
A highly selective fluorescent nanoprobe based on AIE and ESIPT for imaging hydrogen sulfide in live cells and zebrafish†
Peisheng
Zhang
ab,
Xuezheng
Nie
a,
Meng
Gao
a,
Fang
Zeng
*a,
Anjun
Qin
a,
Shuizhu
Wu
*a and
Ben Zhong
Tang
a
aCollege of Materials Science & Engineering, State Key Lab of Luminescent Materials & Devices, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: shzhwu@scut.edu.cn; mcfzeng@scut.edu.cn; Fax: +86 20 22236363; Tel: +86 20 22236262
bKey Laboratory of Theoretical Organic Chemistry and Functional Molecule of Ministry of Education, Hunan Provincial Key Laboratory of Controllable Preparation and Functional Application of Fine Polymers, Hunan Province College Key Laboratory of QSAR/QSPR, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan, Hunan 411201, China
First published on 3rd November 2016
Abstract
The rational design of effective tools capable of monitoring hydrogen sulfide (H2S) is of great importance to fully understand its physiological and pathological functions. Herein, a self-assembled fluorescent nanoprobe with both aggregation-induced emission (AIE) and excited-state intramolecular proton transfer (ESIPT) characteristics for H2S detection has been successfully developed via a modified nanoprecipitation method. The nanoprobe features high water dispersibility, a large Stokes shift (ca. 100 nm), good biocompatibility as well as high selectivity towards H2S over other putative interferents. Live cell imaging experiments demonstrate that the nanoprobe, with good cell-membrane permeability, can facilitate the visualization of exogenous and endogenous H2S levels. Moreover, the nanoprobe has also been found capable of detecting H2S in zebrafish. This nanoprobe with AIE and ESIPT characteristics can provide a novel method for the development of fluorescent probes for monitoring biological processes.
Introduction
Hydrogen sulfide (H2S), the third gaseous transmitter following carbon monoxide (CO) and nitric oxide (NO), plays a pivotal role in various pathophysiological processes, such as regulating intracellular redox status,1 antioxidation,2 apoptosis,3 anti-inflammation,4etc. However, acatastatic levels of H2S may induce an increased risk of many diseases such as Alzheimer’s disease,5 Down’s syndrome6 and diabetes.7 Therefore, selective detection and imaging of H2S in live cells and in vivo is undoubtedly crucial for better understanding of its biological and pathological roles.Recently, a large number of fluorescent sensors for H2S detection in live cells have been reported based on three main strategies including the reduction of azide or nitro groups,8 nucleophilic attack,9 and CuS precipitation.10 However, most of these sensors are made up of small molecules, which may suffer from less satisfactory water solubility, or undergo aggregation caused quenching (ACQ) in aqueous or physiological media, thereby greatly reducing their sensing performance.11 In addition, the “self-absorption” effect with a small Stokes shift of less than 40 nm also hampers their use in biosystems.12 The recently developed fluorogens with both aggregation-induced emission (AIE) and excited-state intramolecular proton transfer (ESIPT) features can effectively solve the above issues.13 AIE-active dyes, such as tetraphenylethene (TPE) and its derivatives, can not only show high emission in the aggregated or solid states, but also offer a high signal-to-noise ratio and excellent photostability.14 On the other hand, ESIPT-based fluorophores possess a high quantum efficiency and large Stokes shift, which greatly reduces self-quenching of the fluorescence.15 The probes based on both AIE and ESIPT mechanisms exhibit emissions with a large Stokes shift and do not require hydrophilic modification, hence they can find potential applications in biological sensing.13b To date, these kinds of fluorescent probes have been developed for the detection of some analytes, such as thiols, lysosomal esterase, palladium cations, perborate, alkaline phosphatase and β-galactosidase.13 Currently, nanoparticle-based fluorescent probes, which are physically or covalently incorporated with analyte-responsive receptors or analyte-reactive moieties, are attracting increasing attention in the field of in vivo fluorescent imaging due to several excellent advantages such as improved water dispersibility, enhanced photostability, high diversity in structure design and so on.16–18
Herein we report an AIE/ESIPT-based fluorescent nanoprobe for the selective detection and imaging of H2S by using a modified nanoprecipitation method. As illustrated in Scheme 1, the nanoprobe was formed by nanoprecipitation of a H2S-responsive TPE derivative (TPE-3) and three phospholipids, followed by surface modification with a Tat peptide to facilitate cell penetration for the nanoprobe. The nanoprobe shows weak fluorescence in the absence of H2S; but with the addition of H2S, significant enhancement of fluorescence can be recorded and observed, thereby affording the nanoprobe as a robust sensing and imaging system for H2S. The nanoprobe displays high water dispersibility, a large Stokes shift (ca. 100 nm), good biocompatibility, etc. Moreover, the nanoprobe can be used to image H2S in living cells and in vivo. To the best of our knowledge, this is the first report of an AIE/ESIPT-based fluorescent nanoprobe enabling in vitro and in vivo imaging of H2S.
