
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phase sequence in diisopropylammonium iodide: avoided ferroelectricity by the appearance of a reconstructed phase†
A.
Piecha-Bisiorek
*a,
A.
Gągor
b,
D.
Isakov
cd,
P.
Zieliński
e,
M.
Gałązka
e and
R.
Jakubas
a
aFaculty of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: anna.piecha@chem.uni.wroc.pl
bW. Trzebiatowski Institute of Low Temperature and Structure Research PAS, P.O. Box 1410, 50-950 Wrocław, Poland
cUniversity of Minho, Centre of Physics, Campus de Gualtar, 4710-057 Braga, Portugal
dUniversity of Oxford, Department of Materials, Parks Road, Oxford OX1 3PH, UK
eThe H. Niewodniczański Institute of Nuclear Physics, PAS, Radzikowskiego 152, 31-342 Kraków, Poland
First published on 9th January 2017
Abstract
Crystals of diisopropylammonium iodide are synthesized, grown and characterized. Two phases: P21/m (Z = 1) and P212121 (Z = 2) are observed. In contrast with analogous compounds no polar phase occurs, despite a critical-like electric behaviour. A phenomenological theory is proposed to describe the thermodynamics of the whole family of diisopropylammonium halides.
Materials with unique optical, electric and magnetic properties are constantly synthesized and studied to respond to the demands of modern technology. Progressive miniaturization of electronic components and equipment, an increase in the operating speed, enhancement of the information storage density and minimization of the electricity sources are the biggest challenges faced by the world's leading laboratories working on the synthesis of new classes of materials. One of the most important groups of polar compounds used on a large scale in industrial production are ferroelectrics and relaxors, particularly in the form of thin layers or composites.1–3
Ferroelectrics are polar substances, in a solid state (crystalline or polymeric) or in a liquid-crystalline state (liquid crystals), in which a spontaneous polarisation (Ps) is present within a certain temperature range. A reversibility of the polarization by an applied electric field distinguishes the ferroelectrics from pyroelectrics and is witness to a spontaneous symmetry breaking origin of the phenomenon. The field causes switching between various domains of the low symmetry phase. The present technological applications of the ferroelectrics are almost entirely restricted to inorganic perovskite materials e.g. lead zirconate titanate (PZT) and barium titanate (BTO).4–6
However, in recent years there has been a significant breakthrough in small-molecular-weight organic single- or two-component polar materials which are characterized by excellent physical properties such as an extremely high value of the spontaneous polarization, a high Curie temperature, good thermal stability, mechanical flexibility, optical transparency and lightweight.7–17 The new materials are also environment-friendly, easy to prepare, relatively non-expensive and non-toxic. The most spectacular example is the well-known croconic acid which belongs to the group of single-component hydrogen-bonded ferroelectrics and possesses as high a spontaneous polarization as about 21 μC cm−2.7
Intense research has been recently focused on ionic materials as potential molecule-based ferroelectrics or piezoelectrics. The most spectacular examples of such type of compounds appeared to be simple organic derivatives of diisopropylammonium chloride18 (DIPAC) and diisopropylammonium bromide (DIPAB),19,20 both characterized by the highest Curie temperature (Tc) among organic ferroelectrics. DIPAB has the highest known spontaneous polarization Ps = 23 μC cm−2 among polar materials. Following the discovery of the diisopropylammonium ferroelectrics, it seems obvious that searching for new polar compounds was extended to an iodide analog, [i-(C3H7)2NH2+][I−], (DIPAI) whose dielectric response resembles those obtained for chloride or bromide analogs. The DIPAI crystals are isomorphic at room temperature to DIPAB-1P and DIPAC analogues (more in the ESI, Table S1†).21,22 They adopt the orthorhombic, chiral P212121 space group with one halogen and one i-(C3H7)2NH2+ ion in the asymmetric unit. The supramolecular structure of these simple ionic salts is built of hydrogen-bonded i-(C3H7)2NH2+ groups with halogen anions forming chains propagating along the aortho direction, Fig. 1(a) and (b). The phase situation of DIPAI at high temperatures differs substantially for those encountered for DIPAB and DIPAC18–20 because the ferroelectric phase does not occur, although some signatures of a ferroelectric phase transition are detected in dielectric permittivity measurements. By heating (up to about 430 K) the DIPAI crystals undergo complex structural rearrangements at 376 K/364 K (heating/cooling scan) – Fig. 2(a) and similarly to the prior diisopropylammonium compounds, shows also a high thermal stability (see Fig. S1†).
