
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses and properties of selenido mercurates with [HgSe2]2− anions in diverse chemical environments†
C.
Donsbach
and
S.
Dehnen
*
Philipps-Universität Marburg, Fachbereich Chemie and Wissenschaftliches Zentrum für Materialwissenschaften, Hans-Meerwein-Straße 4, D-35043 Marburg, Germany. E-mail: dehnen@chemie.uni-marburg.de
First published on 8th December 2016
Abstract
By aminothermal treatment of ternary CsxHgySez phases, or of Cs2Se with HgSO4, respectively, in 1,2-diaminoethane (en) and en/water mixtures at 150 °C, four new cesium selenido mercurate compounds Cs2[HgSe2] (1), [Cs2(H2O)2][HgSe2] (2), Cs[Cs3(H2O)0.5][HgSe2](Se2) (3) and Cs19(Hen)(H2en)(Se2)[HgSe2]2[HgSe3][Hg2Se5]2 (4) were synthesized. The precursor phases were prepared by fusion of Cs2Se and HgSe at 600 °C. 2 comprises an unprecedented 1D anionic selenido mercurate substructure. 3 and 4 are rare examples of mercurate compounds with different anions, one of which comprises a new type of a dinuclear chalcogenido mercurate anion. The structures of all four compounds were identified by single crystal X-ray diffraction. Micro X-ray fluorescence spectroscopy (μ-XFS) was used to confirm the heavy atom compositions, and optical absorption measurements were performed to determine the optical band gaps.
Introduction
Ternary chalcogenido mercurate phases have been discussed in the recent past as being interesting materials for sensing and in diagnostic applications of hard radiation detection and diagnostics, as these applications require a specific combination of properties.1 For example, for an efficient interaction with radiation, high atomic numbers are crucial. Furthermore, dark current and electronic noise are only suppressed if the band gap is not too small (≥1.6 eV), and the crystals should exhibit large enough size and hardness. All of these preconditions seem to be achievable in A/Hg/E phases (with A being an alkali(ne earth) metal and E being a chalcogenide), while many other materials have drawbacks at one or the other point.2Due to these intriguing properties, numerous A/Hg/E compounds have been generated in the past two decades, most of them by the Kanatzidis group, who have enormously developed the field of heavy metal–chalcogen compounds in general by applying high-temperature and flux-syntheses. We have recently shown that chalcogenido metalates can be accessed at much lower temperatures under solvothermal or ionothermal reaction conditions.3 This way, both solvates and solvent-free phases have been observed.4,5
In the course of our most recent investigations, we have obtained a series of cesium selenido mercurates by aminothermal extraction of the corresponding parent phases, CsxHgySez, with 1,2-diaminoethane (en). All of them comprise [HgSe2]2− units, but their integration in the crystal structure is different for all cases. In this report, we describe and discuss the syntheses, the structural variety and the optical absorption properties, thereby complementing the existing series of Cs/Hg/Se phases.6–8
Discussion
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (2, 3; x/y/z = 2/1/2), maintained at 150 °C for 4 days. The ternary precursor phases are prepared by fusion of Cs2Se and HgSe at 600 °C in the respective stoichiometric ratio. This way, 1–3 were obtained in 30–50% crystalline yield. The aminothermal reaction of Cs2Se and HgSO4 in a 2:1 ratio in dry en yielded 4 in very good yield (approximately 80%), yet along with Cs2SO4 as a side product. The formation of further byproducts HgSe and Cs2Hg3Se4 is observed in different amounts in each of these reactions. The overall synthetic route is illustrated in Scheme 1. Further details are given in the Experimental section.
:1) (2, 3; x/y/z = 2/1/2), maintained at 150 °C for 4 days. The ternary precursor phases are prepared by fusion of Cs2Se and HgSe at 600 °C in the respective stoichiometric ratio. This way, 1–3 were obtained in 30–50% crystalline yield. The aminothermal reaction of Cs2Se and HgSO4 in a 2:1 ratio in dry en yielded 4 in very good yield (approximately 80%), yet along with Cs2SO4 as a side product. The formation of further byproducts HgSe and Cs2Hg3Se4 is observed in different amounts in each of these reactions. The overall synthetic route is illustrated in Scheme 1. Further details are given in the Experimental section.
