
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli responsive self-healing polymers: gels, elastomers and membranes
Adérito J. R.
Amaral
 and
George
Pasparakis
*
and
George
Pasparakis
*
UCL School of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK. E-mail: g.pasparakis@ucl.ac.uk
First published on 11th October 2017
Abstract
In recent years, the development of responsive polymers has expanded significantly owing to the constant development of new chemistries and controlled polymerization methods, which allow for the synthesis of complex and well-defined polymer architectures with predictable structure-to-function behaviour. These polymers exhibit multifarious changes of their properties by the application of physicochemical stimuli, be these reversible sol–gel transformations, hydrophilic–hydrophobic transitions, shape and volume changes, or assembly–disassembly ordering. The coupling of these responsive properties with suitable chemistries has generated novel polymeric materials with on-demand healing properties that can be precisely modulated and fine-tuned. In this review article, we discuss the interplay of key chemical reactions, which (usually) take place on crosslink points or on structural elements of polymeric materials with responsive modalities that drive the healing/remending mechanisms in a highly controllable manner. We examine representative emerging examples of bulk healable polymers in the form of polymer solids (elastomers, rigid polymers and hydrogels) and membranes that are designed for various uses in biomaterials science, bioelectronics, sensors, actuators and coating technologies, and analyse their potential for real-world applications.
 Adérito J. R. Amaral | Adérito Amaral obtained his M.Sc. degree in Biomedical Engineering at the University of Coimbra (Portugal) in 2012. Currently, he is a Ph.D. candidate at University College London (UCL School of Pharmacy), working on responsive polymers as cell surface modifiers and 3D healable microenvironments. His research interests include polymer–cell interactions and bioresponsive hydrogels for cell-based therapies and tissue engineering. |
 George Pasparakis | George Pasparakis is an EPSRC Early Career Fellow at the UCL School of Pharmacy. In 2009, he was awarded a Ph.D. from the School of Pharmacy, University of Nottingham, UK. He also holds a Ptychion in Materials Science from the University of Patras, Greece (2005). His current research interests include nanomedicinal formulations for cancer therapy, drug delivery systems, design of “smart” materials for biomedical applications, biophotonics, polymer chemistry, and pharmaceutical technologies. |
1. Introduction
In the past few decades, a plethora of functional polymer networks with stimuli-responsive properties has been widely explored in numerous research areas spanning from biomedical technologies, to bioelectronics, and engineering applications.1–4 These materials are commonly formed through physical blending, or by covalent or physical crosslinking of the polymer chains in the form of solvated networks (i.e. hydrogels), bulk monoliths or coatings. In recent years, significant research efforts have focused on the possibility to develop materials with self-healing properties in order to: (1) facilitate remotely controlled (self-)healing properties upon damage/fracture that fully restore the mechanical properties of the pristine material, (2) repair micron sized cracks/cuts with minimum external intervention, and (so far to a lesser extent) (3) to prolong the overall life-cycle of the polymers.5,6Two main strategies exist to synthesize self-healing materials: (1) the impregnation of catalysts in the form of capsules or fibres that are activated at the fracture point and elicit re-formation of new polymer networks, and (2) the incorporation of dynamic reversible bonds in the polymer matrix either in the form of crosslink points or as (hetero-)bifunctional building blocks (i.e. monomers, oligomers, etc.). For the first category, the reader can refer to excellent review articles here,7–10 as the present article mainly focuses on the second category.
The rapid development of responsive self-healing polymers is mainly driven by the utilization of dynamic (reversible) chemical bonds that can be broken and reformed under relatively mild physicochemical conditions/stimuli, which often overlap with the inherent responsiveness of the bulk materials.11,12 Therefore, the healing behaviour of these materials is intriguingly often accompanied by (spatiotemporally controlled) microphase separation, gel–sol transition, volume/shape change, or photochromic response, which in turn can be exploited in various contexts, such as for real-time monitoring of the damaged/healed area, acceleration of the healing times and re-enforcement of the mechanical properties.13 Further advances in the field of organic polymer chemistry, such as the utilization of controlled polymerization methods and the development of (bio-)orthogonal chemical strategies,14–16 enabled the synthesis of intricate polymer structures with remarkable response versatility and precision by multiple stimuli.
