
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Free radical and RAFT polymerization of vinyl esters in metal–organic-frameworks†
Jongkook
Hwang
 ,
Hui-Chun
Lee
,
Markus
Antonietti
and
Bernhard V. K. J.
Schmidt
*
,
Hui-Chun
Lee
,
Markus
Antonietti
and
Bernhard V. K. J.
Schmidt
*
Department of Colloid Chemistry, Max-Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: bernhard.schmidt@mpikg.mpg.de
First published on 29th September 2017
Abstract
Free radical and RAFT polymerization of vinyl esters with different molecular dimensions are conducted in the nanochannels of metal–organic-frameworks (MOFs). The combination of MOFs with the RAFT technique enables the synthesis of highly isotactic poly(vinyl ester)s with a controlled molecular weight and narrow molecular weight distribution, and stereocontrolled isotactic-block-atactic vinyl ester block copolymers.
Poly(vinyl ester)s (PVEs) are an industrially important class of polymers with a broad range of applications such as adhesives, paints and coatings.1 Moreover, PVEs are utilized as precursors for poly(vinyl alcohol)s (PVAs) which attract considerable attention due to their water solubility, non-toxicity and biocompatibility that are suitable for various bio- and medical-applications.2 To meet the growing demand for functional PVEs, the ability to realize precision polymers having the needed properties and structures is an essential prerequisite.
Control over the primary structures of PVEs, e.g., molecular weight (MW) and – less often considered – tacticity, is an essential yet challenging task to control the properties of polymers. Because vinyl esters lack conjugated substituents, their propagating radicals have high reactivity and low stability, resulting often in uncontrollable chain transfer and side reactions. Although a few reversible deactivation radical polymerization (RDRP) techniques including iodine transfer-,3 cobalt mediated-,4 organostibine mediated-,5 iron complex catalyzed-radical polymerization6 and reversible addition fragmentation transfer (RAFT)7 have provided PVEs with relatively controlled MW and polydispersity (Đ), only negligible or no effect on stereostructures was induced. Indeed, the tacticity control of PVEs can only be achieved by using monomers with bulky/polar substituents and adding Lewis acids or fluoroalcohols into the reaction medium.8 Nevertheless, these approaches sometimes require expensive chemicals or specific conditions at a very low temperature (−78 °C) which are not appropriate for most of the RDRP processes, and thus are mostly limited to free radical polymerization, affording on this end polymers with uncontrolled MW and relatively high Đ. In addition, common methods effectively produce syndiotacticity-rich PVEs only. In this context, the development of an efficient method that simultaneously controls the MW, Đ and tacticity – preferably towards isotacticity which is not accessible by conventional methods – of PVEs is relevant.
Recently, metal–organic-frameworks, MOFs, crystalline porous materials constructed by joining metal-ion-containing nodes with organic ligands, have shown promise as versatile hosts for controlled polymerization or as polymerization catalysts.9 Their unique structural characteristics such as highly ordered and chemically homogeneous micropores (nanochannels), controllable pore size and pore functionality can provide steric discrimination to promote specific size and shape effects on the monomer arrangements/conformation, possibly enabling the production of vinyl polymers with controlled MW, stereo- and regio-structures, reaction sites, monomer sequence and chain alignment.10,11–13 Despite this remarkable progress, there are still several issues to be addressed. First, the previously described effect of the MOF nanochannels on tacticity was relatively small. e.g., poly(vinyl acetate) obtained from the MOF shows an increase in mesodiads of only 8% when compared with that of the bulk counterpart.13 Second, the understanding of detailed aspects of polymerization in MOFs (e.g., effect of the monomer or nanochannel size on polymerization) is still limited. Few systematic studies based on a specific monomer have been reported.12,13 Third, the polymerization of vinyl monomers in the MOF still remains restricted to free radical polymerization. Although propagating radicals in the MOF are remarkably stabilized and show a living-like characteristic, the inherent drawbacks of free radical polymerization inevitably lead to relatively broad molecular weight distribution (MWD) and limited control over macromolecular architectures or end groups.