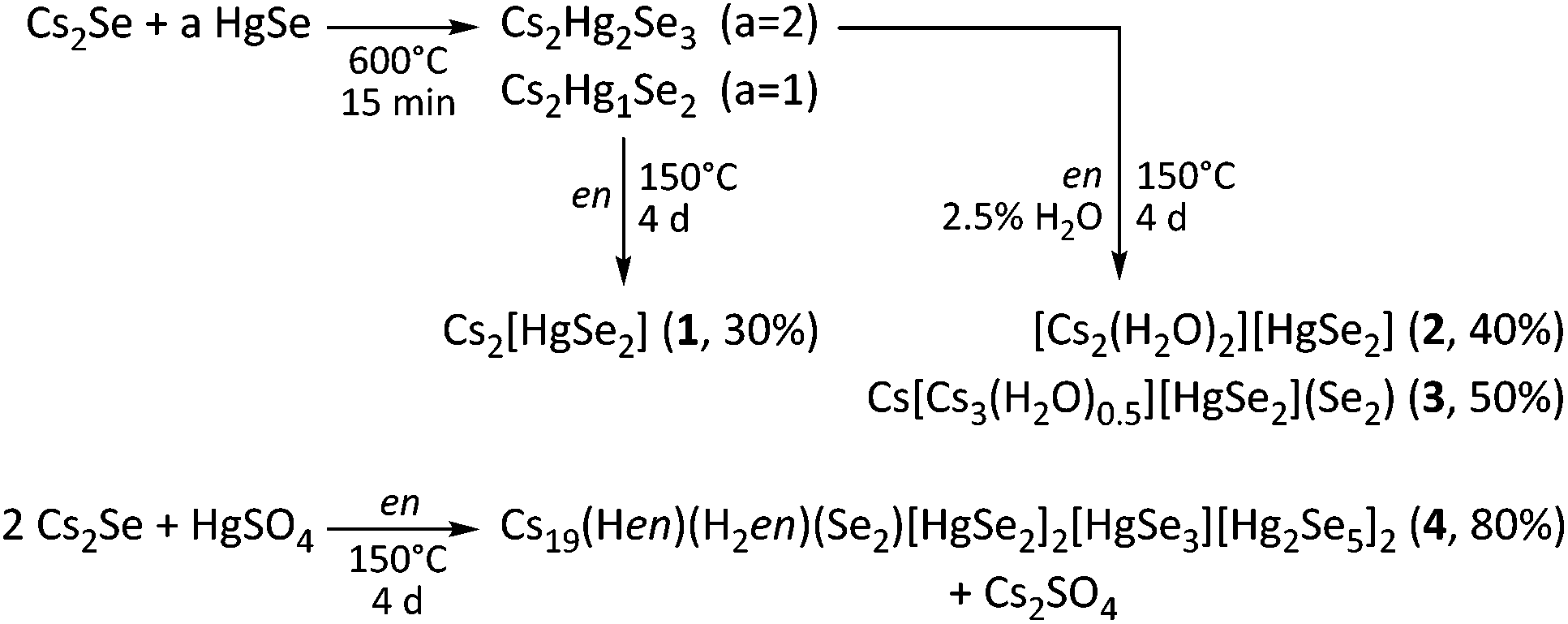 | ||
| Scheme 1 Overview of the reaction paths in the synthesis of compounds 1–4. | ||
Crystal structure description
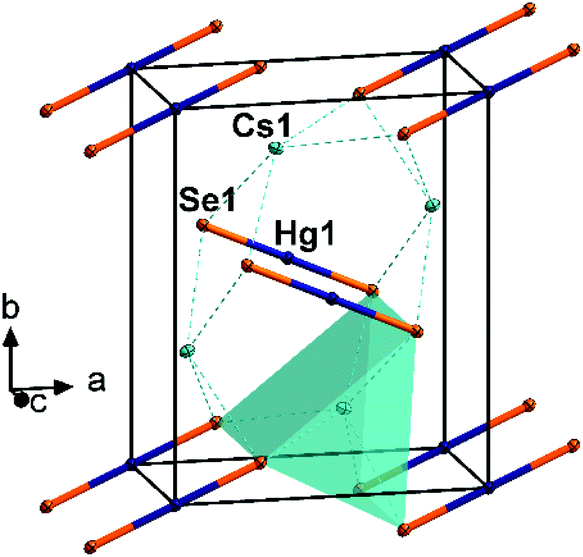 | ||
Fig. 1 Fragment of the crystal structure of 1, viewed approximately along 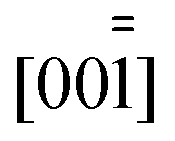 . The coordination polyhedron of one Cs atom is shown. All atoms are drawn with displacement ellipsoids at the 50% probability level. . The coordination polyhedron of one Cs atom is shown. All atoms are drawn with displacement ellipsoids at the 50% probability level. | ||
Furthermore, alkali metal chalcogenido mercurates with discrete [HgE2]2− anions have only been reported as their non-isostructural sulfur analogs, Na2[HgS2] and K2[HgS2].11
§ The Hg–Se bond length of 2.687(4) Å found in K2[HgSnSe4] is in perfect agreement with the observed bonding situation in 2.
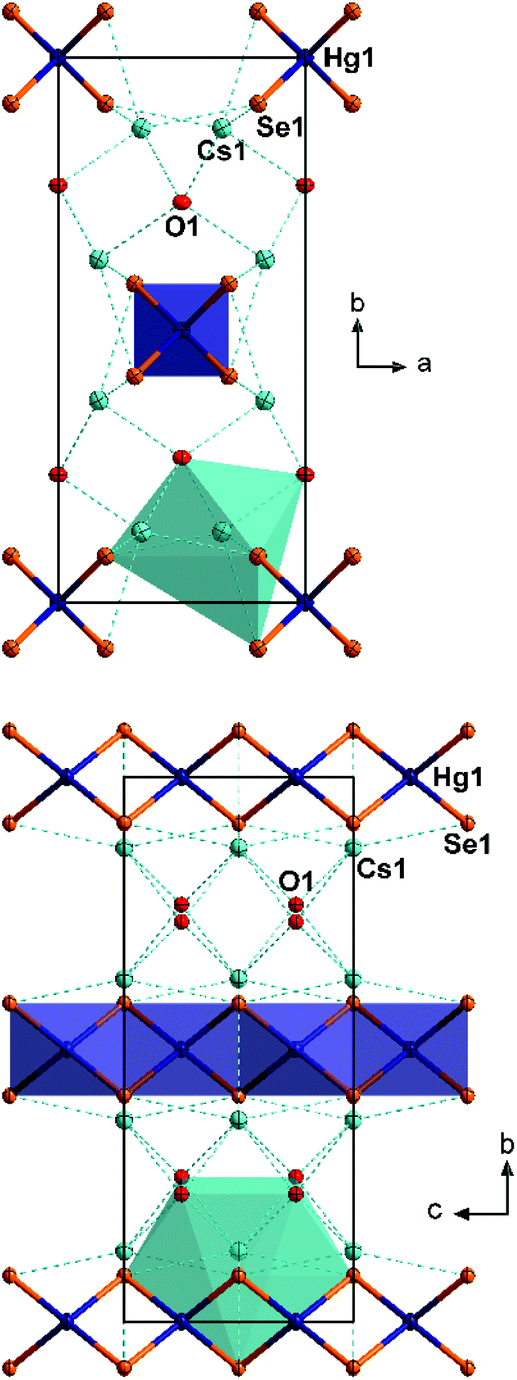 | ||
| Fig. 2 Fragments of the crystal structure of 2, viewed along 〈001〉 (top) and 〈100〉 (bottom). The coordination polyhedra around the Hg atoms of the central strand and around one Cs atom are shown. All atoms are drawn with displacement ellipsoids at the 50% probability level. | ||
In the crystal structure of 2, the collinear anionic strands extend along 〈001〉, four along the unit cell edges, and one passing through ½,½,0. This way, non-interacting layers of anionic strands are formed in the ac plane (smallest Se⋯Se distance 5.476(2) Å), separated by the Cs+ cations, which are located slightly above and below the layers. The Cs+-shielded layers are further separated in the c direction by crystal water molecules. This results in a coordination of the Cs+ cations by four water molecules (Cs⋯O 3.132(11)–3.562(8) Å) and five selenide ligands (Cs⋯Se 3.656(2)–3.761(1)), and a total coordination number for Cs+ is nine. The Cs⋯Se distances are in the typical range (3.489–3.921 Å).10