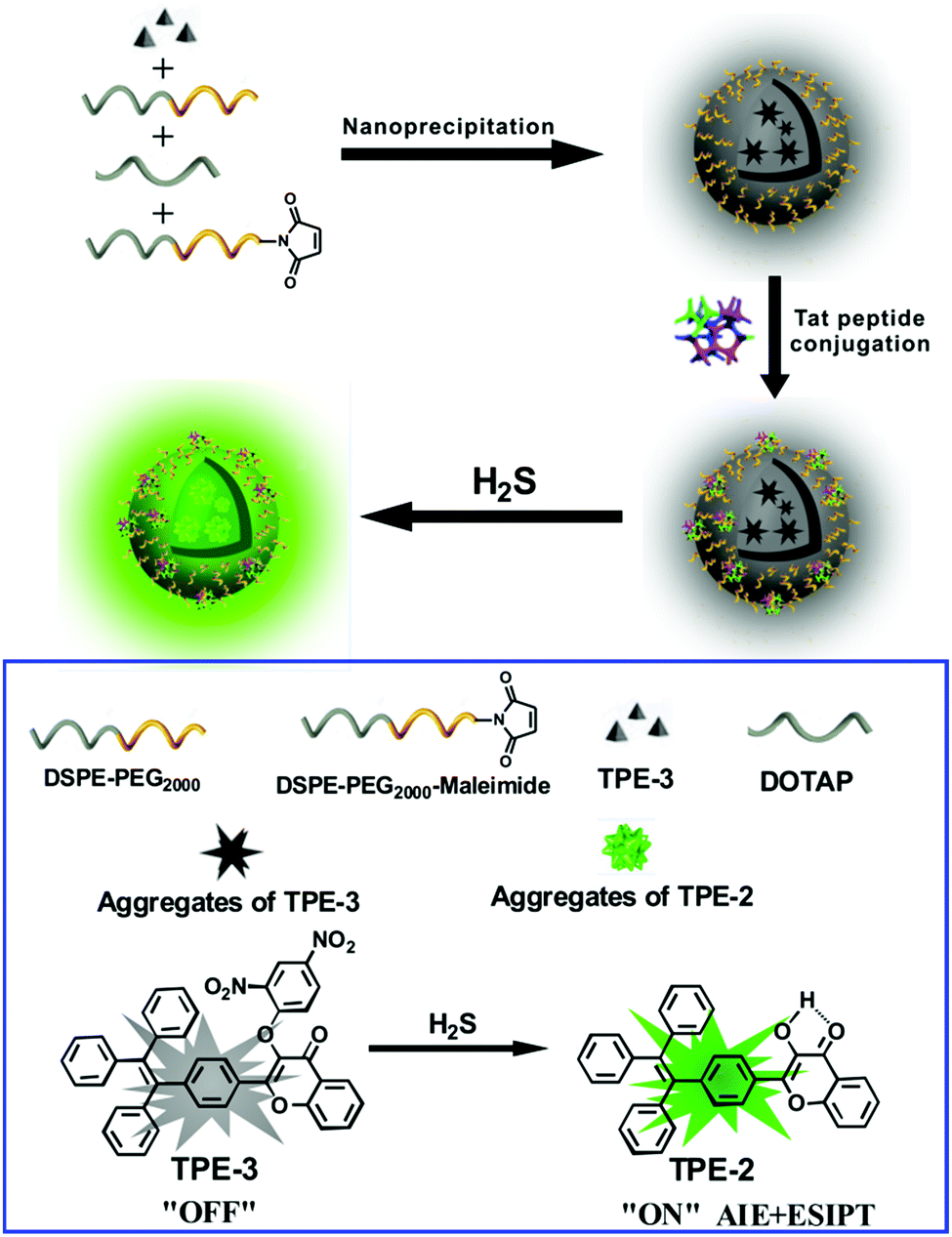 | ||
| Scheme 1 Schematic illustration for the fabrication of a Tat peptide-conjugated nanoprobe and its fluorescent response to H2S. | ||
Experimental section
Materials
Tetrakis(triphenylphosphine)palladium (Pd(PPh)4), tetrabutyl ammonium bromide (TBAB), bromotriphenylethylene, 2-hydroxyacetophenone, 4-formylphenylboronic acid, 2,4-dinitrofluorobenzene and titanium(IV) chloride (99%, TiCl4) were purchased from J&K Chemical Ltd. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly-ethylene glycol)-2000] (DSPE-PEG2000), 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-Mal) were purchased from Avanti Polar Lipids, Inc. The cell penetrating peptide derived from transactivation of transcription proteins–HIV-1 Tat (49–57) with the C-terminus modified with cysteine (RKKRRQRRRC) was a commercial product customized by GenicBio, China. N,N-Dimethyl-formamide (DMF) was dried with CaH2 and vacuum distilled. The water used in the experiments was double-distilled water which was further treated using ion exchange columns and then by a Milli-Q water purification system. All other chemicals were of analytical grade.Characterization
1H NMR spectra were recorded on a Bruker Avance 600 MHz NMR spectrometer. 13C NMR spectra were recorded on a Bruker Avance 500 MHz NMR spectrometer. Mass spectra were obtained using a Bruker Esquire HCT Plus mass spectrometer. UV-vis spectra were recorded on a Hitachi U-3010 UV-vis spectrophotometer. Fluorescence spectra were recorded on a Hitachi F-4600 fluorescence spectrophotometer. The particle size and distribution were determined through dynamic light scattering (DLS) on a Malvern Nano-ZS90 particle size analyzer at a fixed angle of 90° at 25 °C. The zeta potentials were determined on a Malvern Nano-ZS90. Transmission electronic microscopy (TEM) experiments were carried out by mounting a drop of the solution onto a carbon-coated copper grid and observation was carried out on a JEM-2010 HR transmission electron microscope (Japan). HPLC analyses were performed on an Agilent 1260 (Diode Array Detector). Fluorescence images were obtained using an Olympus IX 71 fluorescence microscope with a DP72 color CCD.Preparation of the compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) as the eluent to give a yellow solid TPE-1 (0.93 g, 78%). 1H-NMR (600 MHz, CDCl3) δ 9.93 (s, 1H), 7.64 (d, 2H), 7.22 (d, 2H), 7.15–7.07 (m, 9H), 7.05–6.96 (m, 6H).
:1, v/v) under stirring for 20 min. Then, TPE-1 (360 mg, 1 mmol) in 10 mL ethanol was introduced slowly to the above solution. The mixture was stirred at room temperature for 3 h, and then heated to 60 °C for 16 h. The red precipitate was filtrated and washed with cold EtOH three times. The washed red solid was dissolved in a mixture of 10 mL ethanol and 1.5 mL of 10% NaOH, and then 1.5 mL of a 30% H2O2 solution was added in a dropwise manner. The reaction mixture was further stirred at room temperature for 1.5 h. The resulting reaction mixture was poured into crushed ice water. The yellow solid thus obtained was filtrated, washed with water and dried. The crude product was purified using column chromatography on silica gel using dichloromethane/petroleum ether (5:1, v/v) as the eluent to give a yellow solid powder TPE-2 (423 mg, 86%). 1H-NMR (d6-DMSO, 600 MHz, ppm): δ 9.63 (s, 1H), 8.10 (d, 1H), 8.01 (d, 2H), 7.79 (t, 1H), 7.73 (d, 1H), 7.46 (t, 1H), 7.29–6.91 (m, 17H). MS(ESI): m/z 524.7 [M + MeOH]+.
:10, v/v) as the eluent to give a yellow solid powder TPE-3 (43 mg, 65%). 1H-NMR (CDCl3, 600 MHz, ppm): δ 8.91 (d, 1H), 8.27 (d, 1H), 8.20 (d, 1H), 7.84 (d, 2H), 7.78 (dd, 1H), 7.62 (d, 1H), 7.48 (dd, 1H), 7.18 (d, 2H), 7.14–7.05 (m, 9H), 7.02 (m, 7H). 13C-NMR (CDCl3, 500 MHz, ppm): δ 171.38, 157.06, 155.40, 154.50, 148.23, 143.08, 142.00, 141.69, 139.56, 138.35, 134.61, 131.95, 131.31, 131.22, 128.75, 127.80, 127.78, 126.92, 126.61, 126.07, 125.77, 123.49, 122.44, 118.27, 117.17. MS(ESI): m/z 659.0 [M + H]+.
:2, v/v) as the eluent to give a yellow solid TPE-1 (0.93 g, 78%). 1H-NMR (600 MHz, CDCl3) δ 9.93 (s, 1H), 7.64 (d, 2H), 7.22 (d, 2H), 7.15–7.07 (m, 9H), 7.05–6.96 (m, 6H).
:1, v/v) under stirring for 20 min. Then, TPE-1 (360 mg, 1 mmol) in 10 mL ethanol was introduced slowly to the above solution. The mixture was stirred at room temperature for 3 h, and then heated to 60 °C for 16 h. The red precipitate was filtrated and washed with cold EtOH three times. The washed red solid was dissolved in a mixture of 10 mL ethanol and 1.5 mL of 10% NaOH, and then 1.5 mL of a 30% H2O2 solution was added in a dropwise manner. The reaction mixture was further stirred at room temperature for 1.5 h. The resulting reaction mixture was poured into crushed ice water. The yellow solid thus obtained was filtrated, washed with water and dried. The crude product was purified using column chromatography on silica gel using dichloromethane/petroleum ether (5:1, v/v) as the eluent to give a yellow solid powder TPE-2 (423 mg, 86%). 1H-NMR (d6-DMSO, 600 MHz, ppm): δ 9.63 (s, 1H), 8.10 (d, 1H), 8.01 (d, 2H), 7.79 (t, 1H), 7.73 (d, 1H), 7.46 (t, 1H), 7.29–6.91 (m, 17H). MS(ESI): m/z 524.7 [M + MeOH]+.