 | ||
| Fig. 1 The crystal structure of DIPAI projected along the aortho = bmono direction at (a) 295 K, phase III; (c) 405 K, phase I. The orthorhombic unit cell may be transformed to monoclinic using (0 0.5 −0.5 −1 0 0 0 0.5 0.5) rotation matrix and (0.25 0 0.25) translation vector; (b) hydrogen bonds in orthorhombic phase III, T = 295 K; (d) hydrogen bonds in the high-temperature phase I, T = 405 K; split positions are occupied with 50% probability. The chains propagate along the aortho = bmono direction. In phase III the H-bonds have similar strength; in I, the in-plane bonds are stronger. | ||
 | ||
| Fig. 2 (a) DSC curves upon cooling and heating runs (2 K min−1, m = 7.828 mg); (b) temperature dependence of the real part of complex dielectric permittivity during the cooling scan, along the aortho axis; (c) temperature dependence of second harmonic intensity of DIPAI powder at the heating (red)/cooling (blue) cycle; (d) domain structure in the monoclinic phase (I) of DIPAI bearing traces of stress-free planar domain boundaries typical of ferroelastic species. | ||
The phase transition is marked by a reconstruction of the cationic substructure, in which the i-(C3H7)2NH2+ ions reorient by approx. 90° along the internal long axis and rotate along the [100] orthorhombic direction. Simultaneously, the i-(C3H7)2NH2+ ions split into two equivalent positions, related by the .m. mirror plane. This implies rotational jumps of each i-(C3H7)2NH2+ ion between these two metastable sites, see Fig. S2.† Both processes change the packing of the chains and induce deformation of the anionic substructure, Fig. 1(c) and (d). After the transition, the crystal structure of DIPAI elongates in the bortho direction from 8.24 Å at room temperature to 8.43 Å at 405 K. Also the distances between I− in the chains increase from 5.76 to 5.97 Å. Unlike in DIPAB-1P in DIPAI, there is a substantial difference between the strength of the N–H⋯I hydrogen bonds between the two phases. In DIPAB the phase transitions have an insignificant impact on the geometry of hydrogen bonds whereas in DIPAI the bonds in the bmono direction weaken after the transformation; the donor to acceptor distance increases from 3.59 Å at rt to 3.85 Å at 405 K; whereas the strength of the ‘in plane’ bonds (bonds propagating in (a,c) plane) remains similar in both phases. Tables S2, S3 and Fig. S3† summarize the details concerning geometry and H-bonds in DIPAI.
DIPAI and DIPAB 1-P are isostructural at room temperature having similar crystal packing presented in Fig. S3.† The larger ions cause the increase in the interatomic distances and increase in the unit cell volume, which in turn affects the strength of the hydrogen bond interactions. Along with increasing ionic radii an extension of all the donor-to-acceptor distances is observed. The bonds across the chains elongate from 2.50 to 2.81 Å whereas those along the chains change from 2.46 to 2.67 Å for DIPAB 1-P and DIPAI, respectively. In DIPAI the phase transition to the disordered P21/m phase is shifted by about 50 degrees towards lower temperatures compared to DIPAB 1-P indicating that the N–H⋯I bonds are much weaker than those of N–H⋯Br. So weak bonds are, apparently, not able to overcome thermally activated rotations of DIPA.
Powder diffraction diagrams performed for DIPAI at high temperatures reveal one, pronounced structural phase transition of the first-order type. The temperature evolution of the area intensity of the (300) diffraction peak as well as the d300 lattice spacing, illustrated in Fig. S4 and S5,† show a step-like anomaly around 376 K during heating. It is accompanied by the splitting of diffraction patterns characteristic of the symmetry change from the orthorhombic P212121 to the monoclinic P21/m. The phase transition is reversible but shows a large thermal hysteresis (ΔT = 12 K). When cooled down, the monoclinic phase vanishes below 364 K. A coexistence of monoclinic and orthorhombic phases is observed at 376 K (heating) and 364 K (cooling).
