
CO2 capture in a carbazole-based supramolecular polyhedron structure: the significance of Cu(II) open metal sites†
Alfredo
López-Olvera‡
a,
Elí
Sánchez-González‡
b,
Alberto
Campos-Reales-Pineda
b,
Andrés
Aguilar-Granda
a,
Ilich A.
Ibarra
*b and
Braulio
Rodríguez-Molina
*a
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior s/n, Ciudad Universitaria, Del. Coyoacán, 04510, Ciudad de México, Mexico. E-mail: argel@unam.mx; brodriguez@iquimica.unam.mx; Fax: +52(55) 5622-4595
bInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, Ciudad Universitaria, Del. Coyoacán, 04510 Ciudad de México, Mexico
First published on 1st November 2016
Abstract
A crystalline, porous Metal–Organic Polyhedron (MOP) 4 was readily assembled starting from 9H-carbazole-3,6-dicarboxylic acid, which was synthesised in only three steps. The single crystal X-ray diffraction analysis of the resulting supramolecule evidenced voids that were occupied by coordinated as well as uncoordinated solvent molecules from the solvothermal reaction. Careful analysis of its desolvation process, activating the sample at 150 °C and 10−5 bar (followed by FTIR), evidenced that all solvents were completely removed, leaving an empty but nevertheless crystalline MOP with coordinatively-unsaturated Cu(II) sites that was studied for gas adsorption using N2 at 77 K, and CO2 at 196 K. Notably, carbon dioxide was found to be strongly adsorbed in the activated 4 (12.1 mmol g−1, 53.2 wt%) with a molar enthalpy of adsorption of ΔH = 65.12 kJ mol−1, revealing a great selectivity over nitrogen, which in turn showed negligible uptake under the same activation conditions. In situ Raman experiments demonstrated the direct interaction of CO2 molecules and coordinatively-unsaturated Cu(II) sites within 4.
Introduction
Carbon dioxide (CO2) emissions, from fossil fuel combustion,1 negatively impact our atmosphere causing the continuous increase of temperature across the planet, which contributes to one of the greatest threats to human civilisation: global warming. Startlingly, last year global CO2 emissions increased up to 35.7 billion tonnes and 90% of these emissions are exclusively related to fossil fuel combustion.2 Clearly, a severe reduction of atmospheric CO2 emissions is critical to diminish the associated risks that global warming presents to our planet. At this time, many international governments are actively encouraging the growth of new CO2 capture technologies.3 Industrially, large-scale carbon capture is achieved by chemical absorption of CO2 (Lewis acid) by aqueous solutions of alkanolamines (Lewis base). However, their major drawbacks include heat instability, corrosion of pipelines and significant costs for solvent regeneration, thus restraining the long-term application and widespread use of these alkaline solutions considerably.4Alternatively, the use of porous solid materials for CO2 capture (adsorption process) is nowadays a very hot research area. Poliakoff5 proposed ‘the twelve principles of CO2 chemistry’ where CO2 sequestration represents one of these principles (maximise integration). Hence, the search for porous materials with high structural stability, adsorption capacity, solvent stability, fast sorption kinetics and mild regeneration conditions represents a major challenge for scientists. Porous metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are amongst the most promising candidates for CO2 capture and separation because their sorption selectivity to CO2 is directly tailored as a function of the chemical composition of the pores.6 Regarding CO2 sequestration there is a vast emphasis on optimising the interactions between MOF materials and CO2 molecules, leading to the discovery of new functional porous materials with enhanced CO2 capture properties.7 Current synthetic approaches to enhance host–guest interactions in MOF systems employ the functionalisation of the pores with basic (Lewis) nitrogen-containing groups such as triazole,8 amines,9 and tetrazole.10 Additionally, the introduction of open (uncoordinated) metal sites11 represents a very exciting post-synthetic modification, and although the generation of uncoordinated metal sites has been usually applied for increasing the affinity of MOFs towards H2,12 significant efforts have also been made in CO2 capture.13
CO2 capture occurs in MOF materials due to the intrinsic and well defined porosity that they commonly show. Thus, the combination of high surface areas and post-synthetic modifications within the pores (vide supra) of these materials is essential to achieve very promising CO2 sequestration results. Conversely, non-porous materials would be anticipated as very poor CO2 capturing materials. However, some non-porous materials that have open metal sites are post-synthetically modified, and hence they can capture CO2. As an example we can mention PCM-14,14 a non-porous coordination polymer constructed from Ca(II) and an organophosphonium ligand (mptbc2− = tris(p-carboxylate)triphenyl-methyl phosphonium). PCM-14 is formed from one Ca(II) metal ion coordinated to one mptbc ligand and three molecules of water (H2O), [Ca(mptbc)(OH2)3], and these water molecules can only be removed under the right activation conditions, generating open metal sites within PCM-14. Interestingly, when this material was activated from room temperature to 150 °C (under 10−10 bar) the three water molecules remained coordinated to the Ca(II) metal centres. However, when activated at 180 °C and 10−10 bar, the removal of the coordinated water molecules was feasible and therefore the generation of uncoordinated metal sites promoted CO2 capture in this material (2.92 wt% at 196 K).