Dynamic, that is, reversible bonding can be covalent, such as the (retro-)Diels–Alder reaction products, disulfide and diselenide bridging, imine bonds, acylhydrazones and reversible boronate ester bonds, among others, or non-covalent, including hydrogen bonds, ionic and hydrophobic interactions, π–π stacking, metal coordination and host–guest interactions (Fig. 1). The nature of these crosslinking bonds in combination with the physicochemical properties of the polymers used determines, in part, the stimulus-responsiveness of the materials as well as their thermodynamic parameters. During the repairing mechanism, the dissociation of the crosslinks temporarily enhances the mobility of the polymer chains and the diffusion near the damaged area, following the formation of new bonds that results in the repair of the network and recovery of the mechanical properties.17 Thus, the optimization of free volume is an important factor to achieve the desirable mobility of the chains, which can also be controlled by external stimuli.
 | ||
| Fig. 1 Chemical strategies utilizing dynamic bonds for the design of stimuli responsive self-healing polymers. | ||
In this review, we present this interplay between bond dissociation of crosslink points with stimuli responsive properties to form responsive healable polymeric networks, and highlight recent examples and potential applications of dynamic polymers. We focus on polymer networks that undergo mechanical healing in response to an applied stimulus in a broader framework of “healing-on-demand”. These systems are introduced according to the particular stimulus responsible to initiate the healing mechanism, moving from temperature to light, redox, pH and electromagnetic field (Table 1).
| Reaction mechanism | Stimulus [ref.] | |||||
|---|---|---|---|---|---|---|
| Temperature | Light | pH | Redox | Electromagnetic | Multi-responsive | |
| Diels–Alder | 21–32 | 112 | 151 | |||
| Michael addition | 33, 41 | 142 | ||||
| Photo-cycloaddition | 67, 68, 77, 78 | |||||
| Acylhydrazone coupling | 39, 40, 64 | 108–112, 170 | 110, 169 | |||
| Alkoxyamine bond | 43 | |||||
| Diselenide bond | 96, 97 | |||||
| Disulfide bond | 34–36, 61–63 | 71, 72, 98 | 170 | 143, 144 | 110, 169 | |
| Trithiocarbonate | 70 | |||||
| Hindered urea bond | 42 | |||||
| Diarylbibenzofuranone | 65 | |||||
| Imine bond (Schiff base) | 64 | 113–117 | 152 | 171 | ||
| Cyclodextrins–guest molecules | 55, 56 | 73, 74, 86–88 | 122, 123 | 135–139 | 153, 154 | 168 |
| Cucurbiturils–guest molecules | 57, 58 | |||||
| Crown ethers | 118–121 | 167 | ||||
| Metal–ligand coordination | 75, 92, 93 | 128, 130, 131 | 159–167 | |||
| π–π stacking | 44–47 | |||||
| Hydrogen bonding | 39, 42–45 | 79 | 111, 112, 114 | 143 | ||
| Electrostatic interaction | 126 | |||||
| Hydrophobic interaction | 90 | 140 | ||||
| Boronate esters | 176, 178, 180 | 175, 177, 179–183 | ||||
2. Thermo-responsive polymer networks
Temperature is a fundamental and broadly accessible stimulus used in the construction of responsive materials. The Diels–Alders (DA) reaction is one of the most dominant synthetic strategies for the development of thermally healable materials as it is synthetically accessible and versatile (it can be combined with various monomer synthons), it does not require the presence of catalysts other than temperature to drive the equilibrium of the reaction and it exhibits adequate tolerance towards oxygen.18–20 In addition, the DA reaction is a self-contained system in that the number of atoms of the reactants and the products is constant (unlike for example, in condensation processes), which simplifies significantly the reaction conditions, and the problem of volume change of the bulk material during healing/remending is relatively limited. In a seminal paper, Wudl et al. presented a novel, two-component, re-mendable polymeric material comprising a 4-arm furan capped precursor that could be readily polymerized with a 3-arm maleimide synthon via a thermally triggered DA type of crosslinking.21 The resulting material was transparent (albeit with a yellow hue) and exhibited excellent mechanical properties comparable with those of commercially available resins (i.e. tensile strength >60 MPa and compression modulus >3 GPa). Interestingly, the material exhibited thermally triggered remending via the retro-DA that reached an efficiency of as high as 50% at 150 °C; the material could recover almost completely, and remarkably it showed adequate oxidation resistance during the thermal cycling. The same group further improved the drawbacks of the approach (e.g. the requirement for organic solvent to elicit adequate miscibility of the monomers, the ill-defined optical transparency and the high temperature required for effective remending) by synthesizing DA monomer components with significantly lower melting points (i.e. a bis-maleimide synthon with a soft ethylenedioxy bridge), which in turn reduced the healing temperature without compromising significantly the mechanical properties.22 Further refinement of their efforts led to the development of a single polymerizable component based on cyclic-type norborene derivative that could be readily (de-)polymerized via the r-DA reaction. This approach elegantly solves the problem of poor monomer miscibility and potentially the need for organic solvents that is encountered in two-component systems.23 This study was inspirational to other researchers who followed a similar approach towards the development of thermally healable materials based on the retro-DA crosslinking. For example, Heo and Sodano combined the DA motif with a shape memory polyurethane (PU) matrix to construct a novel material that could re-mend itself without the need for external forces, owing to its unique shape memory capacity.24 The researchers used furfuryl alcohol (FA) and N-(2-hydroxyethyl)-maleimide (HEM) as the furan–diene couple that could be readily reacted (due to the bis-hydroxyl functionality) with a PU precursor comprising triethanolamine and hexamethylene diisocyanate (Fig. 2a). The resulting crosslinked polymer exhibited excellent healing efficiencies, which were maintained above 50% (defined as the percentile of maximum