:10, v/v) as the eluent to give a yellow solid powder TPE-3 (43 mg, 65%). 1H-NMR (CDCl3, 600 MHz, ppm): δ 8.91 (d, 1H), 8.27 (d, 1H), 8.20 (d, 1H), 7.84 (d, 2H), 7.78 (dd, 1H), 7.62 (d, 1H), 7.48 (dd, 1H), 7.18 (d, 2H), 7.14–7.05 (m, 9H), 7.02 (m, 7H). 13C-NMR (CDCl3, 500 MHz, ppm): δ 171.38, 157.06, 155.40, 154.50, 148.23, 143.08, 142.00, 141.69, 139.56, 138.35, 134.61, 131.95, 131.31, 131.22, 128.75, 127.80, 127.78, 126.92, 126.61, 126.07, 125.77, 123.49, 122.44, 118.27, 117.17. MS(ESI): m/z 659.0 [M + H]+.
Preparation of Tat peptide-functionalized nanoprobes
TPE-3 (2 mg), DSPE-PEG2000 (2 mg), DSPE-PEG2000-Mal (2 mg) and DOTAP (30 mg) were dissolved in 2 mL of THF solution, followed by mixing with 20 mL of water. The mixture was then sonicated for 60 seconds using an ultrasonic cell crusher at 15 W output (SJIA-150W, Ningbo Scientz Biotechnology Co., Ltd). After filtration using a 0.2 μm syringe driven filter, the suspension was stirred vigorously at room temperature overnight to yield the unfunctionalized nanoparticles. Subsequently, the modified HIV-1 Tat peptide (1 mg mL−1, 0.5 mL) was mixed with the above nanoparticle solution. After reaction overnight at room temperature, the solution was dialysed against MilliQ water for 2 days to eliminate the excess peptides. Finally, the Tat peptide-functionalized nanoprobes were collected and stored at 2–8 °C for further use.Cell viability assay
The cytotoxicity of the nanoprobe against HeLa cells (human cervical cancer cells) was assessed using an MTT assay according to ISO 10993-5. In this experiment, HeLa cells were incubated in DMEM medium supplemented with 10% fetal bovine serum (FBS) and allowed to grow for 24 h at 37 °C with 5% CO2. After removal of the medium, cells were treated with the nanoprobe of varied concentrations and then incubated for an additional 24 h. For each independent experiment, the assays were performed in eight replicates. And the statistic mean and standard derivation were utilized to estimate the cell viability.Cell culture and imaging
HeLa cells were incubated in DMEM medium supplemented with 10% fetal bovine serum (FBS, Invitrogen) at 37 °C in a humidified incubator containing 5% CO2.For imaging of exogenous H2S in live cells, the HeLa cells were incubated in DMEM media containing the nanoprobe (final nanoprobe concentration 0.01 mg mL−1) at 37 °C under 5% CO2 for 2 h, and then treated with NaHS (the donor of H2S with a concentration of 0 μM, 50 μM and 100 μM respectively) for another 30 minutes. After that, the culture dishes were washed with PBS three times to remove the culture medium, then subject to an Olympus IX71 inverted fluorescence microscope equipped with a DP72 color CCD for cell imaging.
For imaging of endogenous H2S in HeLa cells, the HeLa cells were incubated with glutathione (GSH, 0 mM, 1 mM and 2 mM respectively) for 60 min at 37 °C under 5% CO2, and then treated with the nanoprobe (0.01 mg mL−1) for another 2 h. After that, the culture dishes were washed with PBS three times to remove the culture medium, and then subjected to fluorescence microscopy imaging.
Fluorescence imaging in zebrafish
In this study, wild-type zebrafish were provided by Key Laboratory of Zebrafish Modeling and Drug Screening for Human Diseases of Guangdong Higher Education Institute, Southern Medical University. All zebrafish experiments were performed in full compliance with international ethics guidelines and according to the procedures stated in our previous report.19 Briefly, 5-day-old larvae were transferred into a 96-well microplate using a disposable transfer pipette, and then the larvae were incubated in 100 μL of E3 media containing the nanoprobe (final concentration: 0.005 mg mL−1) for 30 min. In contrast, the control group larvae were only incubated in 100 μL of E3 media. Then, the media solution was removed and the fish were washed three times with 100 μL E3 media to remove the remaining nanoprobe. The fish were incubated with E3 media containing NaHS (the donor of H2S, 0 μM, 50 μM and 100 μM respectively) for another 30 min at 28 °C before the removal of the media solution. After that, the fish were washed three times with 100 μL E3 media and observed on an Olympus IX71 inverted fluorescence microscope equipped with a DP72 color CCD. All the photographs were taken under identical exposure conditions.Results and discussion
Preparation of the Tat peptide-functionalized nanoprobe
For the preparation of the Tat peptide-functionalized nanoprobe, the H2S recognition probe (TPE-3) was first synthesized, as shown in Scheme S1 (ESI†). The structures of the intermediates and the final product (TPE-3) were characterized using 1H NMR and mass spectroscopy (Fig. S1–S3, ESI†). Subsequently, a modified nanoprecipitation method was utilized for the fabrication of the Tat peptide-functionalized nanoprobe,17b as shown in Scheme 1. In brief, a THF solution containing TPE-3, DSPE-PEG2000, DOTAP and DSPE-PEG2000-Mal was mixed with water under sonication. Upon evaporation of THF, the hydrophobic TPE-3 molecules were embedded in the hydrophobic inner part of the formed micelle-like particles; while the hydrophilic PEG chains served as the exterior part and extended into the aqueous phase. Afterwards, the Tat peptide, which facilitates cell penetration for the nanoparticles, was further incorporated onto the nanoparticles to obtain the Tat peptide-modified nanoprobe. No precipitation from the nanoprobe dispersion can be observed after storage at 4 °C for two months, suggesting the excellent colloidal stability of the prepared Tat peptide-functionalized nanoprobe.Fig. 1A demonstrates a high-resolution transmission electron microscopy (HR-TEM) image for the nanoprobe. It is clearly seen that most of the nanoparticles are discrete spheres with ca. 60.0 nm of diameter. Dynamic light scattering (DLS) measurements indicate that the nanoprobe particles exhibit an average hydrodynamic diameter of 68.5 nm (Fig. 1B). In addition, the zeta potential of the self-assembled nanoprobe was determined to be +48.3 mV (Fig. S4, ESI†).
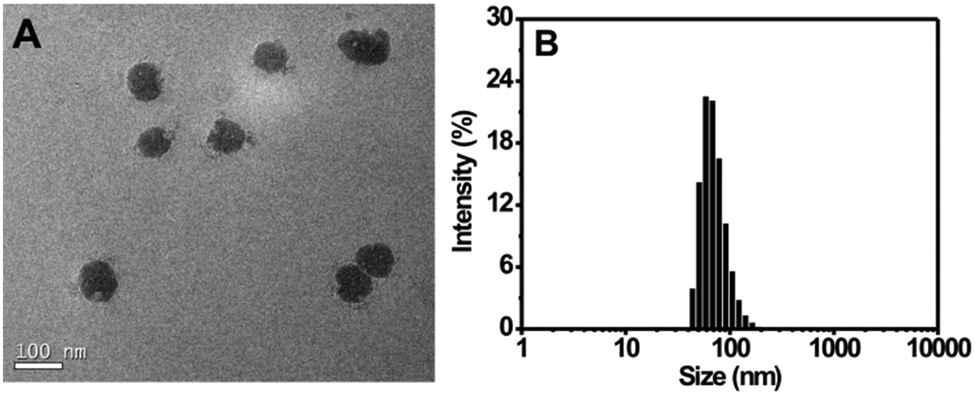 | ||
| Fig. 1 (A) HR-TEM image of the nanoprobe. (B) Size distribution of the nanoprobe as determined using DLS. | ||
Fluorescent sensing of H2S in totally aqueous media
It is a well known fact that AIE-active molecules are nonemissive when dissolved in good solvents, but show strong fluorescence in poor solvents or in the solid state due to the restricted intramolecular rotation (RIR) mechanism.20 In this study, TPE-2 (the reaction product of the probe and H2S) was synthesized to verify its AIE properties (Scheme S1, ESI†). The photoluminescence (PL) properties of TPE-2 were investigated using DMSO/water mixtures of varied water contents as the solvents. As shown in Fig. S5A (ESI†), TPE-2 is nonemissive in pure DMSO, but, with increasing water percentage in DMSO/water mixtures from 0 to 80 vol%, the fluorescence intensity gradually increases. As the percentage of water is further increased to 99 vol%, a sharp enhancement in the fluorescence intensity at 550 nm can be observed (Fig. S5B, ESI†). Also, at 99 vol% water content, the fluorescence intensity is nearly 26-fold higher than that in the pure DMSO, indicating an obvious AIE effect.The absorption and emission properties of the nanoprobe towards H2S were studied in PBS buffer solution (pH 7.4). As revealed in Fig. S6 (ESI†), the nanoprobe dispersion shows an absorption peak at around 365 nm. At a H2S concentration of 200 μM, the absorption at around 450 nm gradually increases with reaction time.