Carbazole-containing materials are very attractive for the following reasons: (i) 9H-carbazole is an inexpensive precursor that can be chemically modified at its 2, 3 or 6, 7 positions to provide a wide range of derivatives; (ii) since it is fully aromatic, its chemical stability is considerably high; (iii) the N–H functional group can also be modified for different applications; (iv) its fused structure can be used in emissive materials enabling new applications.15 In our groups, we are interested in the design and synthesis of metal–organic polyhedra (MOPs) constructed from carbazole–carboxylate ligands because the flexibility that carbazole offers to be chemically modified opens a vast range of new and emerging applications; for example, carbazole-based materials can be very emissive and therefore highly desirable for the construction of photovoltaic cells, among other possibilites.16 Therefore, we report here the synthesis, characterisation and crystal structure description of a carbazole-based supramolecular polyhedron (4) constructed from a carbazole–carboxylate ligand (H2L1, Scheme 1) and Cu(II) metal centres along with its CO2 capture properties.
 | ||
| Scheme 1 9H-Carbazole-3,6-dicarboxylic acid (H2L1) (3) ligand used for the synthesis of the metal–organic polyhedron discussed here. | ||
Experimental section
Synthetic preparation
Carbazole, potassium iodide, potassium periodate, glacial acetic acid, sodium sulphite, di-tert-butyl dicarbonate, 4-(dimethylamino)pyridine, n-butyllithium (2.5 M in hexanes), sodium hydroxide, dichloromethane, hydrochloric acid (conc.), ethanol and N,N′-dimethylformamide were used as purchased, without the need for further purification. Tetrahydrofuran was purified prior to use by drying with metallic sodium followed by column distillation and hexane was purified by flash column chromatography.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 N,N′-dimethylformamide/ethanol. The solution was then heated in a scintillation vial at 120 °C for 8 h to yield prismatic blue-green crystals of the target material, which were isolated by decanting away from the mother liquor and dried in open vials for 6 h (yield 9.2 mg, 67%).
:1 N,N′-dimethylformamide/ethanol. The solution was then heated in a scintillation vial at 120 °C for 8 h to yield prismatic blue-green crystals of the target material, which were isolated by decanting away from the mother liquor and dried in open vials for 6 h (yield 9.2 mg, 67%).
Single-crystal X-ray crystallography
The precipitated crystals from the solvothermal reaction were preserved in their mother liquor for microscopy observation. For diffraction experiments, one crystal was removed from the solvent, mounted in a Cryoloop® and immediately covered with Paratone® oil while it was cooled down for analysis. Data collection was performed at 150 K on a Bruker-APEX-II CCD diffractometer with Mo Kα-radiation, λ = 0.71073 Å. The structure was solved by direct methods and refined using SHELXL-2014. Some highly disordered solvent molecules could neither be located nor refined due to their rapid volatility during the acquisition, so their diffused electron densities were removed from the dataset using the SQUEEZE protocol of PLATON17 and they are not represented in the unit cell. Relevant crystallographic parameters are compiled in Table S1 in the ESI.†Adsorption isotherms for N2 and CO2
N2 and CO2 isotherms (up to 1 bar) were recorded on a Belsorp mini II and a Belsorp HP (High Pressure) analyser, respectively, under high vacuum in a clean system with a diaphragm and turbo pumping system.Adsorption microcalorimetry for CO2
CO2 adsorption microcalorimetry experiments were carried out at 196 K using approximately 100 mg of sample (4). The evolved heat was measured using a Tian-Calvet microcalorimeter (CA-100, ITI). This instrument measures the CO2 isotherm and the enthalpy of adsorption for CO2 simultaneously using a point by point introduction of CO2 gas to the sample. Prior to any adsorption experiment, the freshly synthesised sample 4 was activated at 150 °C and 10−5 bar for 1 h.Results and discussion
Synthesis and characterization
The carbazole dicarboxylic ligand 3 used for the solvothermal synthesis of the MOP reported here was synthesized using a new methodology involving three steps with good yields (Scheme 2). First of all, an oxidative iodination over the positions 3 and 6 allowed us to obtain the di-iodinated carbazole precursor 1 (yield 85%). | ||
| Scheme 2 Synthesis of ligand 3 for the solvothermal synthesis of the MOP. | ||