To overcome these limitations, here we introduce free radical and RAFT polymerization of vinyl esters in the MOF, [Zn2(bdc)2ted]n (bdc = benzene-1,4-dicarboxylic acid and ted = triethylenediamine),14 which has uniform one-dimensional nanochannels of 7.5 × 7.5 Å2 along the c axis (Scheme 1). The MOF nanochannel is utilized as a nanoreactor to regulate the stereostructures (i.e., increase in isotacticity), while RAFT further provides access to PVEs with narrow MWD and controlled molecular architectures. The combination strategy allows the synthesis of highly isotactic PVEs with controllable MW and low Đ, which in turn enables the preparation of stereocontrolled isotactic-b-atactic vinyl ester block copolymers for the first time.
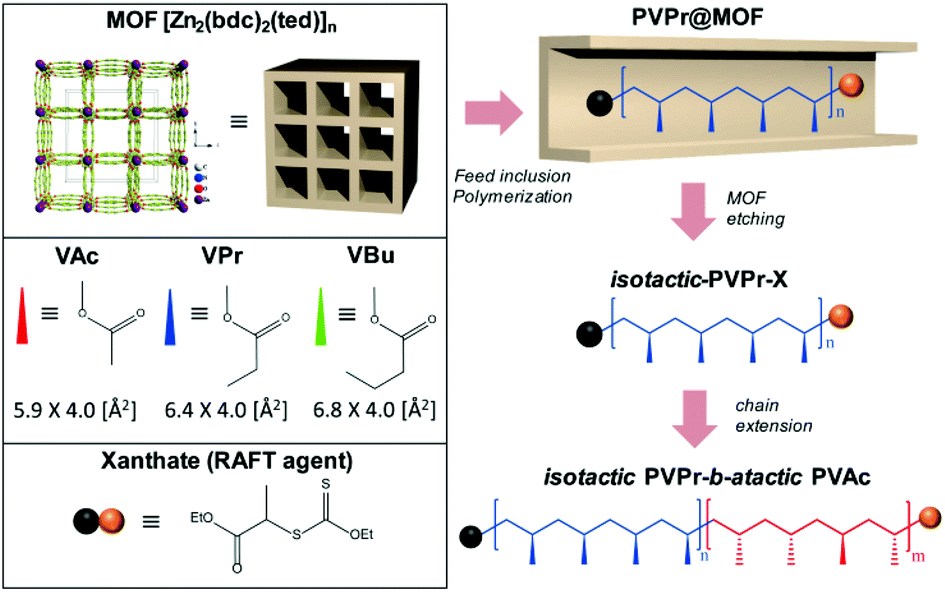 | ||
| Scheme 1 Free radical and RAFT polymerization of vinyl esters in the MOF and sequential preparation of isotactic-b-atactic block copolymers. | ||
We first studied the monomer size effect on free radical polymerization by using vinyl esters with different molecular dimensions (length × width), i.e., vinyl acetate (VAc) (5.9 × 4.0 Å2), vinyl propionate (VPr) (6.4 × 4.0 Å2), and vinyl butyrate (VBu) (6.8 × 4.1 Å2). The monomer/AIBN/(RAFT agent) solution was adsorbed in the nanochannels of the MOF by wetness impregnation. The excess monomer external to the MOF was removed under reduced pressure. The monomer loaded MOF (monomer@MOF) was heated at 60 °C for 48 h under a nitrogen atmosphere to form a polymer@MOF composite. Subsequently, the MOF host was decomposed in an aqueous Na2EDTA solution to liberate the polymers from the frameworks. The results of f-/r-PVE in the MOF are summarized in Table 1, where f and r denote free radical and RAFT polymerization, respectively.
| Sample | Adsorbed monomerc [number per unit cell] | Conv.c [%] |
M
n, SEC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d [g mol−1] d [g mol−1] |
Đ | Tacticity, mm:mr:rr (m)e [%] |
|---|---|---|---|---|---|
|
a [AIBN]:[monomer] = 1:471 at 60 °C for 48 h.
b [AIBN]:[RAFT agent]:[monomer] = 1:3:471 at 60 °C for 48 h.
c Determined by TGA.
d Determined by SEC against PS calibration.
e Determined by 1H NMR of PVA in dimethyl sulfoxide (DMSO)-d6 at ambient temperature.