Fig. 2A shows the fluorescence titration experiment of the nanoprobe dispersion upon addition of varied amounts of H2S. It can be seen from the figure that the nanoprobe dispersion itself exhibits a weak fluorescence at 550 nm; however, a strong emission band at 550 nm appears upon the addition of H2S. Furthermore, the addition of H2S into the nanoprobe solution induces an obvious fluorescent color change from colorless to green (inset of Fig. 2B). The above results clearly indicate the occurrence of a chemical reaction between the nanoprobe and H2S. In addition, by plotting the emission intensity at 550 nm versus H2S concentration, a good linear relationship was also established in the range of 0–40 μM (Fig. S7, ESI†), which covers the physiological levels of H2S (10–600 μM).8a,21 Moreover, the detection limit for the nanoprobe has been determined to be 0.09 μM (Fig. S6, ESI†).
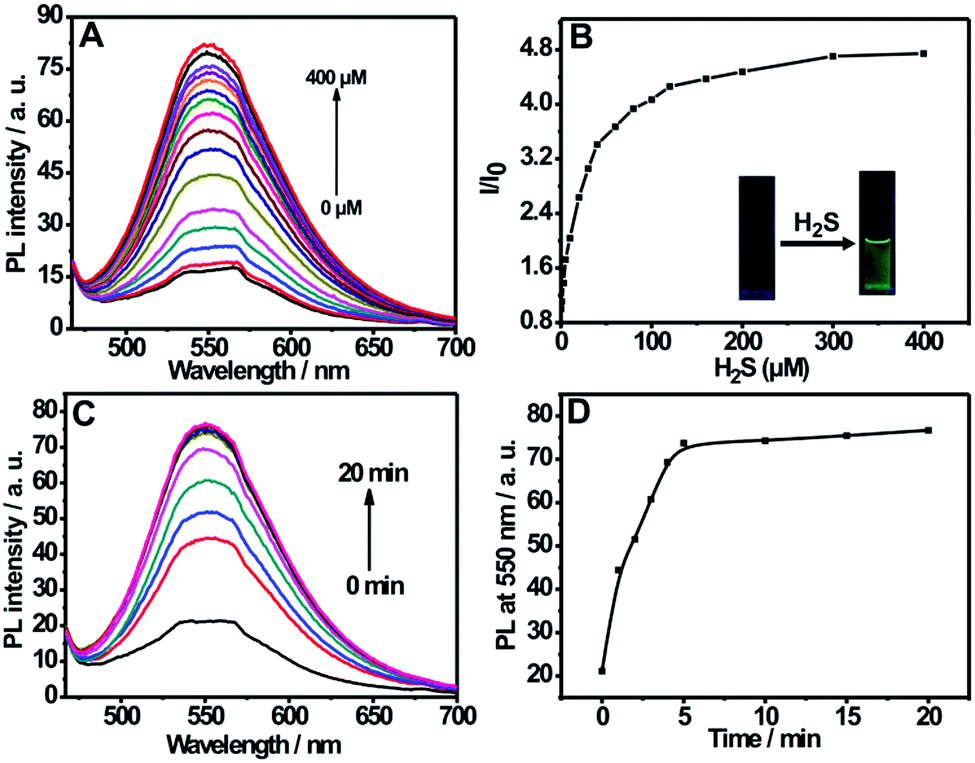 | ||
| Fig. 2 (A) Fluorescence emission spectra of the nanoprobe dispersion (0.01 mg mL−1) in the presence of different concentrations of H2S (0–400 μM) in PBS buffers (pH 7.4, 10 mM). (B) The plot of fluorescence intensity ratio (I/I0) versus H2S concentrations, I0 is the fluorescence intensity of the nanoprobe in the absence of H2S; the inset displays the photographs for the nanoprobe dispersion in the absence and presence of H2S under a hand-held UV lamp. (C) Time-dependent fluorescence spectra and (D) plot of fluorescence intensity at 550 nm (I550) versus time for the nanoprobe dispersion upon addition of 200 μM H2S, λex = 452 nm. | ||
The response time is an important factor for reaction-based probes. The time-dependent variation in fluorescence for the nanoprobe dispersion in response to H2S was recorded. As depicted in Fig. 2C and D, the fluorescence intensity at 550 nm increases with the reaction time and then levels off at about 5 min, indicating that the nanoprobe can serve as a rapid analytical sensor for H2S detection. In addition, the suitable pH range of H2S sensing was investigated (Fig. S8, ESI†). No remarkable fluorescence change was observed between pH 5.0 and 12.0 in the absence of H2S. Upon the addition of H2S, the sensing system exhibits fairly stable fluorescent intensities from pH 7.0 to 12.0, suggesting that the nanoprobe is operational in most biological environments. Moreover, the effect of pH on the size of the self-assembled nanoprobe has also been evaluated, as shown in Fig. S9 (ESI†). No obvious size changes of the nanoprobe were observed between pH 3.0 and 9.0, indicating that the nanoprobe is fairly stable in various pH environments.
The selectivity of the nanoprobe towards H2S was evaluated by utilizing a number of putative interferents, and the results are presented in Fig. 3A. It is clear that the fluorescence intensity ratio (I/I0) variation for the nanoprobe significantly increases upon the addition of H2S. In comparison, no obvious change in the fluorescence intensity ratio can be observed in the presence of other analytes such as metal ions (Na+, Mg2+, Ca2+, Cu2+), anions (F−, Cl−, Br−, I−, NO2−, NO3−, H2PO42−, SO32−, SO42−, S2O32−, CO32−), thiols (Cys, GSH, Hcy) and ROS (H2O2, ClO−, O2−, ˙OH, 1O2). Furthermore, competition experiments using these interferents were also performed (Fig. 3B). It is obvious that the co-existence of anions and thiols has a negligible interfering effect on H2S sensing. The above results indicate that the nanoprobe could potentially be used to detect H2S in biological environments.