A closer look at the structures of 2 and [K2(H2O)][HgSe2] reveals a notable structure directing effect of crystal water on the structure of alkali metal chalcogenido mercurates of the nominal composition “A2HgE2”.12 As crystal water is present in both cases, the aggregation of [HgSe2]2− to 1D strands seems to be favored in this environment – in contrast to the situation in 1. In 2, each of the μ2-bridging Se2− ligands show coordinative interaction with four Cs+ cations whereas each of the terminal Se ligands in 1 coordinate to six Cs+ cations. A similar situation is observed in [K2(H2O)][HgSe2], with each of the μ2-bridging Se2− ligands coordinating to four K+ cations, and each of the terminal Se2− ligands coordinating to five K+ cations. Their coordination spheres are saturated by additional interaction with water molecules. In summary, the cationic framework forms channels that stabilize the 1D anions within.
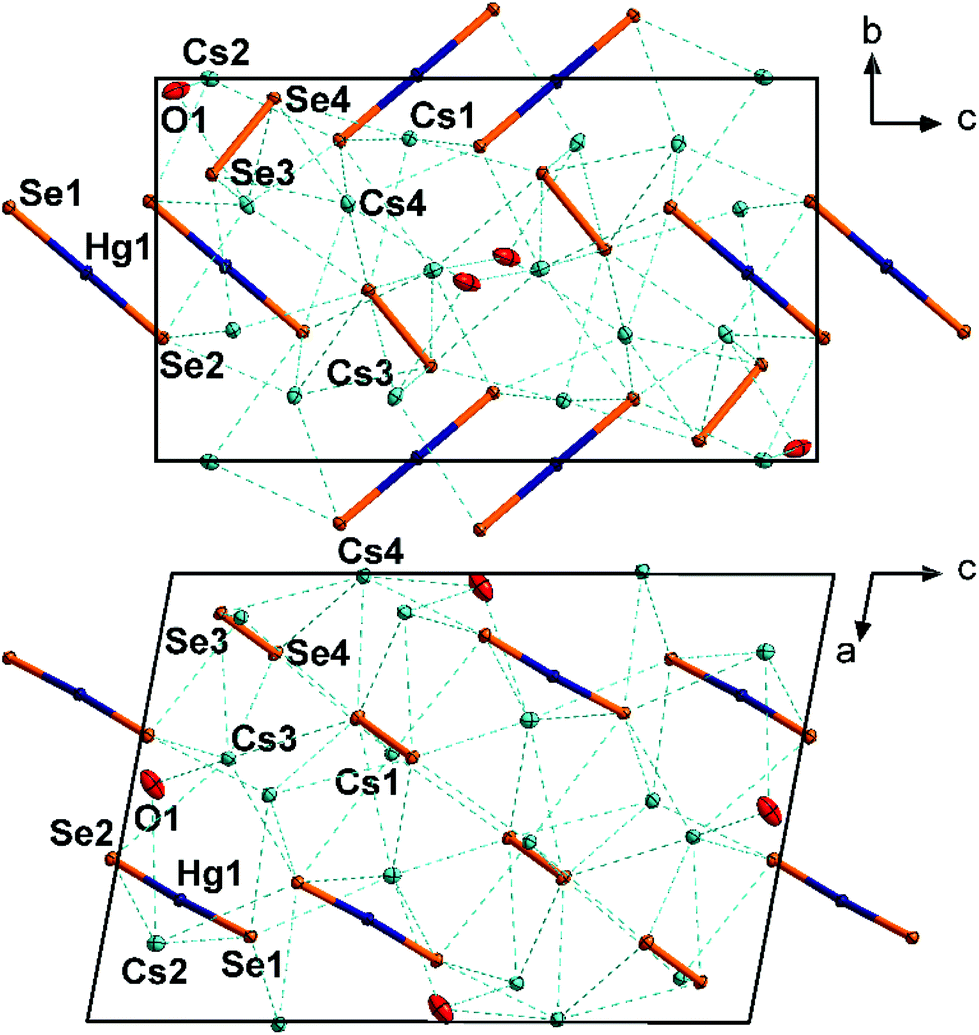
3 crystallizes in the monoclinic space group P21/n with four formula units per unit cell (Fig. 3). The Hg atoms are coordinated by two Se atoms almost linearly (177.31(2)°). A slight deviation from linearity is unusual, since attractive interaction with any neighboring Se atom seems to be absent (nearest Se⋯Hg distances are above 4.0 Å). However, the nearest Cs⋯Hg distance is 3.803(1) Å and might therefore induce weak repulsive interactions. The Hg–Se distance of 2.417(1) is in good agreement with the distances in 1 and [K(crypt-222)]2[HgSe2] (see above). The [HgSe2]2− anions are arranged in two sets of different orientations, approximately along 〈111〉 and 〈1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1〉, respectively. The diselenide anions are oriented almost collinearly with regard to the mercurate anions. The diselenide Se–Se bond length of 2.387(1) Å is in good agreement with that in known cesium polyselenides such as Cs4(Se2)(Se3)·en (2.394(3)–2.404(3) Å).19 All cations are separated by Cs+ cations, which in turn are coordinated by six Se atoms (Cs1, octahedral coordination), seven Se atoms and one additional water molecule (Cs2, irregular coordination), or six Se atoms and 0.5 water molecules (Cs3, Cs4, irregular coordination). The Cs⋯Se distances (3.443(1)–3.912(1) Å) are as expected.