|
|||||
| f-PVAc in MOF | 3.6 | 63 | 42500 |
2.17 | 30:50:20 (55) |
| f-PVPr in MOF | 2.8 | 43 | 20400 |
1.71 | 36:49:15 (61) |
| f-PVBu in MOF | 2.4 | 32 | 17500 |
1.51 | 25:49:26 (50) |
| r-PVAc in MOF | 3.6 | 65 | 21700 |
1.25 | 30:50:20 (55) |
| r-PVPr in MOF | 2.8 | 47 | 14300 |
1.34 | 36:50:14 (61) |
The amount of the adsorbed monomer in the MOF decreases slightly with increase of the molecular dimension of the monomer, as determined by thermogravimetric analysis (TGA) (Fig. S1;†Table 1). Powder X-ray diffraction patterns of polymer@MOF composites are in good agreement with that of the pristine MOF, indicating that microstructures of the MOF are well-retained without structural deformation during polymerization (Fig. S2a†). The relative peak intensity changed, which results from the introduction of the PVEs in the nanochannels of the MOF.11,15 To further support the incorporation of PVEs, N2 gas physisorption experiments were performed for the MOF and polymer@MOF composites (Fig. S2b†). All the isotherms correspond to the Type-I curve with a steep gas uptake at very low P/P0 ∼0.02, suggesting that uniform micropores are dominant. The decrease in the amount of N2 adsorption and the pore volume of polymer@MOF indicates that the nanochannels of the MOF are partially occupied by the PVEs.16 Such a decrease becomes more noticeable as the monomer size decreases from VBu to VAc, implying that the amount of the encapsulated polymer in the MOF increases in the order of PVBu < PVPr < PVAc, which is consistent with the conversions for the polymerization of VBu (32%), VPr (43%), and VAc (63%).
Monomer conversion is strongly affected by monomer size. Only a slight increase in monomer size leads to significant decrease in conversion. Previously, a similar behavior was observed in the polymerization of vinyl monomers in MOFs with different nanochannel sizes (4.3–10.8 Å).13 As the size of the nanochannels narrowed, the polymer yields and conversion decreased because of the reduced monomer mobility in the narrow nanochannels, which was confirmed by solid state nuclear magnetic resonance (NMR) spectroscopy. Likewise, the mobility of the large monomer (i.e., VBu) is more strongly restricted by the given nanochannels than that of the small monomer (i.e., VAc), which results in the relatively poor reactivity and low conversion of VBu. For comparison, when a bulkier monomer, vinyl pivalate, was employed for polymerization in the MOF, no polymeric products were obtained possibly due to the poor mobility of the monomer, although the adsorbed amount of monomer per unit cell (∼2 molecules) is sufficient for polymerization.
The MWDs become narrower with an increase of monomer size. The Đ decreases markedly from 2.17 for f-PVAc, 1.71 for f-PVPr and 1.51 for f-PVBu, which are much smaller than those prepared in bulk (Table S1†). The conventional free radical polymerizations of vinyl esters underwent uncontrollable chain transfer and termination due to the high reactivity of propagating radicals, which led to extremely broad MWD (Fig. S3†). In contrast, the propagating radicals in the MOF are remarkably stabilized and the termination reactions are largely suppressed because of effective entrapment in the nanochannels.11–13 These effects become prominent in the large monomer (VBu), enabling the preparation of f-PVBu with Đ as low as ∼1.5.
To further understand the free radical polymerization process in the MOF, the MW dependence on reaction time and thus conversion was investigated. The representative size exclusive chromatography (SEC) profiles of f-PVPr with different reaction times show that the MW has no correlation with conversion in the given nanochannels of the MOF (Fig. 1a). The MW reaches a high value ∼20000 g mol−1 at an early stage of polymerization and remains constant, whereas the conversion increases steadily with reaction time. The primary radicals may be slowly generated in the initiation step and undergo a fast propagation reaction to consume the monomers confined in a compartment (note that the reactant only partially occupies the nanochannels). Thus, an increase of monomer size can lead to a decrease of MW, because of the relatively low monomer loading (i.e., small compartment) and low mobility of the large monomer in the nanochannels.
