrossRef CAS PubMed.
- E. J. W. Crossland, M. Kamperman, M. Nedelcu, C. Dukati, U. Wiesner, D.-M. Smilgies, G. E. S. Toombes, M. A. Hillmyer, S. Ludwigs, U. Steiner and H. J. Snaith, Nano Lett., 2009, 9, 2807–2812 CrossRef CAS PubMed.
- H. Y. Hsueh, Y. C. Huang, R. M. Ho, C. H. Lai, T. Makida and H. Hasegawa, Adv. Mater., 2011, 23, 3041–3046 CrossRef CAS PubMed.
- H. Y. Hsueh, Y. C. Ling, H. F. Wang, L. Y. Chien, Y. C. Hung, E. L. Thomas and R. M. Ho, Adv. Mater., 2014, 26, 3225–3229 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- J. H. Wolf and M. A. Hillmyer, Langmuir, 2003, 19, 6553–6560 CrossRef CAS.
- R. A. Pai, R. Humayun, M. T. Schulberg, A. Sengupta, J. N. Sun and J. J. Watkins, Science, 2004, 303, 507–510 CrossRef CAS PubMed.
- M. Glassner, J. P. Blinco and C. Barner-Kowollik, Polym. Chem., 2011, 2, 83–87 RSC.
- J. Rao, S. De and A. Khan, Chem. Commun., 2012, 48, 3427–3429 RSC.
- R. Majdoub, T. Antoun, B. L. Droumaguet, M. Benzina and D. Grande, React. Funct. Polym., 2012, 72, 495–502 CrossRef CAS.
- J. Rzayev and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 13373–13379 CrossRef CAS PubMed.
- H. Mao, P. L. Arrechea, T. S. Bailey, B. J. S. Johnson and M. A. Hillmyer, Faraday Discuss., 2005, 128, 149–162 RSC.
- S. Li, J. Biomed. Mater. Res., 1999, 48, 342–353 CrossRef CAS PubMed.
- A. S. Zalusky, R. Olayo-Valles, C. J. Taylor and M. A. Hillmyer, J. Am. Chem. Soc., 2001, 123, 1519–1520 CrossRef CAS.
- A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761–12773 CrossRef CAS PubMed.
- H. Tsuji and Y. Ikada, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 59–66 CrossRef CAS.
- B. L. Droumaguet, R. Poupart and D. Grande, Polym. Chem., 2015, 6, 8105–8111 RSC.
- C. Cummins, P. Mokarian-Tabari, J. D. Holmes and M. A. Morris, J. Appl. Polym. Sci., 2014, 131, 40798–40810 CrossRef.
- C. Cummins, P. Mokarian-Tabari, P. Andreazza, C. Sinturel and M. A. Morris, ACS Appl. Mater. Interfaces, 2016, 8, 8295–8304 CAS.
- X. B. Wang, T. Y. Lo, H. Y. Hsueh and R. M. Ho, Macromolecules, 2013, 46, 2997–3004 CrossRef CAS.
- F. Wang, L. H. Yu, X. B. Wang and R. M. Ho, Macromolecules, 2014, 47, 7993–8001 CrossRef.
- D. Grande, J. Penelle, P. Davidson, I. Beurroies and R. Denoyel, Microporous Mesoporous Mater., 2011, 140, 34–39 CrossRef CAS.
- L. Lei, Y. Xia, X. Chen and S. Shi, J. Appl. Polym. Sci., 2014, 131, 39638–39653 Search PubMed.
- J. Oh and M. Seo, ACS Macro Lett., 2015, 4, 1244–1248 CrossRef CAS.
- L. Chen and M. A. Hillmyer, Macromolecules, 2009, 42, 4237–4243 CrossRef CAS.
- T. S. Bailey, J. Rzayev and M. A. Hillmyer, Macromolecules, 2006, 39, 8772–8781 CrossRef CAS.
- E. A. Jackson, Y. Lee and M. A. Hillmyer, Macromolecules, 2013, 46, 1484–1491 CrossRef CAS.
- I. Rashkov, N. Manolova, S. M. Li, J. L. Espartero and M. Vert, Macromolecules, 1996, 29, 50–56 CrossRef CAS.
- M. Scherer, PhD Thesis, University of Cambridge, 2012.
- W. Jacubowski, B. K. Denizli, R. R. Gil and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 32–39 CrossRef.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- R. M. Ho, M. C. Li, S. C. Lin, H. F. Wang, Y. D. Lee, H. Hasegawa and E. L. Thomas, J. Am. Chem. Soc., 2012, 134, 10974–10986 CrossRef CAS PubMed.
- A. Baruth, M. Seo, C. H. Lin, K. Walster, A. Shankar, M. A. Hillmyer and C. Leighton, ACS Appl. Mater. Interfaces, 2014, 6, 13770–13781 CAS.
- E. J. W. Crossland, P. Cunha, S. Ludwigs, M. A. Hillmyer and U. Steiner, ACS Appl. Mater. Interfaces, 2011, 3, 1375–1379 CAS.
- H. Ukei, T. Hirose, S. Horikawa, Y. Takai, M. Taka, N. Azuma and A. Ueno, Catal. Today, 2000, 62, 67–75 CrossRef CAS.
- V. Karmore and G. Madras, Ind. Eng. Chem. Res., 2002, 41, 657–660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 2D SAXS of as-made, 1D SAXS of as-made and NaOH-etch, 2D heteronuclear single quantum coherence spectra, etching experiment details, PLA etch rate kinetics, PS-b-PLA2 synthesis details and characterization, 1H NMR, 1D SAXS of HEX-as-made and HEX-TFA-etch, and top-view SEM of HEX-TFA-etch. See DOI: 10.1039/c6qm00266h |
| This journal is © the Partner Organisations 2017 |