Subsequently, we carried out the N-protection using BoC2O to remove the reactivity of the nitrogen atom in the carbazole obtaining intermediate 2 (yield 85%). Finally, a metalation with n-butyllithium followed by the addition of dry ice (CO2) gave a solid intermediate that was immediately treated with HCl to remove the protecting group and afford the desired ligand 3 (yield 79%). The isolated intermediates and the final product were fully characterized by spectroscopic techniques as detailed in the Experimental section and the ESI.†
X-ray single crystal studies
The solvothermal reaction of Cu(NO3)2·5H2O with H2CDC13 (Scheme 2) in DMF/EtOH afforded the supramolecular polyhedron [Cu2(CDC)2(DMF)(H2O)]6·7DMF·4H2O (4) as blue-green crystals; CDC (9H-carbazole-3,6-dicarboxylate). The single-crystal X-ray structural determination confirms that 4 crystallises in the monoclinic space group P21/n and has a structure based on the dinuclear building block, paddlewheel clusters, [Cu2(O2CR)4] (Fig. 1), two of which are present in the unit cell. Furthermore, we noticed that although the heterocyclic ligand is planar, the carbazole rings in each paddlewheel adopt a conformation slightly above or below a dividing plane, allowed by minor changes in the orientation of the coordinating carboxylate group, rendering a slightly distorted polyhedron. | ||
| Fig. 1 Ball and stick view of 4 with all the carbazole ligands involved in hydrogen bonds (dashed lines) and with a slightly distorted conformation. The insert highlights the paddlewheel SBU. Uncoordinated solvents were removed for clarity purposes. | ||
The solid state array of this polyhedron was dominated by hydrogen bonds of the N–H⋯O type, where the donor is the nitrogen of the carbazole moieties and the acceptors are either coordinating DMF in neighbouring MOPs or uncoordinated solvent molecules (dashed lines in Fig. 1). Using these intermolecular interactions that propagate along all directions, the molecules finally adopted a layered array in which the major difference is the orientation of the polyhedron, as presented in Fig. 2, giving rise to a crystalline packing that is isostructural to a similar polyhedron reported earlier.18
 | ||
| Fig. 2 Ball and stick view along the a–b plane of the layered array of the MOP 4. Uncoordinated solvent molecules were removed to highlight the potential cavity of the polyhedron. | ||
The analysis of the molecular structure evidenced six coordinated DMF molecules in the outer part of the discrete assembly along with six coordinated water molecules within the cavity. This cavity was also occupied by solvent molecules (water and DMF) that could be possibly eliminated to allow access to permanent porosity, as described in the following sections. As an early observation, when the crystals were exposed to open air under the microscope they lost their prime quality within a few minutes, revealing a fragility that was characterized in detail as discussed in the following sections.
Thermal studies
In order to characterize the stability of the crystalline sample a thermogravimetric analysis (TGA) of 4 was carried out under N2. This study showed a continuous weight loss of 13% over a temperature range of 25–180 °C (see Fig. S7, ESI†). MOP 4 reaches a plateau above 180 °C with the structure decomposing at 270 °C (see Fig. S7, ESI†). Powder X-ray diffraction (PXRD) measurements on sample 4 confirmed a good fit observed between the experimental PXRD data and the calculated single-crystal data (see Fig. S8, ESI†).Infrared spectroscopy
As previously described, one of the most interesting features of 4 is the di-copper(II) paddlewheel clusters (Fig. 1). These clusters have been extensively studied19 since terminal-coordinated ligands (such as DMF and H2O) can be removed to afford square planar coordinated Cu(II) metal centres, where the oxidation state of copper is unchanged and the crystallinity of the materials is retained, e.g. HKUST-1.20 Typically the generation of coordinatively unsaturated metal sites has been applied to significantly increase the interaction between H2 and MOFs12 and most recently, considerable actions have been directed towards CO2 capture.21 Thus, the generation of open Cu(II) metal sites within 4, that is the fate of coordinated H2O molecules, was studied with FTIR spectroscopy. Then, FTIR spectra were acquired at room temperature (25 °C) on two different samples. First, the FTIR spectrum of 4 at 25 °C, non-activated (Fig. 3, black line), exhibits a broad adsorption band at 3250 cm−1 corroborating the presence of non-coordinated H2O inside the material. | ||
| Fig. 3 FTIR spectra of 4 at 25 °C. As-synthesised (black line) and activated at 150 °C under vacuum (10−5 bar, red line). | ||
At 3730 cm−1, a characteristic sharp peak attributed to metal-coordinated H2O12,22,23 is discernible in the spectrum (Fig. 3, black line). Additionally, the DMF ν(CO) band coordinated to the metal centre24 appears at 1655 cm−1 in the spectrum (Fig. 3, black line).