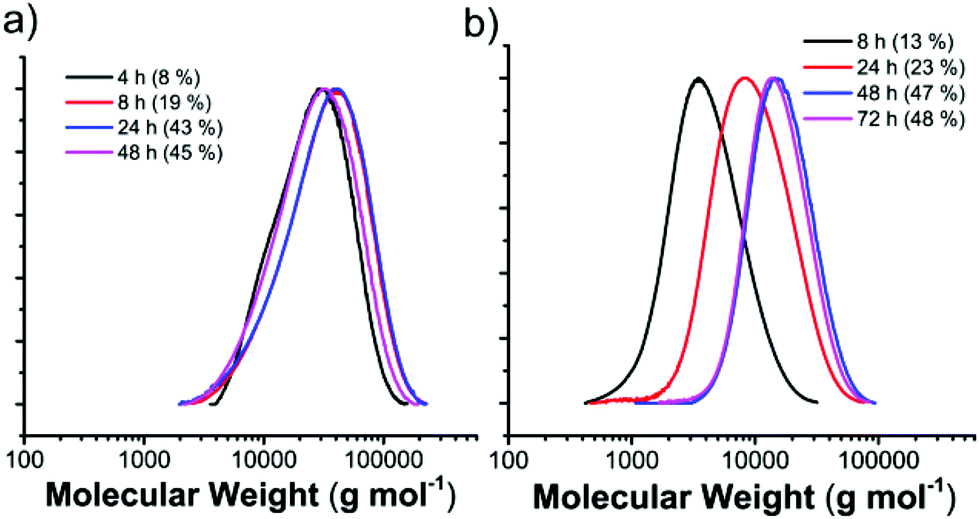 | ||
| Fig. 1 MWDs (obtained via SEC in THF with PS calibration) of (a) PVPr prepared by free radical polymerization in the MOF and (b) PVPr prepared by RAFT polymerization in the MOF for various reaction times (conversions). | ||
The relation between the monomer size and the stereostructure was studied, where the polymers obtained from the MOF (Table 1) have a higher isotacticity than those obtained from the bulk (Table S1†). The fractions of isotactic (mm)-, heterotactic (mr)- and syndiotactic (rr)-triads in each polymer were determined by 1H NMR spectroscopy after the saponification of the PVEs to PVAs (Fig. 2a). Compared with the bulk counterparts, a substantial increase in mesodiads (m) was observed in f-PVAc by 8%, in f-PVPr by 14% and in f-PVBu by 5% (Fig. 2b). Because there is no specific interaction between nanochannel walls and adsorbed monomers, the change in tacticity mostly depends on the monomer arrangement in a confined space. Polymerization in the confined nanochannels preferably induces the formation of less sterically bulky isotactic moieties,12,13 because the isotactic polymer has a smaller conformational diameter than its corresponding syndiotactic polymer.17 However, we found that the mesodiad fraction does not always increase with the increasing monomer size, but has a certain maximum value of 61% of f-PVPr in the MOF. Unlike the others, the tacticity change of f-PVBu is very small, probably due to the relatively large steric repulsion among the butyl substituents which could induce the formation of syndiotactic units, compromising the increase of isotactic units induced by nanochannels of the MOF. However, the detailed aspects of such behavior still remain elusive and will be the subject of future work. Notably, polymerization in the MOF is the only way known to increase the isotacticity of PVEs. Previous approaches such as the use of bulky monomers, protic solvents or additives can only afford syndiotactic-rich PVEs so far, e.g., fluoroalcohols form hydrogen bonds with the carbonyl group of the monomer and increase the effective size of side groups (steric hindrance), thereby inducing syndiotactic-specific polymerization. In that regard, the polymerization of vinyl esters in the MOF is an effective method to control the isotacticity of PVEs and it thus enables the preparation of PVPr with higher isotactic fractions (61% mesodiads).