1〉, respectively. The diselenide anions are oriented almost collinearly with regard to the mercurate anions. The diselenide Se–Se bond length of 2.387(1) Å is in good agreement with that in known cesium polyselenides such as Cs4(Se2)(Se3)·en (2.394(3)–2.404(3) Å).19 All cations are separated by Cs+ cations, which in turn are coordinated by six Se atoms (Cs1, octahedral coordination), seven Se atoms and one additional water molecule (Cs2, irregular coordination), or six Se atoms and 0.5 water molecules (Cs3, Cs4, irregular coordination). The Cs⋯Se distances (3.443(1)–3.912(1) Å) are as expected.
 | ||
| Fig. 3 Fragment of the crystal structure of 3, viewed along 〈100〉 (top) and 〈010〉 (bottom). All atoms are drawn with displacement ellipsoids at the 50% probability level. | ||
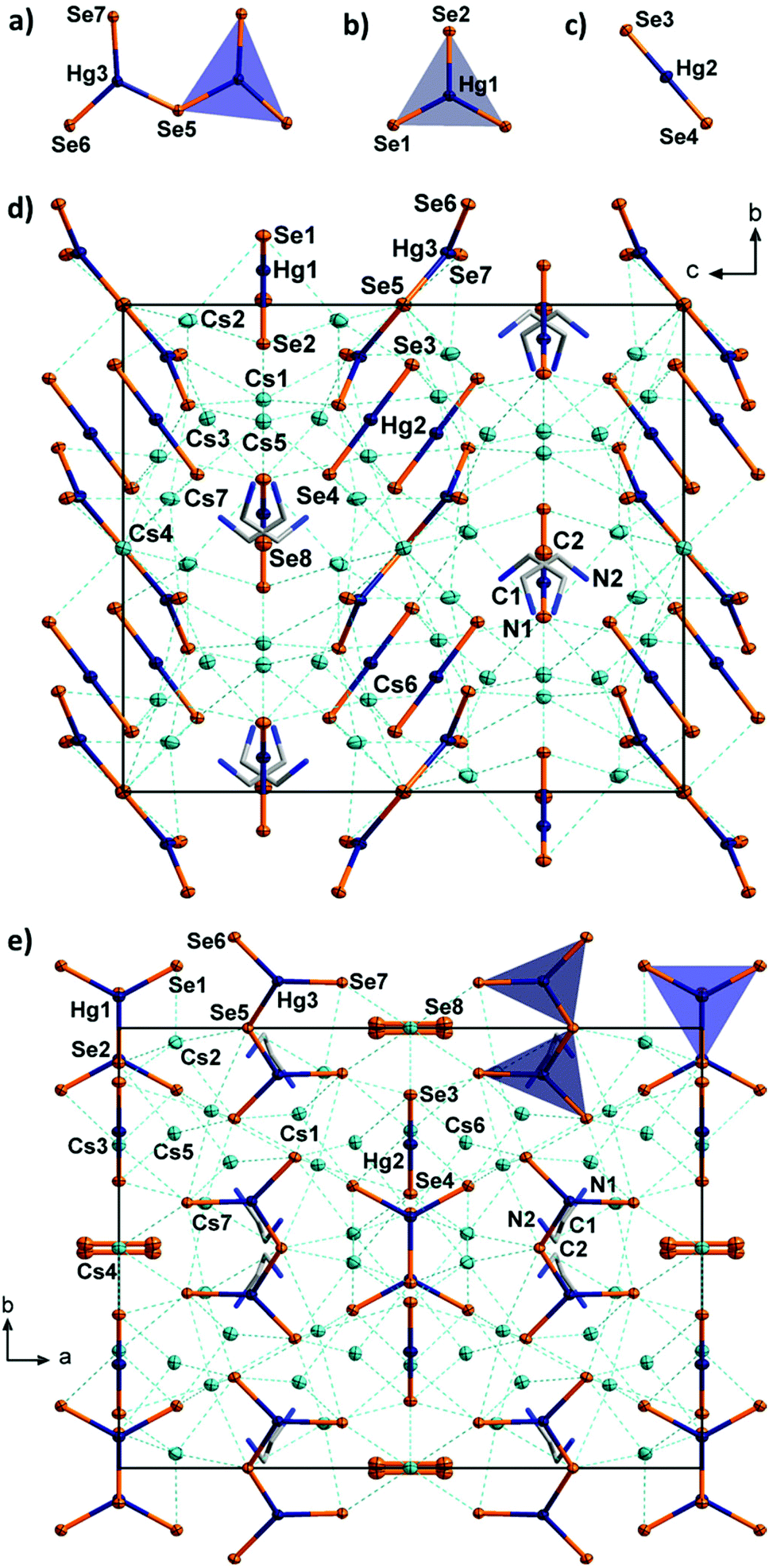 | ||
| Fig. 4 Molecular selenido mercurate anions in 4, [Hg2Se5]6− (a), [HgSe3]4− (b), [HgSe2]2− (c), and fragments of the crystal structure of 4, viewed along 〈100〉 (d), and 〈001〉 (e), respectively. The coordination envorinments of one [HgSe3]4− anion and one [Hg2Se5]6− anion are shown as grey and blue triangles, respectively. The Cs, Hg and Se atoms are drawn with displacement ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
The unit cell of 4 contains eight linear [HgSe2]2− anions with typical structural parameters (Hg–Se 2.419(2)–2.420(2) Å; Se–Hg–Se 179.89(7)°). Furthermore, four slightly distorted, trigonal planar [HgSe3]4− anions are present in the unit cell (Hg–Se 2.559(2)–2.589(3) Å; Se–Hg–Se 118.32(4)–123.37(7)°). Although the Se–Hg–Se angles vary slightly, these anions are absolutely planar (sum of angles 360.0°). This molecular selenido mercurate anion was recently reported as its potassium salt monohydrate, with similar bond lengths (2.552(2)–2.610(2) Å) but with more notable distortion (Se–Hg–Se 115.03(6)–125.87(6)°, sum of angles 359.97°).12
Furthermore, eight symmetry equivalents of an unprecedented third anionic selenido mercurate species, [Hg2Se5]6−, are observed in the unit cell of 4. To the best of our knowledge, this dinuclear molecular anion is the first discrete multinuclear chalcogenido mercurate anion without polychalcogenide ligands.17d,20 The [Hg2Se5]6− anion is formed by two trigonal planar [HgSe3] fragments that are linked via a μ2-bridging Se atom. Hence, it can be formally regarded as being composed of one of the two other molecular anions, [HgSe2]2− and [HgSe3]4−, each, which may be the reason for this uncommon co-existence. It can be furthermore considered as a fragment of the anionic 1D-chains that were recently found in [K2(H2O)][HgSe2]12 (but different from the chains observed in 2). The two [HgSe3] subunits do not face each other, but are arranged in an almost coplanar manner (dihedral angle 20.04(4)°), which is different from the heavily inclined arrangement in [K2(H2O)][HgSe2] (dihedral angle 77.65(2)°). The Hg–Se distances (2.524(2)–2.542(2) Å for terminal