The phase transition anomalies have been also investigated by measurements of electric permittivity. The observed dielectric response (Fig. 2(b)) turned out to be surprisingly close to those attributed to pure ferroelectric materials.5 The temperature-dependent dielectric constant of DIPAI reveals relatively large dielectric anomaly, which shows a peak as high as 1150 at the lowest measured frequency (135 Hz) at about 364 K upon cooling. This is well consistent with the DSC data that confirm the occurrence of a reversible phase transition. The dielectric dispersion studies between 135 Hz and 2 MHz reveal that the fundamental relaxation process in DIPAI above 364 K takes place in the kHz frequency region (see Fig. 3). Such a dielectric response indicates the presence of a critical, or quasi-critical, slowing down of the macroscopic relaxation time which is often encountered in the ferroelectric crystals of the order–disorder type. The dynamic parameters of this process (τ, τ0, Ea) are presented in the ESI (part 3).†
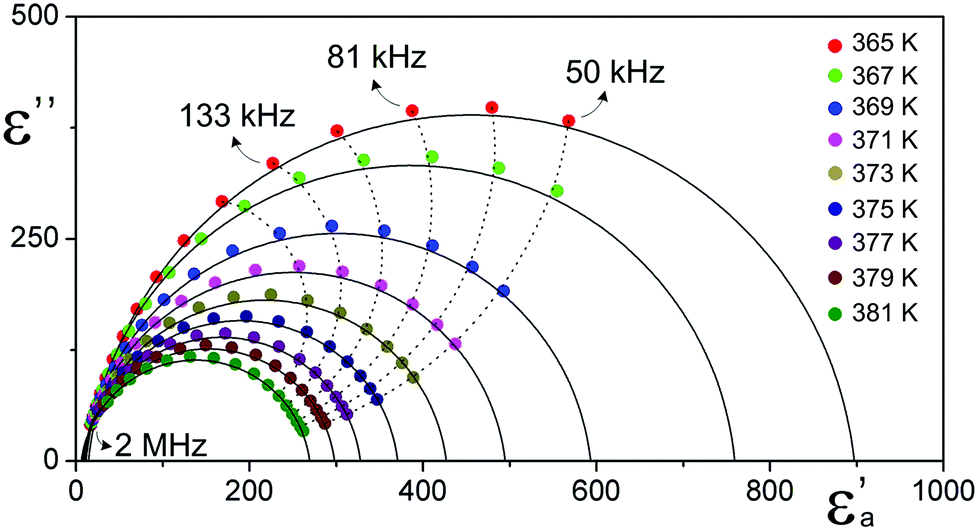 | ||
| Fig. 3 Cole–Cole plots, ε′′ vs. ε′ for selected temperatures showing the relaxation character of the dielectric dispersion in the phase (I) of DIPAI. | ||
These results suggest that the system approaches the expected ferroelectric phase transition driven by an order–disorder mechanism but is suddenly brought off out of this path towards the highly reconstructed orthorhombic phase. The dielectric results (Fig. S6 and S7†) illustrate the temperature dependence of the complex dielectric permittivity during two heating/cooling cycles carried out on the same sample along the a-axis. It is noteworthy that there are significant differences both between heating and cooling during one cycle and between two full cycles. The measurement carried out on a non-treated sample during heating (Fig. S6(a) and (b)†) suggests an additional anomaly that occurs about seven degrees before the expected phase transition. While cooling (Fig. S6(c) and (d)†) a dubious behaviour, especially for dielectric losses, is also seen. The successive measurement cycle (Fig. S7(a–d)†) differs slightly from those observed previously. It may suggest that in the case of DIPAI, as well as DIPAB-1P, we deal with an irreversible phase change. The optical second harmonic generation (SHG) measurements were undertaken in order to confirm the centrosymmetric-to-polar phase transition in DIPAI. SHG is a second-order nonlinear optical process which is forbidden in a medium with inversion symmetry and therefore may be used as a powerful tool for the detection of the polar state of materials.23,24Fig. 2(c) shows the temperature dependence of the SHG intensity I(T) during the heating/cooling cycle in DIPAI powder with powder size in the range of 140–200 μm. The SHG signal diminishes during the heating and drops to 0 at 376 K thus revealing the structural change from polar/acentric (P212121) to centrosymmetric (P21/m). During subsequent cooling, the intensity of the second harmonic is almost completely restored after the passage of 364 K, repeating the intensity I(T) observed during heating. A weak drop in the intensity values after the heating/cooling cycle is due to a small decrease in the optical transparency of the DIPAI powder after heating above the phase transition temperature.