load of the healed sample to the maximum load of the initial sample) for three consecutive cracking and healing cycles. It was demonstrated that the material could recover mechanically without the application of external forces, owing to the shape memory effect facilitated by the triethanolamine and the hexamethylene diisocyanate components, simply by heating the samples above the glass transition temperature of the polymer that also enabled the activation of the retro-DA reaction to take place. A similar approach based again on the combination of PU components blended with DA precursors was proposed by Bowman et al.:25 in their approach, the PU polymer network served as a structural component that prevented significant shape/volume change during the triggering of the DA-driven healing via capillary flow above the gelation temperature (Fig. 2b). The concept of capillary flow to mediate the healing of micron sized cracks and holes is conceptually powerful in that it eliminates the need for gravitational or mechanical forces to facilitate re-joining of the damaged interfaces. In addition, these studies showed the orthogonality of the DA coupling with other polymerization reactions, which allow further conceptualization of even more intricate self-healing systems. The DA reaction is also well-known to take place reversibly between anthracene and maleimide derivatives, and Yoshie et al. demonstrated elegantly the principle by synthesizing an anthryl-telechelic poly(ethylene adipate) polymer precursor that could be directly crosslinked with a tris-maleimide derivative (though with the use of organic solvent to form the crosslinked specimen).26 The resulting polymers had a tensile modulus of up to 25.9 MPa and elongation percentage at break point up to 1077%. Remending of previously cut samples took place by heating at 100 °C for up to 7 days (to re-initiate the formation of the DA crosslink adducts) resulted in up to 46% recovery of the tensile modulus and 79% of the elongation break point. In a similar study, Liu and Hsieh reported tri-functional maleimide and furan monomers with relatively low melting points and good miscibility with common organic solvents, such as ethanol, tetrahydrofuran, acetone, etc.27 The crosslinked polymer showed good recovery properties as it could be re-mended by heating to 120 °C to initiate the reverse the DA reaction in order to break the crosslink adducts followed by heating at 50 °C to re-initiate the DA reaction; nearly complete healing was observed within 24 hours of healing as evidenced by electron microscopy in previously cut thin film samples. Variations of DA to reduce the mending temperature close to room temperature or at least below the material degradation temperature were later reported. Barner-Kowollik and co-workers elegantly departed from the classic furan–maleimide coupling and conceived the formation of a cyanodithioester tetrafunctional derivative capped with cyclopentadiene, which could be readily crosslinked by a reversible thermal trigger.28 The material exhibited acceptable remending capabilities within the range of 40–120 °C. In the same context of altering the DA components chemically to further lower the healing temperature, the Lehn group has discovered a range of fulvene synthons that undergo DA coupling at remarkably low temperatures (−10 to 50 °C).29 By capping a linear or a 4-arm oligo poly(ethylene glycol) (PEG) with fulvene moieties, followed by mixing with a bis(dicyanofumarate) crosslinker in acetone, it was possible to obtain yellow-hued transparent elastomers with healing properties at room temperatures.30 Polyhedral oligomeric silsesquioxane (POSS) nanocomposites have also been reported based on DA chemistry and presented thermally triggered healing behaviour.31,32 | ||
| Fig. 2 (a) Formation of a DA adduct through the reaction of FA and HEM; images of the thermal healing process of the resulting PU network (after heating up to 135 °C and slowly cooling down to 70 °C). Adapted with permission from ref. 24. Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) Optical microscopy images of a gel demonstrate the capillary-based healing of a damaged area after heating to 120 °C for 15 min followed by 30 min at 70 °C (250 μm thick film). Adapted with permission from ref. 25. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Despite the apparent advantages of the DA reaction in designing healable polymer materials, there are pragmatic limitations which somehow narrow the potential applications. For example, the toxicity of the furan derivatives is a prohibitive factor in applying these materials in the biomedical and bioelectronics fields, and additionally, the relatively high temperatures required for the retro reaction may be limiting the combination range of DA synthons with other polymer networks of relative low degradation temperatures. Therefore, alternative reaction systems have been explored in the context of healable networks. Michael addition motifs constitute an appealing reaction cascade owing to their orthogonality and compatibility with radical polymerization methods. Konkolewicz et al. synthesized healable and malleable polymer networks by a classical free radical polymerization (FRP) reaction of a novel type of crosslinker (2-((3-(2-(acryloyloxy)ethoxy)-3-oxopropyl)thio)ethyl acrylate) that could undergo reversible Michael addition at 90 °C.33 The polymer network exhibited nearly complete recovery upon cutting and healing due to the reversibility of the reaction and the reconstitution of the polymer network. In the same context, Klumperman et al. reported on a healable epoxy network that could heal at temperatures as low as 60 °C owing to the well-known, albeit less studied in the field of healable materials, disulfide exchange reaction, simply by incorporating a disulfide bridge on the backbone of an epoxy polymer network.34 Recently, disulfide bonds were also incorporated into polymer networks via the inverse vulcanization reaction of elemental sulfur and 1,3-diisopropenylbenzene (DIB)35 or methacrylated POSS;36 the materials exhibited good healing efficiency when heated at 100 °C after 5 minutes, or at 120 °C during 3 hours, respectively.