Se ligands, 2.665(1) Å for the μ-bridge) are only slightly longer than those found in [K2(H2O)][HgSe2] (2.510(1) Å for terminal Se ligands, 2.609(1) Å for the μ-bridge). The Se–Hg–Se angles (109.82(4)–129.13(5)°) deviate significantly from ideally 120° (cf., the [HgSe3]4− anions in 4 and K2[K2(H2O)][HgSe3]) due to the unsymmetrical structure of the [Hg2Se5]6− anion.12 Yet, the [HgSe3] subunits are still nearly planar (sum of angles around Hg is 359.8°). Besides the three selenido mercurate anions, the complicated crystal structure of 4 further comprises four equivalent diselenide anions per unit cell.
The almost planar [Hg2Se5]6− anions and the linear [HgSe2]2− anions form two sets of non-bonded layers parallel to the ab plane (Fig. 4). The anions within each of the sets are oriented approximately parallel to (012) and (01![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ), respectively. These layers are intercalated with another set of layers, formed by the planar [HgSe3]4− anions and the diselenide anions both of which are arranged coplanarly and parallel to (001). All of the diselenide anions are further oriented along 〈100〉. The anions are separated by Cs+ cations and en molecules. The latter are disordered over two positions, which inhibited the localization of protons on the difference fourier map. We assume that three quarters of the amine groups are protonated for charge balance, as further Cs atoms were not observed. Proton transfer and even more complex degradation of en are not untypical for reactions involving chalcogenidometalates in en. For this, it is most likely that the protons originate from the solvent itself.21 The Cs+ cations are situated in different coordination polyhedra, with Cs⋯Se distances of 3.481(2)–4.030(1) Å, and with coordination numbers five (Cs1, Cs3, Cs5, Cs7) in distorted square pyramids or six (Cs2, Cs4, Cs6) in octahedral coordination, respectively.
), respectively. These layers are intercalated with another set of layers, formed by the planar [HgSe3]4− anions and the diselenide anions both of which are arranged coplanarly and parallel to (001). All of the diselenide anions are further oriented along 〈100〉. The anions are separated by Cs+ cations and en molecules. The latter are disordered over two positions, which inhibited the localization of protons on the difference fourier map. We assume that three quarters of the amine groups are protonated for charge balance, as further Cs atoms were not observed. Proton transfer and even more complex degradation of en are not untypical for reactions involving chalcogenidometalates in en. For this, it is most likely that the protons originate from the solvent itself.21 The Cs+ cations are situated in different coordination polyhedra, with Cs⋯Se distances of 3.481(2)–4.030(1) Å, and with coordination numbers five (Cs1, Cs3, Cs5, Cs7) in distorted square pyramids or six (Cs2, Cs4, Cs6) in octahedral coordination, respectively.