Later, a sample of 4 was activated at 150 °C under vacuum (10−5 bar) for 1 h. The activated sample was cooled down to 25 °C and a FTIR experiment was carried out (Fig. 3, red line). This FTIR spectrum showed a major reduction in the intensity of the broad absorption band (ν = 3250 cm−1) and the sharp peak at 3730 cm−1 is lost from the spectrum (Fig. 3, red line). In Fig. 4, the decrease in the intensity of the ν = 3730 cm−1 band is emphasised, which confirms the removal of the coordinated water to the Cu(II) metal centres at 150 °C and 10−5 bar.
 | ||
| Fig. 4 FTIR spectra of 4 at 25 °C. As-synthesised (black line) and activated at 150 °C under vacuum (10−5 bar, red line), emphasising the fading of coordinated H2O molecules. | ||
It is also possible to observe the reduction of the intensity of the band at 1655 cm−1 (Fig. 3). As occurred previously for the characteristic sharp peak of coordinated H2O, the intensity reduction for this band (ν = 1655 cm−1) on the activated sample indicates the removal of coordinated DMF molecules to the Cu(II) metal centres. Fig. 5 highlights the loss of these DMF molecules within the material.
 | ||
| Fig. 5 FTIR spectra of 4 at 25 °C. As-synthesised (black line) and activated at 150 °C under vacuum (10−5 bar, red line), emphasising the fading of coordinated DMF molecules. | ||
Adsorption experiments
N2 sorption analyses were conducted in order to study the porosity in 4. A sample of as-synthesised 4 was activated at 150 °C and 10−5 bar for 1 h and a N2 adsorption–desorption experiment (at 77 K) was performed. The very low uptake of N2, even at the cryogenic temperature of 77 K (see Fig. S9, ESI†), suggested two possible scenarios: (i) there is no access to any permanent porosity within 4 or (ii) MOP 4, selectively, does not adsorb N2 even at 77 K. Indeed, we managed to elucidate its remarkable selectivity of CO2 over N2 at cryogenic temperatures (vide infra).We activated the sample at 150 °C and 10−5 bar for 1 h since these conditions provided access to uncoordinated Cu(II) metal sites as demonstrated previously by FTIR (vide supra). A PXRD was carried out on the activated sample 4 to investigate the crystallinity of the material. Surprisingly, retention of the crystallinity of 4 after the activation process was observed (see Fig. S10, ESI†). Although the crystalline phase of the activated sample showed to be different from the as-synthesised phase (see Fig. S10, ESI†), it is remarkable that a crystalline phase is obtained since, typically, these MOP materials loose completely their crystallinity after being activated,25 due to their non-rigid lattice. In an effort to determine the crystal structure of the activated sample, a freshly synthesised single crystal of 4 was mounted on a goniometer head and heated at 150 °C under a stream of dry N2. After cooling down to 150 K, a single crystal X-ray diffraction experiment was attempted. Unfortunately, this single crystal did not diffract, presumably due to the harsh activation conditions and thus, we were not able to elucidate the crystal structure of the activated sample of 4.
Motivated by the very interesting results that Doonan recently reported25 in a series of hetero-bimetallic Pd(II)–Cu(II), Ni(II), Zn(II) MOPs, we decided to follow their proposed careful activation protocol, which guaranteed the complete removal of the coordinated solvents (H2O and DMF) and access to permanent porosity.25 Thus, a sample of 4 was soaked in dry acetone for 7 days, followed by supercritical CO2 drying and finally heating at 50 °C under vacuum for 3 h. Then, after the activation was completed, another N2 sorption experiment (77 K) was carried out. Unfortunately, the N2 isotherm obtained was identical to the previous one showing a very low uptake. Thus, it was not possible for us to determine any permanent porosity for 4 using N2.