Acylhydrazones are another interesting class of ligands that have been explored in self-healing materials; first introduced by Lehn and co-workers,37 who reported healable polymers, the so-called “dynamers” with remendable properties.38 Inspired by these studies, Schubert and co-workers employed acylhydrazone-rich methacrylate crosslinkers to synthesize covalently bonded polymer networks by copolymerization with commercially available methacrylate monomers.39 The polymer films produced showed good healing capacity driven by a thermal annealing process that was required to trigger the metathesis exchange reaction at the damaged interfaces (Fig. 3). Recently, Qin et al. further expanded the use of acylhydrazone crosslinkers in thermoresponsive (poly(N-isopropylacrylamide) (PNIPAM)-based) polymer networks, which elegantly proves the healing efficiency of these systems with a variety of monomers and physicochemical conditions.40
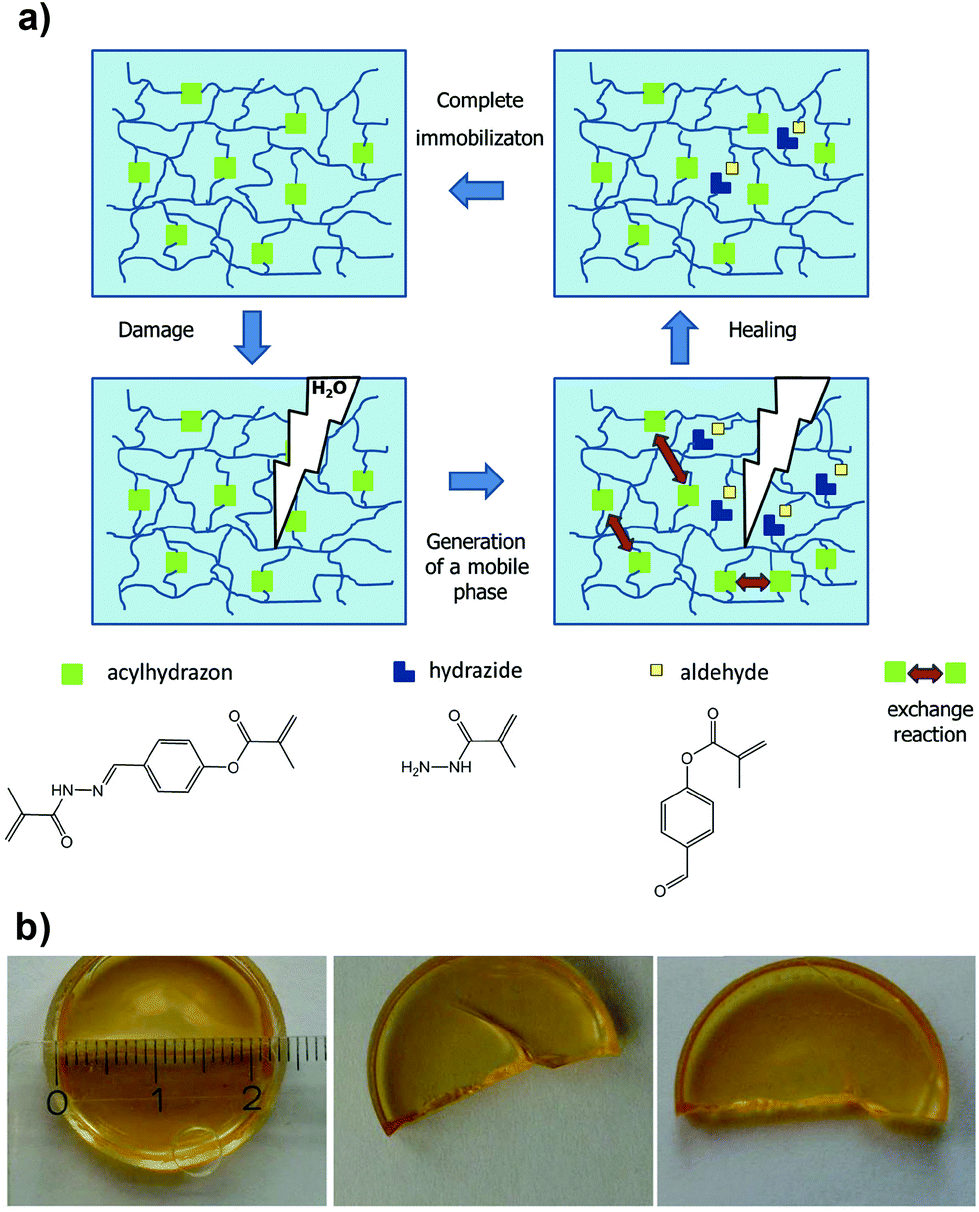 | ||
| Fig. 3 (a) Schematic illustration of the thermally triggered healing mechanism based on acylhydrazone bonds. (b) Photographs of the initial polymer film after being cut and healed (64 h at 100 °C). Adapted with permission from ref. 39. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
An alternative, but perhaps equally robust as the aforementioned, reaction motif was reported by Tang et al. based on the polymerization of aroylacetylenes and azides under mild and catalyst-free (that is without the addition of Cu(I) catalysts) reaction conditions that produce high molecular weight poly(aroyltriazole) products.41 This approach is powerful in that the alkyne–azide reaction is inherently orthogonal and can form irreversible triazole bridges that can withstand different chemical environments. Another appealing class of reversible bonding that also addresses the problem of high healing temperatures, which is mostly encountered in the (retro-)DA system, is the dynamic urea bond formation between isocyanate and amino derivatives. Cheng et al. introduced “hindered urea bonds” (HUB) by varying systematically the steric hindrance of the amino segments in order to facilitate optimum amidation and amidolysis equilibria