Fig. 2(d) (see also Fig. S9† and film DIPAI.wmv) shows the polarization microscopy patterns recorded perpendicular to the naturally grown crystal plates. The parallel strips suggest a system of stress-free domain boundaries separating regions related by a simple shear strain, i.e. a system characteristic of ferroelastic species. Here we deal with an ‘inversely’ improper ferroelastic because the unit cell is reduced (instead of being doubled) when passing from the orthorhombic (high-symmetry) to the monoclinic (low-symmetry) structure. The macroscopic strain involves a variation of the angle between the planes (011) and (0−11) (counted in the orthorhombic phase). E.g. the angle amounts to 120.47° at 295 K and to 118.95° at 405 K. The shear plane must, thus, lie parallel to the aortho = bmono but is not necessarily perpendicular to the crystal plates. What is seen in Fig. 2(d) and S9† are the traces of the section of the shear planes with the surface of the plate.
Importantly, given the presented results (see Fig. 4), DIPA ferroelectrics are characterized by the different sequences of phase transitions. DIPAC possesses a reversible ferroelectric anomaly observed by differential scanning calorimetry at the cooling cycle from a high-temperature paraelectric phase (P21/m) to a low-temperature ferroelectric phase (P21) at 436 K,18 while in the bromide derivative there are two different structural polymorphs (1-F (F = ferroelectric) and 1-P (P = piezoelectric)) already at room temperature.19,20 The phase situation observed for the 1-F analog resembles the one detected in a chloride derivative (one reversible phase transition at 426/418 K (heating/cooling)). On the other hand, the variable-temperature X-ray diffraction analysis of the DIPAB 1-P polymorph indicates the presence of irreversible structural transformation from a chiral space group (orthorhombic P212121) to a polar monoclinic phase (P21) at 417 K (seen only at the first heating). Then the 1-P analog passes through the next reversible phase transition at 425 K to a paraelectric phase (P21/m). The successive heating/cooling cycles in the 1-P derivative correspond to the sequence of phase transitions as described for the 1-F polymorph.
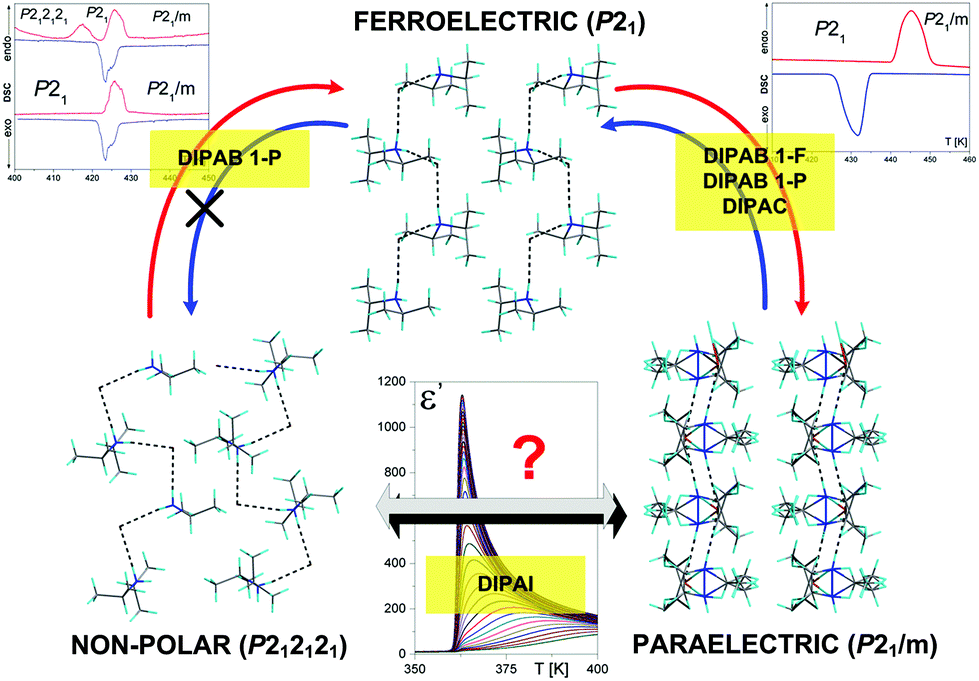 | ||
| Fig. 4 Schematic of phases in diisopropylammonium compounds (ferroelectrics). | ||
The direct jump of DIPAI from the monoclinic phase P21/m (I) to the orthorhombic phase P212121 (III) at 364 K, without the expressed presence of an intermediate P21 state, is seen in Fig. 2(b). The symmetry relationships of the phases of DIPAB, DIPAC and DIPAI can be summarized in a 3D space of the symmetry-adapted order parameters as depicted in Fig. 5 (see also part 5 in the ESI†). All the space groups involved are subgroups of the space group Cmcm. The straight dashed arrows correspond to the symmetry reductions involving, each, the elimination of a second order