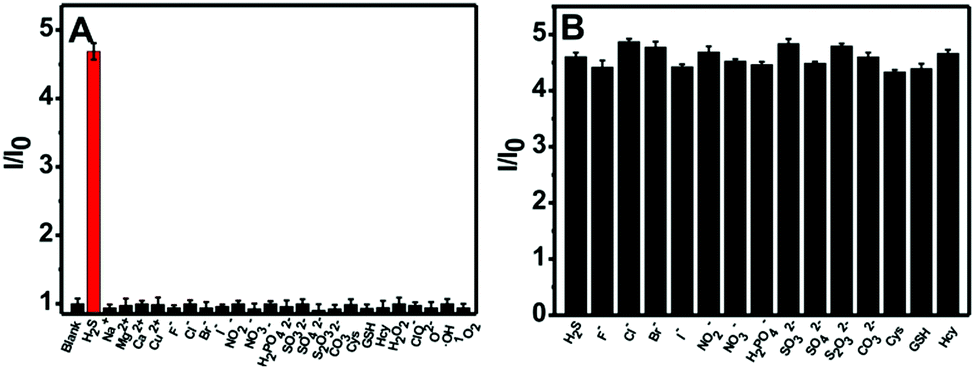 | ||
| Fig. 3 (A) Fluorescence intensity ratio (I/I0) at 550 nm for the nanoprobe dispersion (0.01 mg mL−1) in the presence of 200 μM H2S or 1 mM metal ions or anions, 200 μM ROS and 5.0 mM thiols respectively. (B) Fluorescence intensity ratio (I/I0) at 550 nm for the nanoprobe (0.01 mg mL−1) in the presence of 200 μM of H2S and one potential interferent (1 mM anions or 5.0 mM thiols respectively). λex = 452 nm. | ||
It has been well documented that the 2,4-dinitrophenyl (DNP) ether moiety can be cleaved by H2S (predominantly existing as HS− at physiological pH) to release hydroxy-containing fluorophores.22 To further confirm the cleavage of the 2,4-dinitrophenyl (DNP) ether moiety by H2S, the reaction solution of TPE-3 with H2S was analyzed using ESI-MS (Fig. S10, ESI†). The peaks at m/z 531.8 and 563.1 corresponding to TPE-2 ([M + K+]+, [M + K+ + CH3OH]+) were found in the MS spectra, which verifies the H2S induced thiolytic cleavage in this process. In addition, the expected cleavage was further confirmed using HPLC (Fig. S11, ESI†), which indicates the direct conversion from TPE-3 into TPE-2, with no detectable intermediate formation.
Fluorescence imaging of exogenous and endogenous H2S in live cells
Based on the prominent spectral features of the nanoprobe, we set out to evaluate its potential applications for H2S imaging in living cells. MTT assays were carried out first to assess the cytotoxicity of the nanoprobe towards HeLa cells. As displayed in Fig. S12 (ESI†), with 24 h incubation, there is no obvious decrease in cell viability after treatment with the nanoprobe for concentrations even up to 0.1 mg mL−1, confirming that the nanoprobe can be used for cell imaging.Next, the nanoprobe was used to track exogenous H2S levels in live HeLa cells and the results are given in Fig. 4. As can be seen, both the control (Fig. 4E) and the cells treated only with the nanoprobe (Fig. 4F) exhibit no fluorescence under the same exposure conditions. In contrast, the cells pretreated with the nanoprobe and then incubated with 50 μM or 100 μM H2S exhibit strong green fluorescence. And those treated with 100 μM H2S (Fig. 4H) display brighter fluorescence than those exposed to 50 μM H2S (Fig. 4G). To quantify the emission intensity of the nanoprobe in cells, the imaging ratio (ratio of intensity to control) was calculated using the software Image J, and the result is given in Fig. 4(II). With an increase in H2S concentration, a significant increment of the emission ratio can be observed. This imaging experiment clearly demonstrates that the nanoprobe is cell membrane permeable and can act as an excellent sensing system for mapping intracellular H2S.
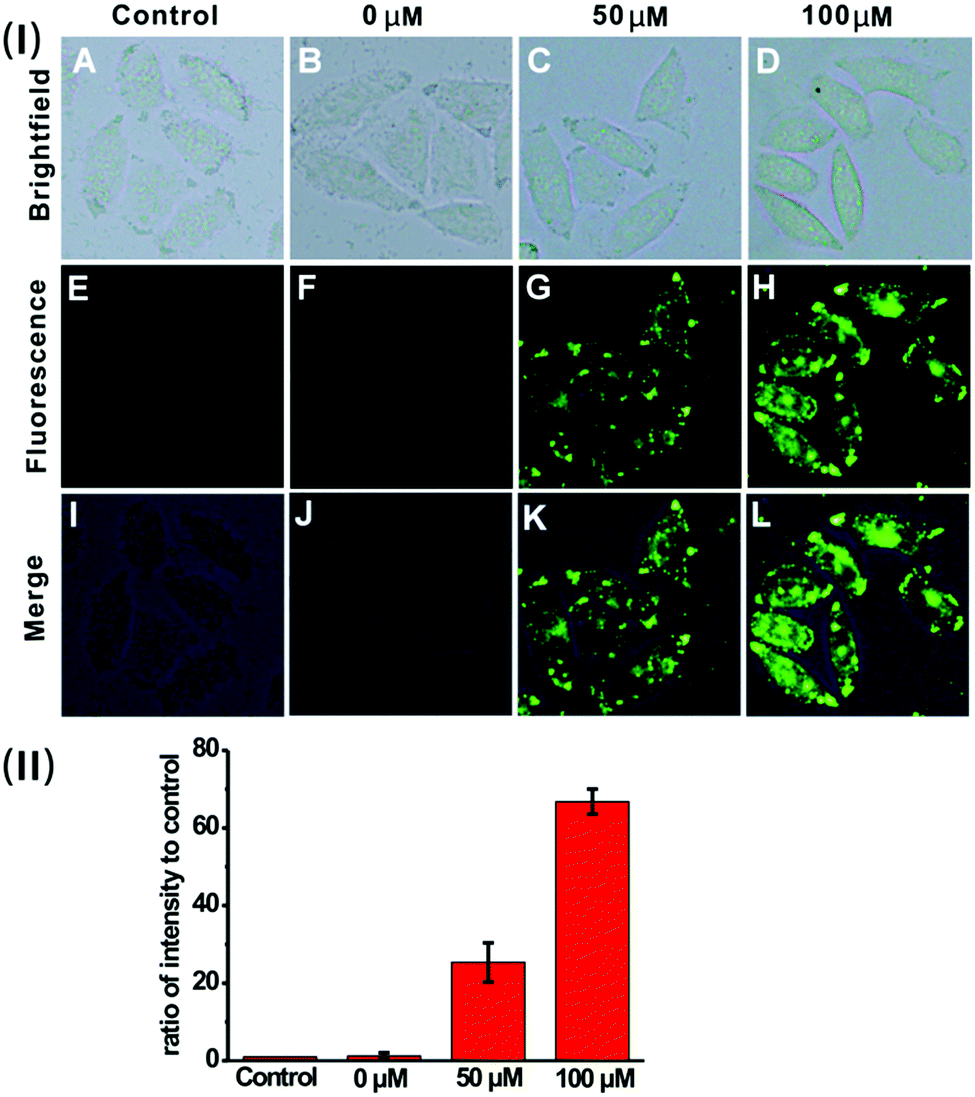 | ||
| Fig. 4 (I) Fluorescence images of untreated HeLa cells (the control) (A, E and I), and HeLa cells pretreated with the nanoprobe (0.01 mg mL−1) and then incubated with H2S at a concentration of 0 μM (B, F and J), 50 μM (C, G and K) and 100 μM (D, H and L) respectively. (II) Quantification of fluorescence intensity per unit area for the image group (E–H). The histograms were plotted as a ratio of intensity to that of the control E, respectively. Values are the mean ± s.d. For n = 5. | ||
We then sought to test the feasibility of the nanoprobe for detecting and imaging the endogenous H2S in live cells. Previous investigation suggests that H2S can be endogenously generated from glutathione (GSH) or cysteine (Cys) by using cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) as the catalysts.23 HeLa cells were pre-treated with GSH (0 mM, 1 mM or 2 mM), and then incubated with the nanoprobe. As displayed in Fig. 5, the cells exhibit much brighter intracellular green fluorescence compared to the control; moreover, the calculated fluorescence intensity ratio for the GSH-treated cells is much higher than that of the control (Fig. 5(II)). These results indicate that the nanoprobe is capable of detecting endogenous H2S produced by the cells.