to drive the healing mechanism within a reasonable temperature window (Fig. 4).42 The proposed concept is highly appealing due to the facile adjustment of the dynamic properties of the bulk polymers simply by adjusting the bulkiness of the adjacent substituents, and also due to the intrinsic capability of the system to form hydrogen bonds, which further enhance the mechanical properties of the bulk materials. Finally, the alkoxyamine C–ON covalent bond has been explored as a thermally activated mechanism for the fabrication of remendable polymer bulk materials, owing to the simultaneous radical type homolysis and recombination of the C–ON bond at temperatures comparable to the retro-DA systems.43 Although this system exhibits very fast remending times, as low as 15 minutes, it still suffers from unavoidable radical termination, which could compromise the robustness of the healing mechanism over multiple cycles.
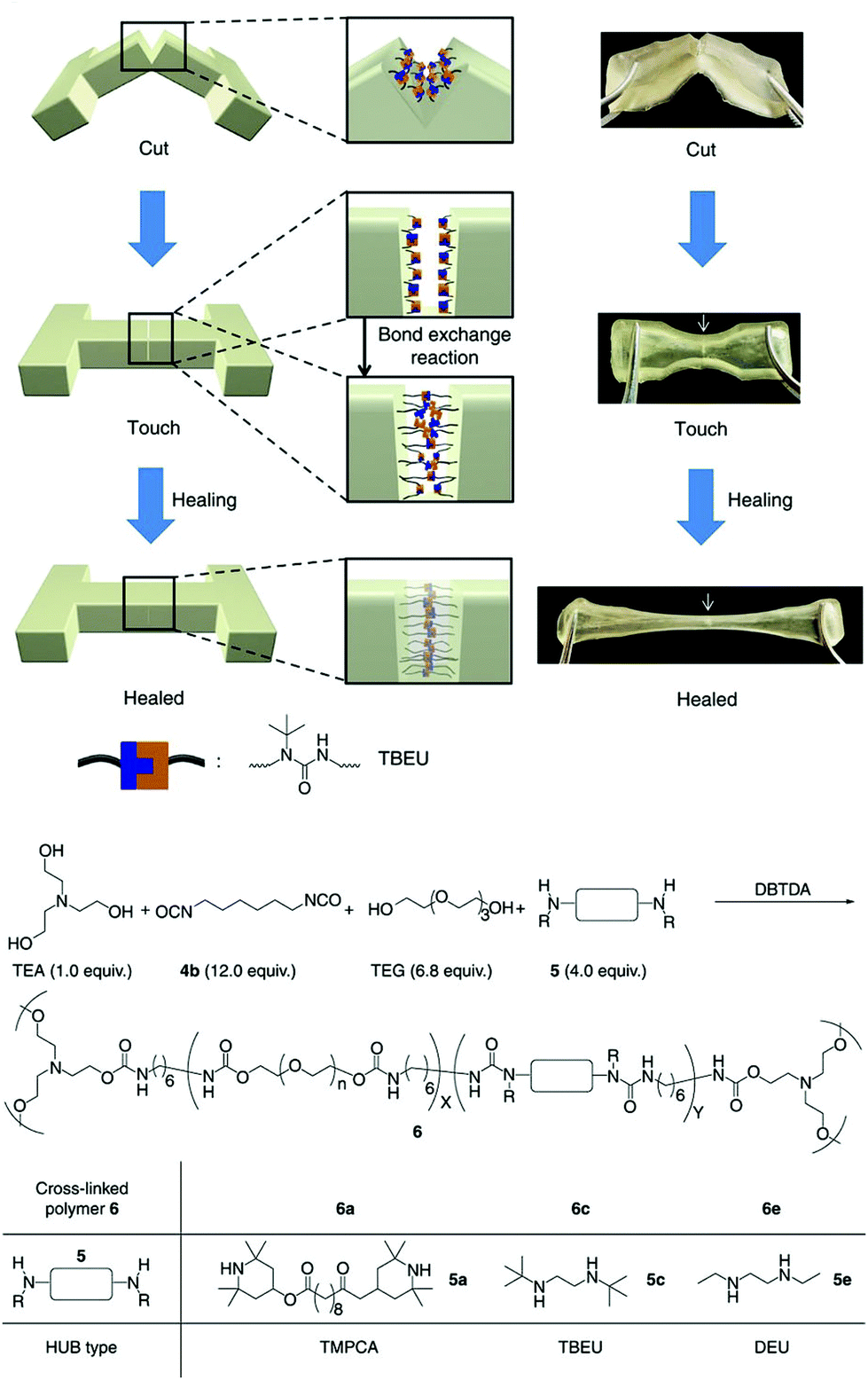 | ||
| Fig. 4 Illustration of the self-healing process (at 37 °C during 12 h), and chemical structures of the components used for the synthesis of HUB-based polymer networks. Adapted from ref. 42 with permission of the Nature Publishing Group. | ||
Although hydrogen bonding and π–π stacking are well-known chemical routes to fabricate hydrogels and elastomers, it was the seminal paper by Leibler et al.,44 which primarily directed the field towards the development of supramolecular assemblies in the context of healable materials. An excellent example of this category was reported by Hayes and co-workers who reported a supramolecular ensemble between two polymer precursors, namely, one with polyimide-rich repeating units and another with urea linked aromatic residues that was end-capped with pyrenyl residues.45 The two precursors were found to form strong π–π stacks between the aromatic residues and the naphthalenediimide segments and urea driven hydrogen bonding, which resulted in a bulk polymer with extremely high viscosity; characteristically, it had a relaxation time of 1 × 1010 s (ca. 300 years) at 30 °C. The researchers elegantly demonstrated the interplay of the π–π stacks and the hydrogen bonding with regard to the mechanical