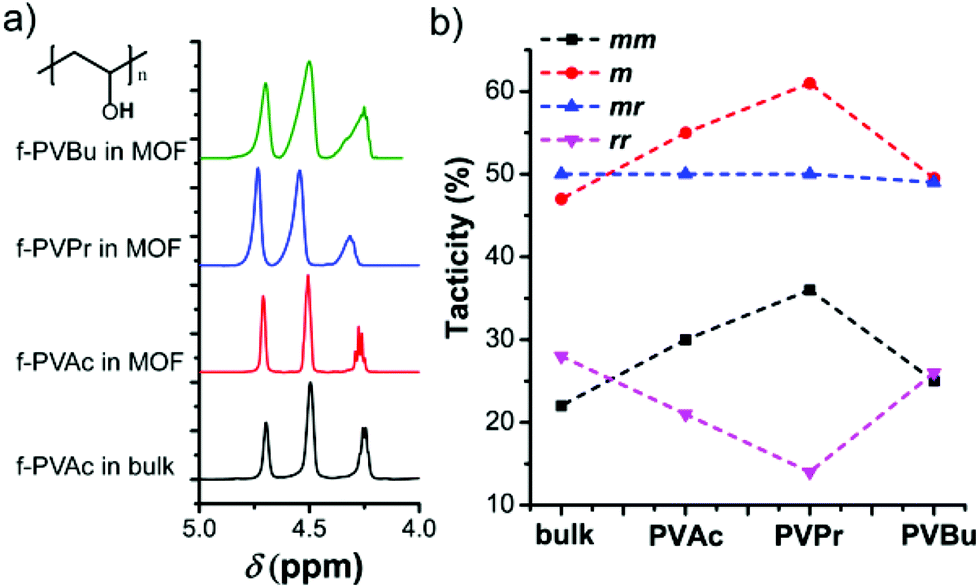 | ||
| Fig. 2 (a) 1H NMR spectra of PVA (obtained by saponification of the corresponding PVE) in DMSO-d6 at ambient temperature and (b) plots of tacticity change with respect to monomer types used for polymerization in the MOF. | ||
Although free radical polymerization in the MOF allowed the synthesis of PVEs with significant isotacticity, it still lacked control over MW, MWD and molecular architectures. One possible solution to overcome these limitations is the combination of the MOF with RDRP techniques. Among the various RDRP techniques, RAFT was chosen because of its ease of use, versatility, and compatibility with a wide range of monomers. It should be noted that nitroxide mediated polymerization18 and atom transfer radical polymerization (ATRP)19 of vinyl esters have proved to be very difficult. Here, a RAFT agent, (S)-2-(ethyl propionate)-(O-ethyl xanthate), was prepared according to the literature procedures20 (Fig. S4†) and used for mediating the polymerization of vinyl esters in the MOF.
The addition of a RAFT agent into the reaction feed of free radical polymerization led to the preparation of isotactic PVAc and PVPr (denoted as r-PVAc/r-PVPr in the MOF) (Table 1). The 1H NMR spectra of the resulting polymers show characteristic peaks of xanthate end groups (Fig. S5 and S6†). The tacticities of r-PVAc and r-PVPr in the MOF are similar to those of f-PVAc and f-PVPr in the MOF, which strongly suggests that RAFT polymerization mostly proceeds within the nanochannels of the MOF.
In sharp contrast to free radical polymerization in the MOF, the MW is determined by monomer conversion (Fig. 1), which is a clear indication for the controlled characteristic of the RAFT process. For instance, during the RAFT polymerization of VPr in the MOF, the MW increases linearly from 3200 to 7600 to 14300 g mol−1, as the monomer conversion (reaction time) increases from 13% (8 h) to 23% (24 h) to 47% (48 h), while maintaining low Đ at 1.3–1.6 (Fig. 1b). The conversion is saturated after polymerization for 48 h, which takes two times longer than f-PVPr in the MOF does. It is common that RAFT polymerization is slower than free radical polymerization in bulk and solution partly because of the adduct intermediate radical. We cannot exclude the possibility that the rate of degenerate transfer, i.e., main equilibrium in the RAFT process (Fig. S7†), can be slowed down by the largely restricted mobility of polymer/radical adducts in the narrow nanochannels. The MWDs of the resulting r-PVAc (1.25) and r-PVPr (1.34) in the MOF are as narrow as those of bulk RAFT counterparts (Fig. S8†), and are significantly narrower than those of f-PVAc (2.17) and f-PVPr (1.71) in MOF (Fig. S3†). All experimental results suggest that the RAFT agent efficiently mediates the controlled polymerization of vinyl ester in the nanochannels of the MOF.