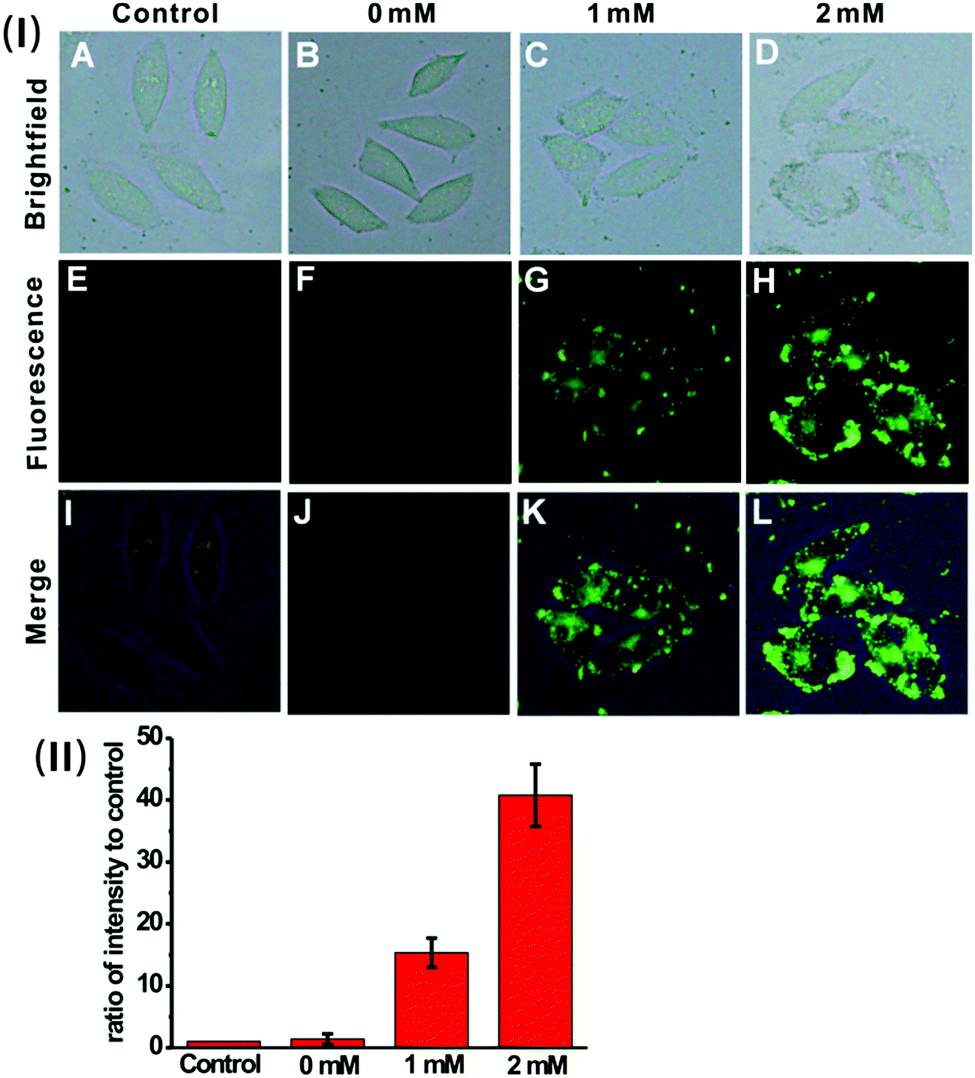 | ||
| Fig. 5 (I) Fluorescence images of untreated HeLa cells (the control) (A, E and I), and HeLa cells incubated with the nanoprobe (0.01 mg mL−1) after being treated with GSH at a concentration of 0 mM (B, F and J), 1 mM (C, G and K) and 2 mM (D, H and L) respectively. (II) Quantification of fluorescence intensity per unit area for the image group (E–H). The histograms were plotted as a ratio of intensity to that of the control E, respectively. Values are the mean ± s.d. For n = 5. | ||
Fluorescence imaging of exogenous H2S in zebrafish
To verify the applicability of the nanoprobe in imaging exogenous H2S in vivo, five-day-old zebrafish larvae were used as the vertebrate model because of their optical transparency during embryonic and larval stages.24Fig. 6 shows the fluorescence images of the zebrafish larvae treated only with the nanoprobe as well as the larvae pre-treated with the nanoprobe and then incubated with 50 μM or 100 μM H2S, respectively. It can be clearly seen that both the control and zebrafish incubated with only the nanoprobe display no fluorescence (Fig. 6E and F). In contrast, as the zebrafish were pretreated with the nanoprobe for a certain time and then incubated with H2S in E3 media for 30 min, bright green fluorescence can be clearly observed, and the green fluorescence becomes brighter with increasing H2S concentration (Fig. 6G, H and (II)). Moreover, the images shown in Fig. 6K and L reveal that the green fluorescence also mainly resides in the digestive system of the zebrafish. These results suggest that the nanoprobe is suitable for the sensing and imaging of H2S in vivo.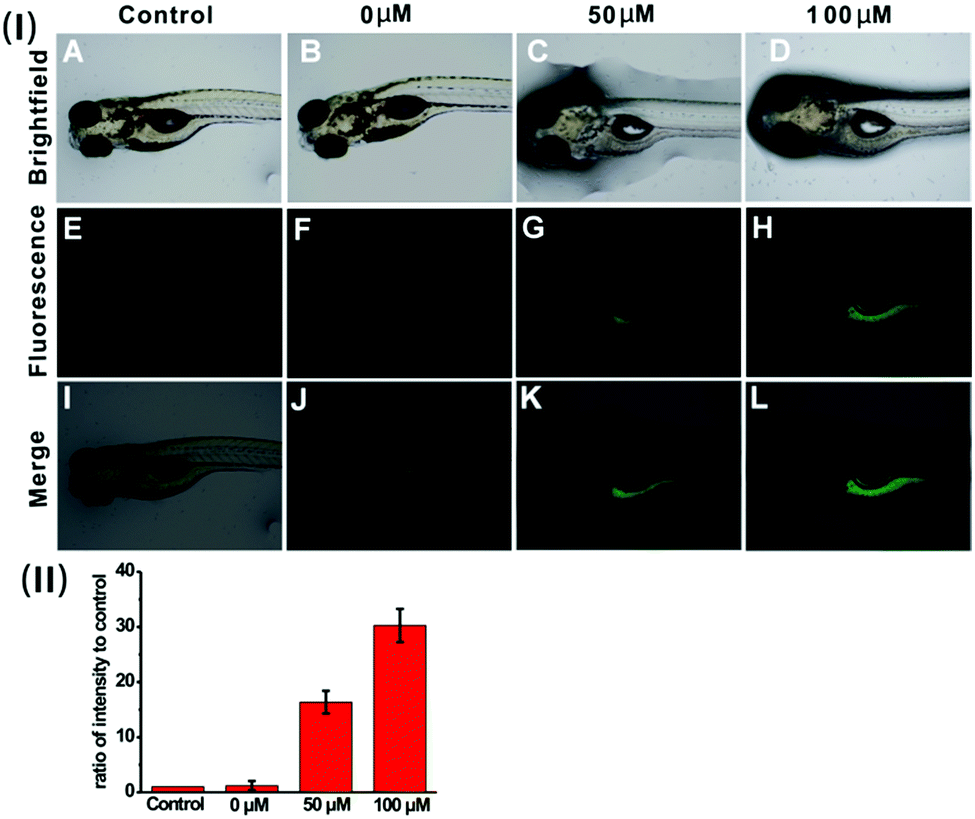 | ||
| Fig. 6 (I) Fluorescent images of untreated zebrafish larvae (the control) (A, E and I), and zebrafish larvae pretreated with the nanoprobe (0.005 mg mL−1) and then incubated with 0 μM H2S (B, F, and J), 50 μM H2S (C, G and K) and 100 μM H2S (D, H and L). (II) Quantification of fluorescence intensity per unit area for the image group (E–H). The histograms were plotted as a ratio of intensity to that of the control E, respectively. Values are the mean ± s.d. For n = 5. | ||
Conclusions
In summary, we have successfully developed an AIE/ESIPT-based fluorescent nanoprobe for the specific detection and imaging of H2S. The nanoprobe displays good water dispersibility, a large Stokes shift (ca.100 nm), good biocompatibility and high selectivity towards H2S over other putative interferents. In addition, cell imaging experiments demonstrate that, upon Tat peptide modification, visualization of the exogenous and endogenous H2S levels in live cells can be achieved for the nanoprobe. Moreover, the nanoprobe has also been found capable of detecting H2S in zebrafish. We anticipate that the nanoprobe may be utilized in real-time monitoring of H2S levels and serves as an efficient tool for studying H2S-related molecular events in biological systems.Acknowledgements
We gratefully acknowledge the financial support from the Science and Technology Planning Project of Guangzhou (Project No. 201607020015), the National Key Basic Research Program of China (Project No. 2013CB834702), NSFC (21474031, 21574044 and 51673066), the Natural Science Foundation of Guangdong Province (2016A030312002) and the Fundamental Research Funds for the Central Universities (2015ZY013).Notes and references
- G. D. Yang, L. Y. Wu, B. Jiang, W. Yang, J. S. Qi, K. Cao, H. Meng, A. K. Mustafa, W. T. Mu, S. M. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS PubMed.
- J. W. Calvert, S. Jha, S. Gundewar, W. J. Elrod, A. Ramachandran, C. B. Pattillo, C. G. Kevil and D. J. Lefer, Circ. Res., 2009, 105, 365–374 CrossRef CAS PubMed.