properties of the polymer, and also showed that the polymer blend exerted adequate healing properties at temperatures above 90 °C. The same research group further exploited this concept and doped the aforementioned polymer matrix with pyrene-coated gold nanoparticles (AuNPs) that were found to enhance the mechanical properties of the film.46 Unfortunately, the researchers have not reported as yet any attempts to remotely activate the healing mechanism by light irradiation although it would be very interesting to see whether AuNPs could, as efficient photothermal antennae, drive the healing mechanism of this unique polymer platform. The combination of aromatic interactions in concert with hydrogen bonding was also demonstrated by Zeng et al. who reported a triblock copolymer based on a poly(ethylene oxide) (PEO) middle block which was end-capped by both ends by a thermoresponsive copolymer of N-isopropylacrylamide-(N-3,4-dihydroxyphenethyl acrylamide), synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization.47 The triblock copolymer exhibited intriguing gelation properties due to the simultaneous hydrogen bonding and the aromatic interactions exerted by the catechol segments; in addition, the gelation could be reversibly tuned by the NIPAM feed close to body temperatures. The gel showed excellent recovery properties as well as intriguing cell-repellent capacity due to the PEO segment, as was shown by in vitro cell adhesion studies. In another study, Liu et al. reported an acrylamide (AAm) based gel composed of N-acryloyl glycinamide (NAGA) and 2-acrylamide-2-methylpropanesulfonic acid (AMPS), which form stable hydrogen bonds that could be disrupted at relatively low temperatures (ca. 60 °C).48 It was possible to physically entrap poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate) (PEDOT/PSS), which rendered the gels conductive (Fig. 5). The resulting material exhibited interesting electrical behaviour and showed adequate healing properties. This approach is appealing in that the healable polyacrylamide polymer matrix can be used as an ink platform for 3D printing applications where thermally driven malleability and robust sol–gel transition are highly required.