Although this supramolecular polyhedron did not show apparent permanent porosity (evaluated with N2 at 77 K), we have demonstrated that it is possible to gain access to Cu(II) open metal sites (FTIR experiments). This gave us the incentive to perform CO2 adsorption experiments on 4 at 196 K. Thus, a freshly synthesised sample of 4 was then activated at 150 °C and 10−5 bar for 1 h. After slowly cooling down to room temperature (approx. 30 °C) the sample holder (high-pressure cell from the Belsorp HP analyser) was immersed in a bath of dry ice and acetone to reach the desired temperature of 196 K. When the isotherm was complete, CO2 was found to be strongly adsorbed in activated 4 at 196 K (12.1 mmol g−1, 53.2 wt%), see Fig. 6, suggesting a high selectivity for CO2.
 | ||
| Fig. 6 Adsorption (red solid circles) and desorption (red open circles) CO2 isotherm for activated 4 (150 °C and 10−5 bar for 1 h) at 196 K. | ||
This CO2 selectivity over N2 at cryogenic temperatures has been previously observed in similar metal–organic materials such as PCM-1414 and CUK-126 and also for the metal–macrocycle framework NMC-1.27 A desorption hysteresis was noted for CO2 (Fig. 6, open red circles). The origin of this hysteresis most likely is due to moderately strong physisorption between CO2 molecules and coordinatively-unsaturated Cu(II) sites. In fact, according to the crystal structure (vide supra, Fig. 1), there are two kinds of open metal sites: six inter-cage and six extra-cage. CO2 molecules can gain access to both kinds of coordinatively-unsaturated Cu(II) sites and also to the interstitial channels which are in between the metal–organic polyhedra (cages). By calculating the molar ratio of the adsorbed CO2 molecules/4, we found that only 3.16 mmol of CO2 is required to fulfil the 12 open metal sites of 4. Then, 12 molecules of CO2 complete the inter-cage and extra-cage sites (0.16 bar approximately, 4th adsorption point in the CO2 adsorption isotherm, Fig. 6) and the rest of the CO2 molecules (total uptake = 12.21 mmol g−1) can diffuse within the cages. To clearly visualise this, a cylindrical probe with a diameter of 3.3 Å (corresponding to the kinetic diameter of CO2) was calculated using the crystal structure array of 4 demonstrating the existence of channels (blue) which suggested an uninterrupted diffusion of CO2 in between the cages (see Fig. 7).
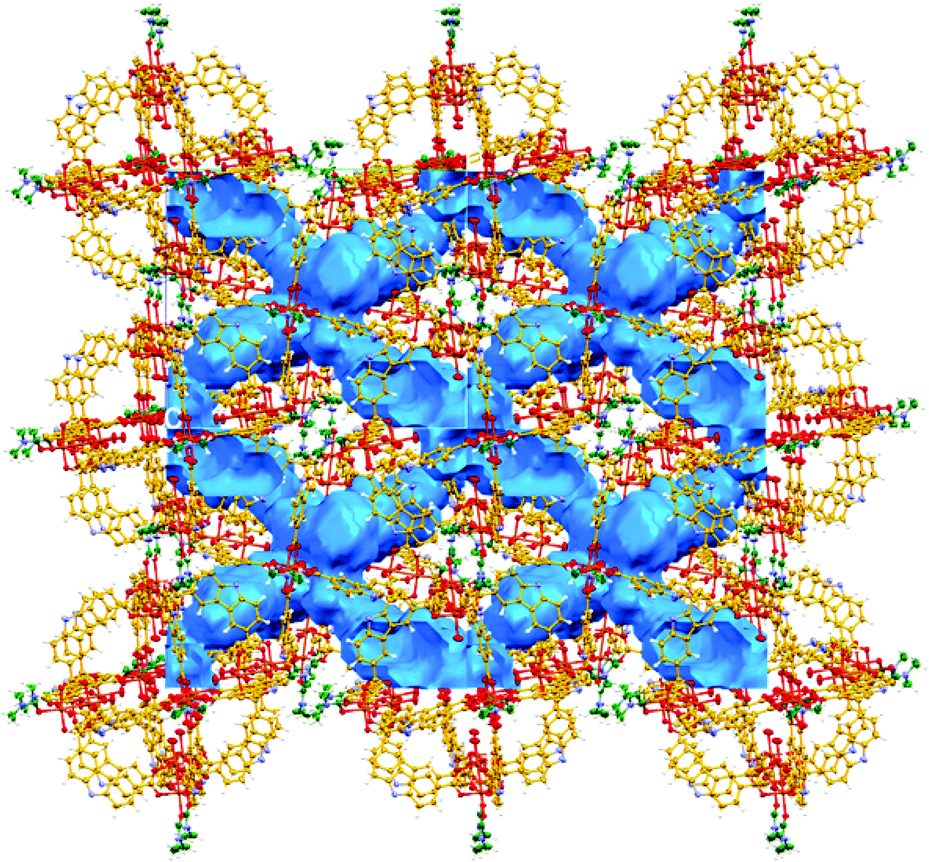 | ||
| Fig. 7 View of the calculated voids in 4 using a probe diameter of 3.3 Å (19.4% of unit cell volume). Blue channels suggest an uninterrupted diffusion of CO2 in between the cages. | ||
When coordinated solvents (H2O and DMF) are removed from the Cu(II) metal centres of 4 (as previously demonstrated by FTIR), CO2 molecules can reach these open metal sites (inter-cage and extra-cage) since there is enough free space both between cages and inside the cages. In order to corroborate this hypothesis, we decided to run in situ Raman spectroscopy experiments. First, Raman spectroscopy was performed on an activated sample (150 °C and 10−5 bar for 1 h) of 4 (Fig. 8). Later, this activated sample was exposed to an atmosphere of CO2 and the increase of the 1389 cm−1 Raman band confirmed the direct interaction of CO2 molecules and the Cu(II) open metal sites as previously reported by Kitagawa28 and Lockard.29