- G. D. Yang, L. Y. Wu and R. Wang, FASEB J., 2006, 20, 553–555 CAS.
- L. Li, M. Bhatia, Y. Z. Zhu, Y. C. Zhu, R. D. Ramnath, Z. J. Wang, F. B. Anuar, M. Whiteman, M. Salto-Tellez and P. K. Moore, FASEB J., 2005, 19, 1196–1198 CAS.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485–1488 CrossRef CAS PubMed.
- P. Kamoun, M.-C. Belardinelli, A. Chabli, K. Lallouchi and B. Chadefaux-Vekemans, Am. J. Med. Genet., 2003, 116A, 310–311 CrossRef PubMed.
- W. Yang, G. Yang, X. Jia, L. Wu and R. Wang, J. Physiol., 2005, 569, 519–531 CrossRef CAS PubMed.
- (a) A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS PubMed; (b) H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. A. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed; (c) J. Zhang and W. Guo, Chem. Commun., 2014, 50, 4214–4217 RSC; (d) T. S. Bailey and M. D. Pluth, J. Am. Chem. Soc., 2013, 135, 16697–16704 CrossRef CAS PubMed; (e) W. Sun, J. Fan, C. Hu, J. Cao, H. Zhang, X. Xiong, J. Wang, S. Cui, S. Suna and X. Peng, Chem. Commun., 2013, 49, 3890–3892 RSC; (f) S. Chen, Z.-J. Chen, W. Ren and H. W. Ai, J. Am. Chem. Soc., 2012, 134, 9589–9592 CrossRef CAS PubMed; (g) F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 2852–2854 RSC; (h) Q. Wan, Y. Song, Z. Li, X. Gao and H. Ma, Chem. Commun., 2013, 49, 502–504 RSC; (i) S. K. Bae, C. H. Heo, D. J. Choi, D. Sen, E. H. Joe, B. R. Cho and H. M. A. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS PubMed; (j) R. Wang, F. Yu, L. Chen, H. Chen, L. Wang and W. Zhang, Chem. Commun., 2012, 48, 11757–11759 RSC; (k) P. S. Zhang, J. Li, B. W. Li, J. S. Xu, F. Zeng, J. Lv and S. Z. Wu, Chem. Commun., 2015, 51, 4414–4416 RSC.
- (a) X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529–10531 RSC; (b) M. D. Hammers and M. D. Pluth, Anal. Chem., 2014, 86, 7135–7140 CrossRef CAS PubMed; (c) Y. Chen, C. Zhu, Z. Yang, J. Chen, Y. He, Y. Jiao, W. He, L. Qiu, J. Cen and Z. Guo, Angew. Chem., Int. Ed., 2013, 52, 1732–1735 CrossRef; (d) C. Liu, J. Pan, S. Li, Y. Zhao, L. Y. Wu, C. E. Berkman, A. R. Whorton and M. Xian, Angew. Chem., Int. Ed., 2011, 50, 10327–10329 CrossRef CAS PubMed; (e) Z. Xu, L. Xu, J. Zhou, Y. Xu, W. Zhu and X. Qian, Chem. Commun., 2012, 48, 10871–10873 RSC; (f) Y. Qian, J. Karpus, O. Kabil, S.-Y. Zhang, H.-L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed; (g) X. Wang, J. Sun, W. Zhang, X. Ma, J. Lv and B. Tang, Chem. Sci., 2013, 4, 2551–2556 RSC.
- (a) K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003–18005 CrossRef CAS PubMed; (b) M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 7390–7392 RSC; (c) F. Hou, J. Cheng, P. Xi, F. Chen, L. Huang, G. Xie, Y. Shi, H. Liu, D. Bai and Z. Zeng, Dalton Trans., 2012, 41, 5799–5804 RSC; (d) L. E. Santos-Figueroa, C. de la Torre, S. El Sayed, F. Sancenón, R. Martínez-Máñez, A. M. Costero, S. Gil and M. Parra, Eur. J. Inorg. Chem., 2014, 41–45 CrossRef CAS; (e) X. Qu, C. Li, H. Chen, J. Mack, Z. Guo and Z. Shen, Chem. Commun., 2013, 49, 7510–7512 RSC.
- (a) Y. Huang, P. S. Zhang, M. Gao, F. Zeng, A. J. Qin, S. Z. Wu and B. Z. Tang, Chem. Commun., 2016, 52, 7288–7291 RSC; (b) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed; (c) J. Chen, F. Zeng, S. Z. Wu, J. Q. Zhao, Q. M. Chen and Z. Tong, Chem. Commun., 2008, 5580–5582 RSC.
- (a) W. Y. Lin, L. Yuan, Z. M. Cao, Y. M. Feng and J. Z. Song, Angew. Chem., Int. Ed., 2010, 49, 375–379 CrossRef CAS PubMed; (b) C. M. Yu, X. Z. Li, F. Zeng, F. Y. Zheng and S. Z. Wu, Chem. Commun., 2013, 49, 403–405 RSC; (c) X. Hou, J. Peng, F. Zeng, C. Yu and S. Z. Wu, Mater. Chem. Front., 2017 10.1039/c6qm00112b.