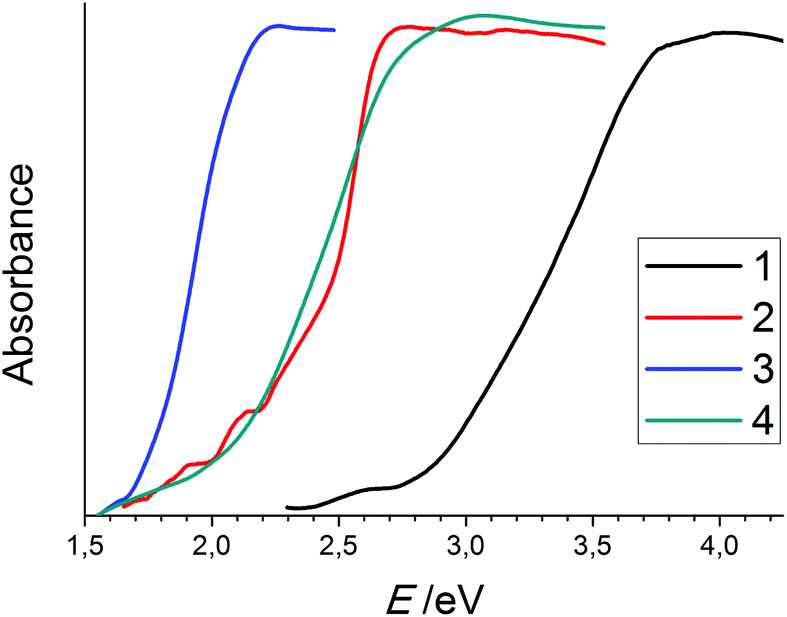 | ||
| Fig. 5 UV-Visible spectra of compounds 1–4. The following Eonset values were determined by means of the tangent method: 2.9 eV (1); 2.4 eV (2); 1.7 eV (3); 2.1 eV (4). Note that the onset of absorption for compound 2 is intrinsically red-shifted due to the beginning of decomposition of the very sensitive compound during sample preparation; for this, only the sharply ascending part of the curve was used to determine the optical gap. | ||
Considering the incorporation of diselenide anions into a selenido mercurate structure, we observed bandgap narrowing with the increasing diselenide ratio. The bandgap of 2.1 eV in 4 is smaller than that in 1 and even 2, though it only comprises selenido mercurate anions of the lowest dimensionality. Yet, the incorporation of a diselenide anion results in a significantly lower bandgap. This effect is even more distinct in the case of 3 (1.7 eV), where one 0D mercurate ion is combined with one diselenide ion.
Experimental
General
All manipulations were performed under strict exclusion of air and moisture (unless stated otherwise) using standard Schlenk and glovebox techniques. En was dried over CaH2 and distilled prior to use. Water was degassed three times at 1 × 10−3 mbar, and subsequently saturated with Ar. Cs2Se was synthesized from the elements in liquid ammonia and HgSe by fusion of the elements in a silica glass ampoule using an oxygen–methane torch. Pure samples of 1–4 suitable for different analyses were prepared by manual selection of the crystals from the raw products due to the presence of side-products. This is not uncommon for solvothermal methods, which represent a certain limitation and drawback of the solvothermal approach.Synthesis of Cs2[HgSe2] (1)
A solid phase with the nominal composition “Cs2Hg2Se3” was prepared by fusion of Cs2Se and HgSe in a 1:2 ratio in a silica glass ampoule with an oxygen–methane torch at about 600 °C, and the resulting solid was pestled after cooling to room temperature. 0.7 g of the raw phase and 2 mL of en were placed in a glass vial inside the PTFE inlay (inner volume 15 mL) of a steel autoclave. The autoclave was closed tightly and heated to 150 °C for 4 days before slowly cooling to room temperature over 1 day. 1 crystallizes as light yellow crystals in approximately 30% yield besides small amounts of microcrystalline Cs2[Hg3Se4], HgSe and unidentified amorphous by-products.
Synthesis of [Cs2(H2O)2][HgSe2] (2) and Cs[Cs3(H2O)0.5][HgSe2](Se2) (3)
Compounds 2 and 3 were obtained during the same reaction. The synthetic procedure is similar to that applied for the synthesis of 1. A precursor phase of the nominal composition “Cs2Hg1Se2” was prepared from Cs2Se and HgSe in a 1:1 ratio via fusion. 0.7 g of the raw phase and 2 mL of en, containing 2.5 vol% of water (0.05 mL), were used in the solvothermal reaction. 2 crystallizes as light yellow sticks alongside dark red blocks of 3 in crystalline yields of approximately 40% and 50%, respectively. Small amounts of amorphous by-products are observed.
Synthesis of Cs19(Hen)(H2en)(Se2)[HgSe2]2[HgSe3][Hg2Se5]2 (4)
For the synthesis of 4, 400 mg of Cs2Se (1.16 mmol, 2 eq.), 172 mg of HgSO4 (0.58 mmol, 1 eq.), and 2 mL of en were placed in an autoclave and heated to 150 °C for 3 days before cooling to room temperature over 1 day. 4 crystallizes as yellow blocks in approximately 80% yield alongside Cs2SO4 and small amounts of microcrystalline Cs2[Hg3Se4], HgSe and unidentified amorphous by-products.Elemental analyses
The heavy atom composition of compounds 1–4 was determined by means of micro X-ray fluorescence spectroscopy (μ-XFS), using a Bruker M4 Tornado with a Rh target X-ray tube and a Si drift detector. Fluorescence photons emitted by the sample were detected during an acquisition time of 120 s, followed by deconvolution of the spectra. The elements were then quantified based on the Se–K, Cs–L and Hg–L radiation. The results are shown in Table 1.| Element | Atom% [calc.] in 1 | Atom% [calc.] in 2 | Atom% [calc.] in 3 | Atom% [calc.] in 4 |
|---|---|---|---|---|
| Cs | 40.4(2) [40.0] | 39.4(3) [40.0] | 43.7(3) [44.4] | 43.0(3) [42.2] |
| Hg | 19.1(1) [20.0] | 20.5(1) [20.0] | 10.9(1) [11.1] | 14.9(1) [15.6] |
| Se | 40.5(1) [40.0] | 40.2(3) [40.0] | 45.4(2) [44.4] | 42.2(2) [42.2] |
Single crystal X-ray diffraction studies