 | ||
| Fig. 8 Raman spectral changes of activated 4 upon exposure to CO2. | ||
The strong physisorption (see Fig. 6) was confirmed by the determination of the isosteric enthalpy of adsorption for CO2 (molar enthalpy of adsorption, ΔH) which was experimentally measured by CO2 adsorption microcalorimetry (see Fig. S14, ESI†) with a value of ΔH = 65.12 kJ mol−1, which is a characteristic value for open-metal site MOF systems.3a,21 After the CO2 adsorption experiment was complete, a PXRD experiment was conducted and it confirmed that the retention of the crystallinity was maintained (see Fig. S11, ESI†), as the activated 4 phase (vide supra).
Therefore, thanks to the accessibility to the Cu(II) unsaturated metal sites it was possible to perform CO2 capture at 196 K, emphasising the relevance of the generation of open metal sites within a MOP material. Finally, we decided to perform three CO2 adsorption cycles on a freshly synthesised 4 sample (see Fig. S12, ESI†). Interestingly, these three CO2 adsorption experiments (196 K) showed consistency since the shape of the CO2 adsorptions was very similar and the maximum CO2 uptake was averaged to be 11.4 mmol g−1 (50.1 wt%). PXRD confirmed a partial retention of the crystallinity on activated 4 after the adsorption cycles of CO2 (see Fig. S13, ESI†). The decrease in the crystallinity of 4 after the cycling experiments can be attributed to the gradual degradation of the sample after consecutive activations.
In addition, to further investigate the state of CO2 molecules in MOP 4, in situ Fourier transform infrared (FTIR) spectroscopy, under an atmosphere of CO2, was performed. The experiments were carried out on an activated sample of 4 (150 °C and 10−5 bar for 1 h) that was exposed to the CO2 atmosphere and directly placed on the spectrometer to collect the first spectra (T = 0, Fig. 9). Then a spectrum was collected every 10 min until no further change was observed. The increase in the CO2 characteristic band (2358 cm−1) clearly indicates that MOP 4 adsorbs CO2. At T = 70 the activated sample of 4 was saturated with CO2 (Fig. 9).
 | ||
| Fig. 9 FTIR spectra of activated 4 under an atmosphere of CO2 from T = 0 min to T = 70 min (saturated), showing the characteristic band for CO2 (2358 cm−1). | ||
Up to this point, we demonstrated that (i) CO2 molecules can directly interact with two kinds of open metal sites (internal and six external) and this interaction was observed by in situ Raman spectroscopy, and (ii) by in situ FTIR the adsorption of CO2 was also corroborated. Indeed, due to the nature of the carbazole ligand that conforms to 4, it may be possible to form hydrogen bonds of the N–H⋯O type, where the donor is the nitrogen of the carbazole moiety (see Scheme 1). Thus, CO2 molecules can interact with these moieties and provide an extra CO2 adsorption site. When analysing the in situ FTIR spectra (under an atmosphere of CO2) the characteristic N–H stretching band (3390 cm−1, carbazole) showed the absence of a shift which clearly indicated no interactions with CO2 molecules (Fig. 10). Therefore, the carbazolate moieties did not provide extra CO2 adsorption sites.