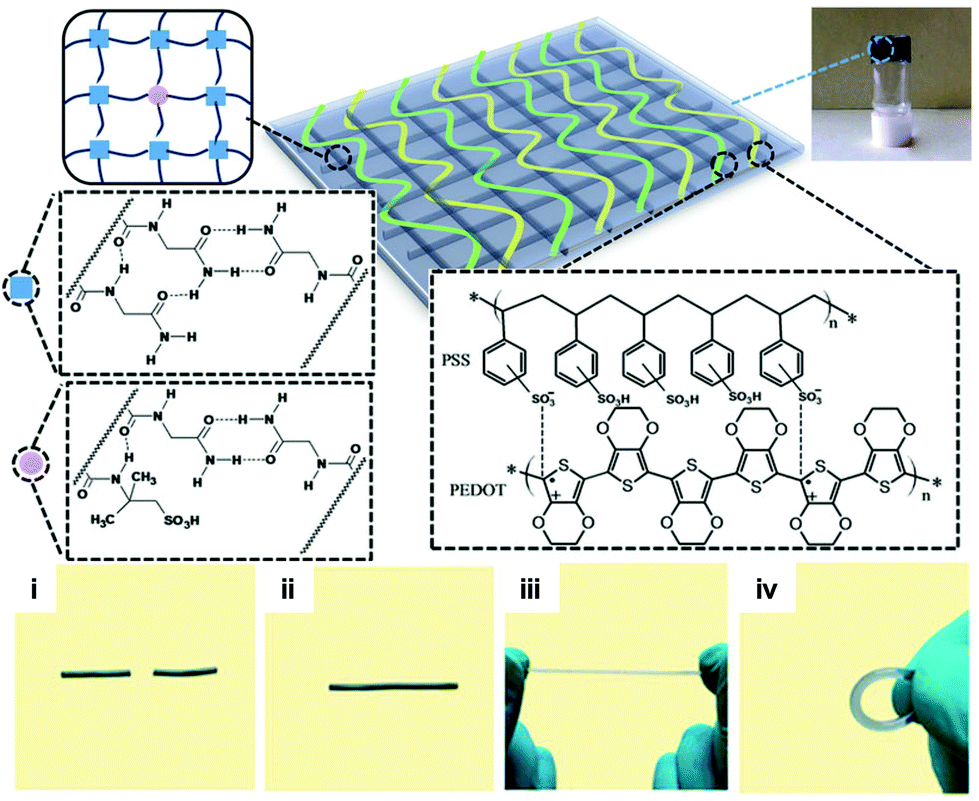 | ||
| Fig. 5 Chemical structure of the supramolecular hydrogel crosslinked by dual amide hydrogen bonds and doped with PEDOT/PSS. Photos show the initial gel cut in two pieces (i), which could be fully repaired upon heating (ii) and subsequently withstand stretching and bending (iii, iv). Adapted from ref. 48 with permission of the Nature Publishing Group. | ||
A very similar approach, again, based on the copolymerization of hydrogen bonding monomers was presented by Cui and del Campo.49 They copolymerized the well-known pH sensitive monomer 2-(dimethylamino)-ethyl methacrylate (DMAEMA) with the hydrogen forming monomer 2-(3-(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl)ureido)ethyl methacrylate; the latter exerts strong hydrogen bonding with itself owing to its rapid dimerization, which acts as reversible crosslink points in the polymer network. The polymer exhibited nearly full recovery upon cutting, but this was only limited to slightly alkaline conditions; conversely, the protonation of the tertiary amino segments of the DMAEMA co-monomer resulted in significant prohibition of the diffusion of polymer chains in acidic medium and hence compromised the healing properties. Nevertheless, the polymer showed adequate healing/recovery capabilities owing to the thermal-responsiveness due to the DMAEMA feed in the polymer chain. Another semi-synthetic approach was reported by Shi et al.50 that reported the formation of healable hydrogels by simple mixing of double stranded DNA and graphene sheets, as depicted in Fig. 6.51,52 The synthesized hydrogels exhibited an impressive storage modulus (G) of 4.6 kPa and exerted intriguing healing properties by relatively mild heating (90 °C) at the interface of the damaged areas, although the recovery rate of the mechanical properties was not reported. Nevertheless, this approach is important since DNA is an interesting building block from various viewpoints, namely, it is an excellent hydrogen-bond synthon, it can be synthesized to encode virtually infinite nucleotide pairs and it can be end-to-end functionalized. On the other hand, graphene constitutes an excellent 2D filler with intriguing electrical and mechanical properties, and consequently, this study could be inspirational for the fabrication of even more complex healable polymers from a functional perspective.