Single crystals for X-ray diffraction analyses were selected under paratone oil using a standard light microscope. Suitable crystals were analyzed on a STOE IPDS-2T (1–3) or IPDS-II (4) diffractometer, respectively, at 100 K using Mo-Kα radiation and a graphite monochromator (λ = 0.71073 Å). Numerical absorption corrections were applied (STOE X-AREA) and the structures were solved via direct methods, followed by full-matrix-least-squares refinement against F2, using SHELXT15, SHELXL15 and the OLEX2 software platform.23 The crystallographic data of 1–4 are summarized in Table 2.| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| CCDC | 1514747 | 1514745 | 1514746 | 1514748 |
| Empirical formula | Cs2Hg1Se2 | Cs2H4Hg1O2Se2 | Cs4H1Hg1O0.5Se4 | C4H19Cs19Hg7N4Se19 |
| Formula weight/g mol−1 | 624.33 | 660.36 | 1057.08 | 5552.89 |
| Crystal color and shape | Yellow needle | Yellow needle | Dark red block | Yellow block |
| Crystal size/mm3 | 0.02 × 0.03 × 0.30 | 0.02 × 0.03 × 0.28 | 0.04 × 0.04 × 0.05 | 0.08 × 0.09 × 0.10 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | Pbam | Cccm | P21/n | Cmcm |
| a/Å | 8.1848(7) | 7.5853(8) | 10.7624(5) | 22.7375(9) |
| b/Å | 10.3302(6) | 16.7226(14) | 8.8940(3) | 17.1603(6) |
| c/Å | 4.7112(3) | 7.0333(6) | 15.6140(7) | 19.7854(5) |
| β/° | — | — | 100.637(3) | — |
| V/Å3 | 398.34(5) | 892.15(4) | 1468.90(11) | 7719.9(5) |
| Z | 2 | 4 | 4 | 4 |
| ρ calc./g cm−3 | 5.21 | 4.89 | 4.78 | 4.78 |
| μ(MoKα)/mm−1 | 37.3 | 33.4 | 30.1 | 31.7 |
| Min/max transmission | 0.0210/0.5636 | 0.0304/0.5927 | 0.3291/0.4951 | 0.0929/0.1449 |
| θ range/° | 3.175–31.771 | 2.436–26.986 | 2.127–29.075 | 1.487–27.144 |
| No. measured refl. | 3653 | 1661 | 15391 |
14727 |
| No. independent refl. | 743 | 532 | 3917 | 4504 |
| R(int) | 0.0521 | 0.1042 | 0.0440 | 0.0562 |
| No. indep. refl. (I > 2σ(I)) | 685 | 479 | 3018 | 3129 |
| No. of parameters | 17 | 22 | 92 | 158 |
| R 1 (I > 2σ(I)) | 0.0334 | 0.0564 | 0.0195 | 0.0418 |
| wR2 (all data) | 0.0800 | 0.1448 | 0.0255 | 0.1023 |
| S (all data) | 1.122 | 1.076 | 0.848 | 0.901 |
| Δρmax, Δρmin/e Å−3 | 2.313/−3.267 | 3.057/−4.479 | 0.894/−0.861 | 3.485/−3.293 |
Optical absorption spectroscopy
UV-visible absorption spectroscopy was performed by analyses of the diffuse reflection of powdered samples. The measurements were carried out using a Varian Cary 5000 dual-beam spectrometer with a Praying Mantis sample holder from Harrick.Conclusions
In summary, we presented four new cesium selenido mercurate compounds synthesized via aminothermal syntheses in 1,2-diaminoethane (en). These comprise unprecedented selenido mercurate anionic substructures (2, 4) and the molecular [HgSe2]2− anion (1), which have not been reported in the solid phase without sequestering agents so far. The bandgaps determined for these low-dimensional cesium selenido mercurates match the theory of the influence of dimensionality reduction, thus complementing and supporting the observations made for another set of chalcogenido mercurates of larger dimensionality. Besides, the influence of water on the ability of cesium cations to stabilize anionic substructures of different dimensionalities was investigated. Owing to their good crystal quality, the compounds may be interesting regarding their potential use in sensing or detection of hard radiation.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of SPP1708. We thank M. Drabek for his help with the synthetic work.Notes and references
- J. Androulakis, S. C. Peter, H. Li, C. D. Malliakas, J. A. Peters, Z. Liu, B. W. Wessels, J.-H. Song, H. Jin, A. J. Freeman and M. G. Kanatzidis, Adv. Mater., 2011, 23, 4163 CrossRef CAS PubMed.
- (a) B. D. Milbrath, A. J. Peurrung, M. Bliss and W. J. Weber, J. Mater. Res., 2008, 23, 2561 CrossRef CAS; (b) D. S. McGregor and H. Hermon, Nucl. Instrum. Methods Phys. Res., Sect. A, 1997, 395, 101 CrossRef CAS; (c) A. Owens, J. Synchrotron Radiat., 2006, 13, 143 CrossRef CAS PubMed; (d) T. E. Schlesinger, J. E. Toney, H. Yoon, E. Y. Lee, B. A. Brunett, L. Franks and R. B. James, Mater. Sci. Eng., 2001, 32, 103 CrossRef.
- (a) S. Santner, J. Heine and S. Dehnen, Angew. Chem., Int. Ed., 2016, 54, 876 CrossRef PubMed; (b) J. Heine and S. Dehnen, Z. Anorg. Allg. Chem., 2012, 638, 2425 CrossRef CAS; (c) S. Dehnen and M. Mellulis, Coord. Chem. Rev., 2007, 251, 1259 CrossRef CAS.