 | ||
| Fig. 10 FTIR spectra of activated 4 under an atmosphere of CO2 from T = 0 min to T = 70 min (saturated), showing the characteristic band for N–H (3390 cm−1). | ||
Conclusions
We have readily synthesised a carbazole-containing polyhedron in four steps. This metal–organic polyhedron (4) features di-copper(II) paddlewheel clusters which are coordinated not only to the carbazole–carboxylate ligands but also to terminal ligands H2O and DMF. By FTIR spectroscopy we demonstrated the removal of these terminal ligands via thermal activation of 4 under vacuum, affording uncoordinated Cu(II) metal sites. Interestingly, MOP 4 showed a remarkable selectivity towards CO2 over N2 at cryogenic temperatures. Thus, by taking advantage of this selectivity, the total CO2 capture was equal to 12.1 mmol g−1 at 196 K. The desorption hysteresis observed from the CO2 isotherm confirmed the moderately strong interaction between CO2 molecules and coordinatively-unsaturated Cu(II) sites, corroborated by the molar enthalpy of the adsorption value for CO2 of ΔH = 65.12 kJ mol−1, highlighting the importance in the generation of open metal sites (150 °C and 10−5 bar for 1 h) for CO2 capture. In situ FTIR corroborated the adsorption of CO2 by 4 and in situ Raman studies showed the direct interaction of CO2 molecules and Cu(II) open metal sites.28,29Acknowledgements
The authors thank Dr Maria del Carmen García Gonzalez (MS), M.Sc. Simón Hernández-Ortega (X-ray; IQ-UNAM), Dr A. Tejeda-Cruz (powder X-ray; IIM-UNAM), Dr Eduardo González-Zamora (MS), and Mónica A. Rincón Guevara (MS). B. R.-M. thanks PAPIIT IA200615 (UNAM, Mexico) for financial support. I. A. I. thanks CONACyT Mexico (212318) and PAPIIT UNAM Mexico (IN100415 and IN101517) for financial support.Notes and references
- (a) S. Chu, Science, 2009, 325, 1599 Search PubMed; (b) R. S. Haszeldine, Science, 2009, 325, 1647 Search PubMed.
- J. G. J. Olivier, G. Janssens-Maenhout, M. Muntean and J. A. H. W. Peters, Trends in global CO2 emissions: 2015 report, 2015.
- (a) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058 Search PubMed; (b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, Z. T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 Search PubMed.
- (a) G. T. Rochelle, Science, 2009, 325, 1652 Search PubMed; (b) F. Karadas, M. Atilhan and S. Aparicio, Energy Fuels, 2010, 24, 5817 Search PubMed.
- M. Poliakoff, W. Leitner and E. S. Streng, Faraday Discuss., 2015, 183, 9 RSC.
- (a) S. Yang, G. S. B. Martin, J. J. Titman, A. J. Blake, D. R. Allan, N. R. Champness and M. Schröder, Inorg. Chem., 2011, 50, 9374 CrossRef CAS PubMed; (b) S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487 Search PubMed; (c) A. J. Nuñez, L. N. Shear, N. Dahal, I. A. Ibarra, J. W. Yoon, Y. K. Hwang, J.-S. Chang and S. M. Humphrey, Chem. Commun., 2011, 47, 11855 Search PubMed; (d) I. A. Ibarra, J. W. Yoon, J.-S. Chang, S. K. Lee, V. M. Lynch and S. M. Humphrey, Inorg. Chem., 2012, 51, 12242 Search PubMed; (e) X. Lin, A. J. Blake, C. Wilson, X. Z. Sun, N. R. Champness, M. W. George, P. Hubberstey, R. Mokaya and M. Schröder, J. Am. Chem. Soc., 2006, 128, 10745 Search PubMed; (f) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80 Search PubMed; (g) O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 522 Search PubMed; (h) W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906 CrossRef CAS PubMed; (i) K. Okada, R. Ricco, Y. Tokudome, M. J. Styles, A. J. Hill, M. Takahashi and P. Falcaro, Adv. Funct. Mater., 2014, 24, 1969 Search PubMed; (j) H. Li, M. R. Hill, R. Huang, C. Doblin, S. Lim, A. J. Hill, R. Babarao and P. Falcaro, Chem. Commun., 2016, 52, 5973 Search PubMed; (k) K. Sumida, N. Moitra, J. Reboul, S. Fukumoto, K. Nakanishi, K. Kanamori, S. Furukawa and S. Kitagawa, Chem. Sci., 2015, 6, 5938 RSC; (l) K. Hirai, S. Furukawa, M. Kondo, H. Uehara, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 8057 Search PubMed.