- (a) M. Gao, Q. Hu, G. Feng, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 3438–3442 RSC; (b) R. Zhang, M. Gao, S. Bai and B. Liu, J. Mater. Chem. B, 2015, 3, 1590–1596 RSC; (c) L. Peng, Y. Zheng, X. Wang, A. Tong and Y. Xiang, Chem. – Eur. J., 2015, 21, 4326–4332 CrossRef CAS PubMed; (d) Z. G. Song, R. T. K. Kwok, E. Zhao, Z. K. He, Y. N. Hong, J. W. Y. Lam, B. Liu and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17245–17254 CrossRef CAS PubMed; (e) L. Peng, M. Gao, X. L. Cai, R. Zhang, K. Li, G. Feng, A. Tong and B. Liu, J. Mater. Chem. B, 2015, 3, 9168–9172 RSC.
- (a) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; (b) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC; (c) R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC; (d) X. Huang, X. Gu, G. Zhang and D. Q. Zhang, Chem. Commun., 2012, 48, 12195–12197 RSC; (e) R. S. Singh, R. K. Gupta, R. P. Paitandi, M. Dubey, G. Sharma, B. Kochb and D. S. Pandey, Chem. Commun., 2015, 51, 9125–9128 RSC; (f) J. Yang, L. Yu, Y. Yu, Z. C. Ren, Q. Peng, S. H. Ye, Q. Q. Li and Z. Li, Mater. Chem. Front., 2017 10.1039/c6qm00014b; (g) X. Y. Yao, X. Ma and H. Tian, J. Mater. Chem. C, 2014, 2, 5155–5160 RSC; (h) X. F. Hou, F. Zeng and S. Z. Wu, Biosens. Bioelectron., 2016, 85, 317–323 CrossRef CAS PubMed; (i) R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2015, 21, 7231–7237 CrossRef CAS PubMed; (j) Q. C. Yao, X. L. Lu and M. Xia, New J. Chem., 2014, 38, 2693–2700 RSC; (k) D. E. Wu, Q. C. Yao and M. Xia, Phys. Chem. Chem. Phys., 2015, 17, 3287–3294 RSC.
- (a) J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed; (b) G. Li, D. Zhu, Q. Liu, L. Xue and H. Jiang, Org. Lett., 2013, 15, 924–927 CrossRef CAS PubMed; (c) T. Kim, H. J. Kang, G. Han, S. J. Chung and Y. Kim, Chem. Commun., 2009, 5895–5897 RSC.
- (a) J. M. Hu and S. Y. Liu, Acc. Chem. Res., 2014, 47, 2084–2095 CrossRef CAS PubMed; (b) J. M. Hu, G. Y. Zhang, Z. S. Ge and S. Y. Liu, Prog. Polym. Sci., 2014, 39, 1096–1143 CrossRef CAS.
- (a) W. Qin, K. Li, G. X. Feng, M. Li, Z. Y. Yang, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2014, 24, 635–643 CrossRef CAS; (b) D. Ding, C. C. Goh, G. X. Feng, Z. J. Zhao, J. Liu, R. R. Liu, N. Tomczak, J. L. Geng, B. Z. Tang, L. G. Ng and B. Liu, Adv. Mater., 2013, 25, 6083–6088 CrossRef CAS PubMed; (c) L. Yan, Y. Zhang, B. Xu and W. J. Tian, Nanoscale, 2016, 8, 2471–2487 RSC; (d) X. Li, K. Ma, S. Zhu, S. Yao, Z. Liu, B. Xu, B. Yang and W. J. Tian, Anal. Chem., 2014, 86, 298–303 CrossRef CAS PubMed; (e) Z. Peng, X. Feng, B. Tong, D. Chen, J. Shi, J. Zhi and Y. P. Dong, Sens. Actuators, B, 2016, 232, 264–268 CrossRef CAS; (f) A. Chowdhury, P. Howlader and P. S. Mukherjee, Chem. – Eur. J., 2016, 22, 7468–7478 CrossRef CAS PubMed; (g) Y. Gao, G. Feng, T. Jiang, C. Goh, L. Ng, B. Liu, B. Li, L. Yang, J. Hua and H. Tian, Adv. Funct. Mater., 2015, 25, 2857–2866 CrossRef CAS.
- (a) M. Chen and M. Yin, Prog. Polym. Sci., 2014, 39, 365–395 CrossRef CAS; (b) C. Yu, M. Luo, F. Zeng, F. Zheng and S. Z. Wu, Chem. Commun., 2011, 47, 9086–9088 RSC.
- P. S. Zhang, X. F. Jiang, X. Z. Nie, Y. Huang, F. Zeng, X. T. Xia and S. Z. Wu, Biomaterials, 2016, 80, 46–56 CrossRef CAS PubMed.
- (a) M. P. Aldred, C. Li, G. F. Zhang, W. L. Gong, A. D. Q. Li, Y. F. Dai, D. G. Ma and M. Q. Zhu, J. Mater. Chem., 2012, 22, 7515–7528 RSC; (b) G. Iasilli, A. Battisti, F. Tatussi, F. Fuso, M. Allegrini, G. Ruggeri and A. Pucci, Macromol. Chem. Phys., 2014, 215, 499–506 CrossRef CAS; (c) Y. Wu, J. Wang, F. Zeng, S. Huang, H. Xie, C. Yu and S. Z. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 1511–1519 CrossRef CAS PubMed.
- (a) G. Yang, L. Wu, B. Jiang, B. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS PubMed; (b) A. Papapetropoulos, A. Pyriochou, Z. Altaany, G. Yang, A. Marazioti, Z. Zhou, M. G. Jeschke, L. K. Branski, D. N. Hern-don, R. Wang and C. Szabo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21972–21977 CrossRef CAS PubMed; (c) K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CAS.
- V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC.
- (a) N. Shibuya, M. Tanaka, M. Yoshida, Y. Ogasawara, T. Togawa, K. Ishii and H. Kimura, Antioxid. Redox Signaling, 2009, 11, 703–714 CrossRef CAS PubMed; (b) S. Singh, D. Padovani, R. A. Leslie, T. Chiku and R. Banerjee, J. Biol. Chem., 2009, 284, 22457–22466 CrossRef CAS PubMed; (c) T. R. Kumar, A. L. Wiseman, G. Kala, S. V. Kala, M. M. Matzuk and M. W. Lieberman, Endocrinology, 2000, 141, 4270–4277 CAS; (d) M. W. Lieberman, A. L. Wiseman, Z. Z. Shi, B. Z. Carter, R. Barrios, C. N. Ou, P. Chevez-Barrios, Y. Wang, G. M. Habib, J. C. Goodman, S. L. Huang, R. M. Lebovitz and M. M. atzuk, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 7923–7926 CrossRef CAS PubMed.
- (a) S. K. Ko, X. Chen, J. Yoon and I. Shin, Chem. Soc. Rev., 2011, 40, 2120–2130 RSC; (b) G. Wu, F. Zeng, C. Yu, S. Wu and W. Li, J. Mater. Chem. B, 2014, 2, 8528–8537 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures, 1H NMR spectra, mass spectra, absorption spectrum, AIE behaviour, fluorescence spectra, detection limit, effect of pH, reaction mechanism and cell viability. See DOI: 10.1039/c6qm00223d |
| This journal is © the Partner Organisations 2017 |