- (a) C. Donsbach and S. Dehnen, Z. Anorg. Allg. Chem., 2016 DOI:10.1002/zaac.201600338; (b) C. Donsbach, G. Thiele, L. H. Finger, J. Sundermeyer and S. Dehnen, Inorg. Chem., 2016, 55, 6725 CrossRef CAS PubMed; (c) G. Thiele, S. Lippert, F. Fahrnbauer, P. Bron, O. Oeckler, A. Rahimi-Imam, M. Koch, B. Roling and S. Dehnen, Chem. Mater., 2015, 27, 4114 CrossRef CAS; (d) E. Ruzin, A. Fuchs and S. Dehnen, Chem. Commun., 2006, 4796 RSC; (e) C. Zimmermann and S. Dehnen, Z. Anorg. Allg. Chem., 2003, 125, 6618 Search PubMed.
- M. K. Brandmayer, R. Clérac, F. Weigend and S. Dehnen, Chem. – Eur. J., 2004, 10, 5147 CrossRef CAS PubMed.
- M. G. Kanatzidis, Chem. Mater., 1990, 2, 353 CrossRef CAS.
- E. A. Axtell III, Y. Park, K. Chondroudis and M. G. Kanatzidis, J. Am. Chem. Soc., 1998, 120, 124 CrossRef.
- R. Stromsky, L. W. Zimmermann, I. Hartenbach and T. Schleid, Z. Kristallogr., 2015, 35, 92 Search PubMed.
- C.-W. Park, D. M. Smith, M. A. Pell and J. A. Ibers, Inorg. Chem., 1997, 36, 942 CrossRef CAS.
- (a) H. Sommer and R. Hoppe, Z. Anorg. Allg. Chem., 1977, 429, 118 CrossRef CAS; (b) P. Böttcher, J. Less-Common Met., 1980, 76, 271 CrossRef.
- K. O. Klepp and K. Prager, Z. Naturforsch., B: Chem. Sci., 1992, 47, 491 CAS.
- G. Thiele, C. Donsbach, R. Riedel, M. Marsch, K. Harms and S. Dehnen, Dalton Trans., 2016, 45, 5958 RSC.
- M. G. Kanatzidis and Y. Park, Chem. Mater., 1990, 2, 99 CrossRef CAS.
- W.-W. Xiong, P.-T. Li, T.-H. Zhou, Y. Zhao, R. Xu and Q. Zhang, J. Solid State Chem., 2013, 204, 86 CrossRef CAS.
- S. M. Islam, J. Im, A. J. Freeman and M. G. Kanatzidis, Inorg. Chem., 2014, 53, 4698 CrossRef CAS PubMed.
- (a) A. Müller, J. Schimanski, U. Schimanski and H. Bogge, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1985, 40, 1277 Search PubMed; (b) T. D. Bailey, R. M. H. Banda, D. C. Craig, I. G. Dance, I. N. L. Ma and M. L. Scudder, Inorg. Chem., 1991, 30, 187 CrossRef CAS.
- (a) J. Adel, F. Weller and K. Dehnicke, Z. Naturforsch., B: Chem. Sci., 1988, 43, 1094 CAS; (b) G. Krauter, F. Weller and K. Dehnicke, Z. Naturforsch., B: Chem. Sci., 1989, 44, 444 CrossRef; (c) R. M. H. Banda, J. Cusick, M. L. Scudder, D. C. Craig and I. G. Dance, Polyhedron, 1989, 8, 1995 CrossRef CAS; (d) S. Magull, B. Neumüller and K. Dehnicke, Z. Naturforsch., B: Chem. Sci., 1991, 46, 985 CrossRef CAS; (e) B. Neumüller, M.-L. Ha-Eierdanz, U. Müller, S. Magull, G. Krauter and K. Dehnicke, Z. Anorg. Allg. Chem., 1992, 609, 12 CrossRef; (f) A. Ahle, K. Dehnicke, K. Maczek and D. Fenske, Z. Anorg. Allg. Chem., 1993, 619, 1699 CrossRef CAS; (g) P. J. Barrie, R. J. H. Clark, R. Withnall, D.-Y. Chung, K.-W. Kim and M. G. Kanatzidis, Inorg. Chem., 1994, 33, 1212 CrossRef CAS; (h) J. C. Bollinger and J. A. Ibers, Inorg. Chem., 1995, 34, 1859 CrossRef CAS; (i) C.-W. Park, D. M. Smith, M. A. Pell and J. A. Ibers, Inorg. Chem., 1997, 36, 942 CrossRef CAS.
- J. C. Bollinger, L. C. Roof, D. M. Smith, J. M. McConnachie and J. A. Ibers, Inorg. Chem., 1995, 34, 1430 CrossRef CAS.
- G. Thiele, L. Vondung, C. Donsbach, S. Pulz and S. Dehnen, Z. Kristallogr., 2014, 640, 2684 CAS.
- (a) J. Li, B. G. Rafferty, S. Mulley and D. M. Proserpio, Inorg. Chem., 1995, 34, 6417 CrossRef; (b) R. C. Haushalter, Angew. Chem., Int. Ed. Engl., 1985, 24, 433 CrossRef.
- (a) G. Thiele, T. Krüger and S. Dehnen, Angew. Chem., Int. Ed., 2014, 53, 4699–4703 CrossRef CAS PubMed; (b) G. Thiele, Y. Franzke, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2015, 54, 11283–11288 CrossRef CAS PubMed; (c) G. Thiele, L. Vondung and S. Dehnen, Z. Anorg. Allg. Chem., 2015, 641, 247 CrossRef CAS.
- J. I. Pankove, in Optical Processes in Semiconductors, Dover Publication, New York, 1975 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnotes |
| † CCDC 1514745–1514748. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00481d |
| ‡ crypt-222 = 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane. |
| § DBN = 1,5-diazabicyclo[4.3.0]non-5-ene. |
| This journal is © the Partner Organisations 2017 |