- (a) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973 Search PubMed; (b) R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173 Search PubMed; (c) W. M. Bloch, R. Babarao, M. R. Hill, C. J. Doonan and C. J. Sumby, J. Am. Chem. Soc., 2013, 135, 10441 Search PubMed; (d) J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 Search PubMed; (e) R. Babarao, C. J. Coghlan, D. Rankine, W. M. Bloch, G. K. Gransbury, H. Sato, S. Kitagawa, C. J. Sumby, M. R. Hill and C. J. Doonan, Chem. Commun., 2014, 50, 3238 Search PubMed; (f) I. A. Ibarra, A. Mace, S. Yang, J. Sun, S. Lee, J.-S. Chang, A. Laaksonen, M. Schröder and X. Zou, Inorg. Chem., 2016, 55, 7219 Search PubMed; (g) R. A. Peralta, A. Campos-Reales-Pineda, H. Pfeiffer, J. R. Álvarez, J. A. Zárate, J. Balmaseda, E. González-Zamora, A. Martínez, D. Martínez-Otero, V. Jancik and I. A. Ibarra, Chem. Commun., 2016, 52, 10273 Search PubMed.
- (a) A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784 Search PubMed; (b) J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2009, 131, 5516 Search PubMed.
- (a) T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973 Search PubMed; (b) R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650 Search PubMed.
- P. Pachfule and R. Banerjee, Cryst. Growth Des., 2011, 11, 5176 Search PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782 Search PubMed.
- (a) M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 Search PubMed; (b) I. A. Ibarra, X. Lin, S. Yang, A. J. Blake, G. S. Walker, S. A. Barnett, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem. – Eur. J., 2010, 16, 13671 Search PubMed.
- J. Park, H. Kim, S. S. Han and Y. Jung, J. Phys. Chem. Lett., 2012, 3, 826 Search PubMed.
- I. A. Ibarra, K. E. Tan, V. M. Lynch and S. M. Humphrey, Dalton Trans., 2012, 41, 3920 Search PubMed.
- J. F. Morin, M. Leclerc, D. Ads and A. Siove, Macromol. Rapid Commun., 2005, 26, 761 Search PubMed.
- N. Blouin and M. Leclerc, Acc. Chem. Res., 2008, 41, 1110 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 Search PubMed.
- J.-R. Li, D. J. Timmons and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 6368 Search PubMed.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159 Search PubMed.
- C. Prestipino, L. Regli, J. G. Viltillo, F. Bonito, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337 Search PubMed.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341 Search PubMed.
- D.-L. Long, R. J. Hill, A. J. Blake, N. R. Champness, P. Hubberstey, C. Wilson and M. Schröder, Chem. – Eur. J., 2005, 11, 1384 Search PubMed.
- R. Yepez, S. García, P. Schachat, M. Sánchez- Sánchez, J. H. González-Estefan, E. González-Zamora, I. A. Ibarra and J. Aguilar-Pliego, New J. Chem., 2015, 39, 5112 Search PubMed.
- C. G. Carson, K. Hardcastle, J. Schwartz, X. Liu, C. Hoffmann, R. A. Gerhardt and R. Tannenbaum, Eur. J. Chem., 2009, 2338 Search PubMed.
- J. M. Teo, C. J. Coghlan, J. D. Evans, E. Tsivion, M. Head-Gordon, C. J. Sumby and C. J. Doonan, Chem. Commun., 2016, 52, 276 Search PubMed.
- S. M. Humphrey, J.-S. Chang, S. H. Jhung, J. W. Yoon and P. T. Wood, Angew. Chem., Int. Ed., 2007, 46, 272 Search PubMed.
- J. Lee, N. W. Waggoner, L. Polanco, G. R. You, V. M. Lynch, S. K. Kim, S. M. Humphrey and J. L. Sessler, Chem. Commun., 2016, 52, 8514 Search PubMed.
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 9005 Search PubMed.
- Y. Chen, H. Wang, J. Lib and J. V. Lockard, J. Mater. Chem. A, 2015, 3, 4945 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR data, TGA data, PXRD data, N2 isotherm data and relevant crystallographic parameters. CCDC 1501023. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00342g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2017 |