One of the main advantages of RAFT is its ability to prepare complex architectures such as block copolymers. Sequential bulk RAFT polymerization with different vinyl esters allowed the synthesis of isotactic (it)-block-atactic (at) vinyl ester block copolymers, which are not accessible otherwise. The chain extension procedure is also important to confirm end group fidelity of the initial block. r-PVPr in the MOF was used as a macro-RAFT agent for chain extension with VAc at 60 °C ([AIBN]:[r-PVPr]:[VAc] = 0.4:1:2500). SEC traces of the initial itPVPr and final itPVPr-b-atPVAc show a clear shift of MWD from Mn 7600 g mol−1 (Đ 1.60) to Mn 12500 g mol−1 (Đ 1.71), which is slightly deviated from the theoretical value (Mn, theo 19500 g mol−1) (Fig. 3). However, considering the relatively high monomer to macro-RAFT agent ratio, such deviation is in a reasonable error range. The formation of the block copolymer was further confirmed by the 1H NMR spectrum, showing the characteristic peaks of two distinct blocks at 2.0–2.1 ppm for PVAc and at 1.0–1.2 ppm for PVPr (Fig. S9†). Although the living characteristics of the presented RAFT polymerization were confirmed by the linear molecular weight increment with conversion and the clear shift of full MWD without bimodalities or tailing after chain extension, it is still elusive whether the RAFT mechanism in the MOF really works in a similar way to conventional RAFT polymerization. The detailed aspects of the RAFT mechanism in the MOF are beyond the scope of the present contribution and are still under investigation.
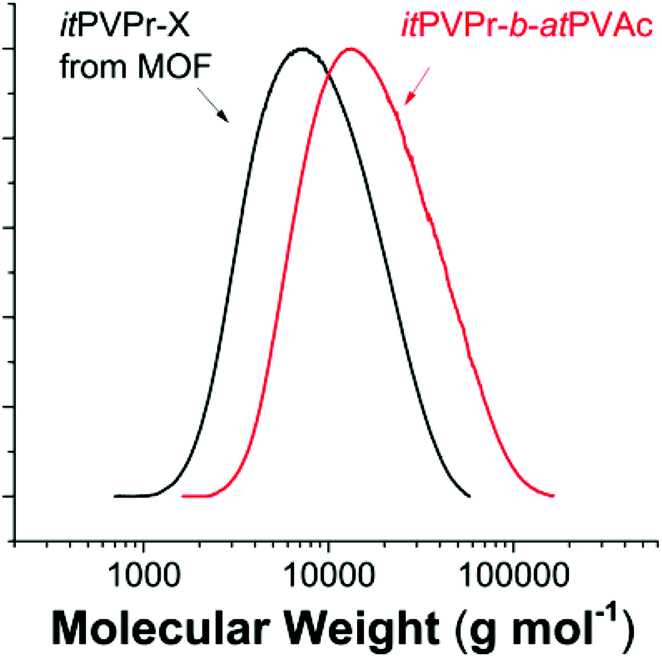 | ||
| Fig. 3 MWDs of the itPVPr macro-RAFT agent and the itPVPr-b-atPVAc block copolymer. | ||
In conclusion, we have demonstrated free radical and RAFT polymerization of various vinyl esters with different monomer dimensions (VAc, VPr, and VBu) in the identical nanochannels of the MOF. Strong correlations between monomer size and polymerization behavior were observed. As the monomer size increased from VAc to VPr to VBu, the conversion, MW, and Đ decreased accordingly. PVPr showed the highest isotacticity (61% mesodiads) among the PVEs. The polymerization system was further combined with the RAFT technique to prepare isotactic PVEs with controllable MW and narrow MWDs. The resulting PVPr was used as a macro-RAFT agent to prepare the stereocontrolled block copolymer, itPVPr-b-atPVAc. The results show that polymerization in the MOF enables simultaneous control over the molecular weight and tacticity of vinyl esters and expands the use of MOF nanochannel-reactors into the area of RDRP techniques for the first time, which might be extended to other types of RDRPs, e.g., ATRP. Therefore, we believe that the combination of the MOF and RDRP techniques enriches macromolecular engineering tools and opens up new possibilities that enable the preparation of unique macromolecules with tailored microstructures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Max Planck Society for funding. The authors acknowledge Marlies Gräwert for SEC measurements. J. H. acknowledges Max Planck Society for a Postdoc scholarship. Open Access funding provided by the Max Planck Society.Notes and references
- S. Harrisson, X. Liu, J.-N. Ollagnier, O. Coutelier, J.-D. Marty and M. Destarac, Polymers, 2014, 6, 1437–1488 CrossRef CAS; H. Rinno, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- M. C. Iovu and K. Matyjaszewski, Macromolecules, 2003, 36, 9346–9354 CrossRef CAS; K. Koumura, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2006, 39, 4054–4061 CrossRef.