operation. Therefore, all the irreducible representations corresponding to the dashed lines are one-dimensional and real. The axes of the scheme, thus, span a 3D space of the order parameters denoted by η1, η2 and η3. One can trace possible symmetry changes between the existing and virtual phases in every compound of the DIPA halides family. Of course, a phase transition which is not a simple symmetry breaking must be discontinuous. This is the case for P212121 (Z = 2) → P21 (Z = 1), whereas the phase transition P21/m (Z = 1) → P21 (Z = 1) can be continuous. The simplest free energy expansion (of a Landau type) invariant with respect to the space group Cmcm is presented in the ESI.†
 | ||
| Fig. 5 Symmetry relations of phases in DIPAB, DIPAC and DIPAI. Phase transition P21/m → P21 (magenta arrow) involves a simple symmetry breaking. Brown arrow (P21 → P212121 (Z = 2)) and green arrow (P21/m → P212121 (Z = 2)) depict reconstructive phase transitions in which some symmetry elements are lost and some other gained. | ||
Conclusions
In conclusion, the DIPAI compound has been synthesized and characterized. The compound exhibits two phases I and III isostructural to the analogous phases of DIPAB and DIPAC. In contrast, however, to the other members of the DIPA halides family the compound DIPAI does not possess the ferroelectric phase II. This finding is confirmed by careful SHG and calorimetric studies conducted at different cooling–heating rates. Surprisingly, the dielectric behavior, when approaching the I → III phase transition, resembles that characteristic of a ferroelectric phase transition, i.e. a strong increase in the dielectric permittivity. The increase does not attain infinity, however, is abruptly broken by a jump into the orthorhombic phase III. This effect is, in fact, related to the system approaching the phase II, but the possibly continuous phase transition is interrupted by the appearance of a deeper minimum of the free energy corresponding to the phase III. The corresponding phenomenological model compatible with the symmetry reduction scheme (Fig. 5) is detailed in the ESI.†Experimental
Synthesis of the complex
All materials used in this work were of reagent grade purity and were used as commercially obtained: diisopropylamine (Sigma-Aldrich, 95.95%) and HI (Merck, 57%).Colorless, needle like, crystals of DIPAI were prepared by the reaction of diisopropylamine with 57% aqueous HI (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After a few days, transparent solids were formed by a slow evaporation from the colorless solution. The obtained polycrystalline material was twice recrystallized in a mixture of methanol and ethanol (1:1). The single crystals of DIPAI were grown at the constant temperature. Chemical analysis gave the following mass percentages (values in brackets are theoretical): C, 31.32 ± 0.1 (31.46); N, 6.25 ± 0.1 (6.11); H 7.31 ± 0.05 (7.04).