- A. Debuigne, J. R. Caille and R. Jérôme, Angew. Chem., Int. Ed., 2005, 44, 1101–1104 CrossRef CAS PubMed.
- S. Yamago, B. Ray, K. Iida, J.-i. Yoshida, T. Tada, K. Yoshizawa, Y. Kwak, A. Goto and T. Fukuda, J. Am. Chem. Soc., 2004, 126, 13908–13909 CrossRef CAS PubMed.
- M. Wakioka, K.-Y. Baek, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2002, 35, 330–333 CrossRef CAS.
- M. H. Stenzel, L. Cummins, G. E. Roberts, T. P. Davis, P. Vana and C. Barner-Kowollik, Macromol. Chem. Phys., 2003, 204, 1160–1168 CrossRef CAS; S.-H. Shim, M.-k. Ham, J. Huh, Y.-K. Kwon and Y.-J. Kwark, Polym. Chem., 2013, 4, 5449–5455 RSC; C. E. Lipscomb and M. K. Mahanthappa, Macromolecules, 2009, 42, 4571–4579 CrossRef.
- K. Yamada, T. Nakano and Y. Okamoto, Polym. J., 1998, 30, 641–645 CrossRef CAS; W. Liu, Y. Koike and Y. Okamoto, Polymer, 2004, 45, 5491–5495 CrossRef; K. Yamada, T. Nakano and Y. Okamoto, Polym. J., 2000, 32, 707–710 CrossRef; K. Yamada, T. Nakano and Y. Okamoto, Macromolecules, 1998, 31, 7598–7605 CrossRef; K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef PubMed.
- H.-C. Lee, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2016, 7, 7199–7203 RSC; T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC; T. Uemura, N. Yanai and S. Kitagawa, Chem. Soc. Rev., 2009, 38, 1228–1236 RSC.
- G. Distefano, H. Suzuki, M. Tsujimoto, S. Isoda, S. Bracco, A. Comotti, P. Sozzani, T. Uemura and S. Kitagawa, Nat. Chem., 2013, 5, 335–341 CrossRef CAS PubMed; Y.-S. Wei, M. Zhang, P.-Q. Liao, R.-B. Lin, T.-Y. Li, G. Shao, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8348 CrossRef PubMed; I. H. Park, R. Medishetty, H. H. Lee, C. E. Mulijanto, H. S. Quah, S. S. Lee and J. J. Vittal, Angew. Chem., Int. Ed., 2015, 54, 7313–7317 CrossRef PubMed; R. Medishetty, I.-H. Park, S. S. Lee and J. J. Vittal, Chem. Commun., 2016, 52, 3989–4001 RSC.
- T. Uemura, K. Kitagawa, S. Horike, T. Kawamura, S. Kitagawa, M. Mizuno and K. Endo, Chem. Commun., 2005, 5968–5970 RSC.
- T. Uemura, Y. Ono, Y. Hijikata and S. Kitagawa, J. Am. Chem. Soc., 2010, 132, 4917–4924 CrossRef CAS PubMed.
- T. Uemura, Y. Ono, K. Kitagawa and S. Kitagawa, Macromolecules, 2008, 41, 87–94 CrossRef CAS.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed.
- K. Moller, T. Bein and R. X. Fischer, Chem. Mater., 1998, 10, 1841–1852 CrossRef CAS.
- C.-G. Wu and T. Bein, Science, 1994, 1013–1015 Search PubMed; J. Hwang, S. H. Woo, J. Shim, C. Jo, K. T. Lee and J. Lee, ACS Nano, 2013, 7, 1036–1044 CrossRef CAS PubMed.
- M. Hunt, D.-W. Jung, M. Shamsheer, T. Uyar and A. Tonelli, Polymer, 2004, 45, 1345–1347 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- M. B. Gillies, K. Matyjaszewski, P.-O. Norrby, T. Pintauer, R. Poli and P. Richard, Macromolecules, 2003, 36, 8551–8559 CrossRef CAS.
- V. K. Patel, N. K. Vishwakarma, A. K. Mishra, C. S. Biswas and B. Ray, J. Appl. Polym. Sci., 2012, 125, 2946–2955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01607g |
| This journal is © The Royal Society of Chemistry 2017 |