:1). After a few days, transparent solids were formed by a slow evaporation from the colorless solution. The obtained polycrystalline material was twice recrystallized in a mixture of methanol and ethanol (1:1). The single crystals of DIPAI were grown at the constant temperature. Chemical analysis gave the following mass percentages (values in brackets are theoretical): C, 31.32 ± 0.1 (31.46); N, 6.25 ± 0.1 (6.11); H 7.31 ± 0.05 (7.04).
:40:42), empirical absorption correction using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. The details of the crystal, data collection and refinement, as well as selected geometrical parameters are presented in Tables S1–S3 (part 2).†
CCDC no. 1521065 and 1521066 contain the supplementary crystallographic data for this paper.
Differential Scanning Calorimetry (DSC) heating traces were obtained using a Perkin Elmer model 8500 differential scanning calorimeter calibrated using n-heptane and indium. Hermetically sealed Al pans with the polycrystalline material were prepared in a controlled-atmosphere N2 glovebox. The measurements were performed between 100 and 430 K. The thermal hysteresis was estimated from the scans performed at various rates (20, 10 and 5 K min−1) extrapolated to a scanning rate of 0 K min−1.
Simultaneous Thermogravimetric Analysis (TGA) and Differential Thermal Analysis (DTA) were performed on a Setaram SETSYS 16/18 instrument in the temperature range 300–700 K with a ramp rate 2 K min−1. The scans were performed in flowing nitrogen (flow rate: 1 dm3 h−1).
The complex dielectric permittivity, ε* = ε′ − iε′′, was measured between 100 and 310 K by using an Agilent 4284A Precision LCR Meter in the frequency range between 135 Hz and 2 MHz. The overall error was less than 5%. The single crystal samples had dimensions ca. 5 × 3 × 0.5 mm3. Silver electrodes were stuck on the opposite faces. The dielectric measurements were carried out under a controlled atmosphere (N2).
The second harmonic generation (SHG) measurements were carried out on a DIPAI powder sample in the range of 140–200 μm size using a Q-switched Nd:YAG laser (LPY604T-10, Litron) with a 7 ns pulse length and 10 Hz repetition rate. The experiment was performed in a transmission mode on a powder sample deposited on a supporting optical glass mounted on the controllable heating cell. The linearly polarized focused beam with λ = 1064 nm and pulse power ∼3 MW was used. Appropriate colour and interference filters were employed to discriminate against the scattered infrared fundamental light and SHG emission was detected by a photomultiplier connected to a gated Boxcar integrator, synchronized with a temperature controller unit.
The ferroelastic domain structure of the DIPAI crystal was studied by means of an Olympus BX53 optical polarization microscope. The samples were placed in a LINKAM THMS-600 heating/cooling stage, where the temperature was stabilized to within 0.1 K.
Acknowledgements
This research was supported by the Ministry of Science and Higher Education (Poland) under grant no. IP 0356/IP2/2015/73 (A. Piecha-Bisiorek).References
- S. T. Han, Y. Zhou and V. A. L. Roy, Adv. Mater., 2013, 25, 5425 CrossRef CAS PubMed.
- J. Li, J. Claude, L. E. Norena-Franco, S. I. Seok and Q. Wang, Chem. Mater., 2008, 20, 6304 CrossRef CAS.
- S. Hong, O. Auciello and D. Wouters, Emerging Non-Volatile Memories, Springer, US, 2014 Search PubMed.
- A. von Hippel, Rev. Mod. Phys., 1950, 22, 221 CrossRef CAS.
- G. F. Jona and G. Shirane, Ferroelectric crystals, Pergamon Press, New York, 1962 Search PubMed.
- M. E. Lines and A. M. Glass, Principles and Applications of Ferroelectrics and Related Materials, Oxford University Press, 2001 Search PubMed.
- S. Horiuchi, Y. Tokunaga, G. Giovannetti, S. Picozzi, H. Itoh, R. Shimano, R. Kumai and Y. Tokura, Nature, 2010, 463, 789 CrossRef CAS PubMed.
- S. Horiuchi, F. Kagawa, K. Hatahara, K. Kobayashi, R. Kumai, Y. Murakami and Y. Tokura, Nat. Commun., 2012, 3, 1308 CrossRef PubMed.
- Y. Liu, Y. Zhang, M.-J. Chow, Q. N. Chen and J. Li, Phys. Rev. Lett., 2012, 108, 078103 CrossRef PubMed.
- A. S. Tayi, A. K. Shveyd, A. C.-H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. D. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485 CrossRef CAS PubMed.
- (a) W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163 CrossRef CAS PubMed; (b) A. Katrusiak and M. Szafrański, Phys. Rev. Lett., 1999, 82, 576 CrossRef CAS; (c) R. Jakubas, U. Krzewska, G. Bator and L. Sobczyk, Ferroelectrics, 1988, 77, 129 CrossRef CAS; (d) G.-C. Xu, X.-M. Ma, L. Zhang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588 CrossRef CAS PubMed; (e) P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625 CrossRef CAS PubMed; (f) B. Zhou, A. Kobayashi, H.-B. Cui, L.-S. Long, H. Fujimori and H. Kobayashi, J. Am. Chem. Soc., 2011, 133, 5736 CrossRef CAS PubMed.
- A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Nat. Chem., 2015, 7, 281 CrossRef CAS PubMed.
- D. Isakov, S. Vasilev, E. De, M. Gomes, B. Almeida, V. Y. Shur and A. L. Kholkin, Appl. Phys. Lett., 2016, 109, 162903 CrossRef.
- H.-Y. Ye, S.-H. Li, Y. Zhang, L. Zhou, F. Deng and R. G. Xiong, J. Am. Chem. Soc., 2014, 136, 10033 CrossRef CAS PubMed.
- D. Semmingsen and J. Feder, Solid State Commun., 1974, 15, 1369 CrossRef CAS.
- A. Piecha-Bisiorek, A. Białońska, R. Jakubas, P. Zieliński, M. Wojciechowska and M. Gałązka, Adv. Mater., 2015, 27, 5023 CrossRef CAS PubMed.
- M. Owczarek, K. A. Hujsak, D. P. Ferris, A. Prokofjevs, I. Majerz, P. Szklarz, H. Zhang, A. A. Sarjeant, C. L. Stern, R. Jakubas, S. Hong, V. P. Dravid and J. F. Stoddart, Nat. Commun., 2016, 7, 13108 CrossRef CAS PubMed.
- D.-W. Fu, W. Zhang, H.-L. Cai, J.-Z. Ge, Y. Zhang and R.-G. Xiong, Adv. Mater., 2011, 23, 5658 CrossRef CAS PubMed.
- A. Piecha, A. Gągor, R. Jakubas and P. Szklarz, CrystEngComm, 2013, 15, 940 RSC.
- D.-W. Fu, H.-L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X.-Y. Chen, G. Giovannetti, M. Capone, J. Li and R.-G. Xiong, Science, 2013, 339, 425 CrossRef CAS PubMed.
- P. Prince, J. A. Miller, F. R. Fronczek and R. D. Gandours, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 336 CrossRef.
- E. P. C. Junk, C. J. Kepert, L. M. Louis, T. C. Morien, B. W. Skelton and A. H. White, Z. Anorg. Allg. Chem., 2006, 632, 1809 CrossRef.
- S. A. Denev, T. T. A. Lummen, E. Barnes, A. Kumar and V. Gopalan, J. Am. Ceram. Soc., 2011, 94, 2699 CrossRef CAS.
- M. Wojtaś, O. Czupiński, Z. Tylczyński, D. Isakov, M. Belsley and R. Jakubas, Chem. Phys., 2014, 441, 1 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of thermal properties, crystal characterization, dielectric and optical properties as well as phenomenological theory of the phase transition. CCDC 1521065 and 1521066. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00583g |
| This journal is © the Partner Organisations 2017 |