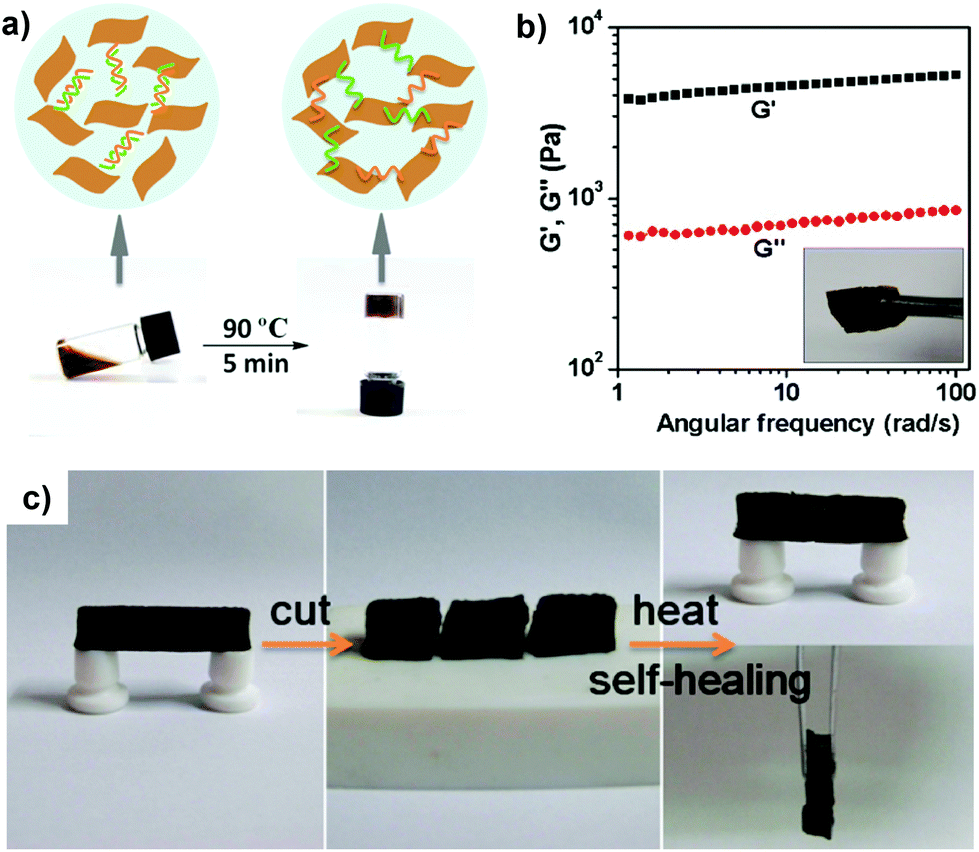 | ||
| Fig. 6 (a) Formation of graphene oxide/DNA hydrogels by thermal processing, and (b) their rheological behaviour. (c) Photos illustrating the self-healing process as a free-standing gel was cut and put in contact for ca. 3 min at 90 °C to recover its original shape. Adapted with permission from ref. 50. Copyright 2010, American Chemical Society. | ||
Bao et al. demonstrated the modification of silicon microparticle (SiMP) electrodes with a polymer thin layer, which establishes hydrogen bonds, and could self-repair at room temperature after mechanical damage, resulting in the maintenance of electrical contacts between the broken particles which in turn increased considerably the lifetime of the anodes made from SiMPs for lithium-ion batteries.53
An intrinsic problem in forming polymer networks by weak interactions, such as π–π stacking and hydrogen bonding, is that they can be easily disrupted especially when the proposed materials perform under aqueous conditions. Also, the crosslinking points usually lack orthogonality and hence the possible inclusion of other hydrogen-bonding-forming molecules – be these drugs, dyes, catalysts, etc. – is often compromised by the deleterious effects on the crosslinking points.
An appealing strategy to overcome these limitations is the construction of crosslinks by host–guest type of interactions with synthons of molecular complementarity with high affinity.54 Macrocyclic hosts as β-cyclodextrins (β-CDs) and cucurbit[n]urils (CBs), which are vital components to form supramolecular materials in aqueous solution, have hydrophobic cavities as well as hydrophilic and polar rims to include specific guest species through the hydrophobic effect. Notable examples of this approach are polymer networks that embed β-CD could host complementary guest molecules, such as cholesterol-type derivatives,55 or other types of hydrophobes, which can further be combined with thermoresponsive polymers56 to form thermally reversible bonds; the latter can further be exploited in applications for healable materials. Alternatively, CB[8], cyclic oligomers of glycoluril that form barrel-shaped cavities of approximately 0.9 nm diameter with high binding constants in water, have been found to form very stable complexes with selective guest molecules (e.g. viologen and naphthyl derivatives). The Scherman group has elegantly demonstrated their applications in healable polymer networks by the combination of RAFT-made polymers with pendant guest molecules that can be instantly crosslinked by the simple addition of CB[8] at varying concentration feeds.57 It was possible to fine control the mechanical properties of the gels and also to facilitate thermally driven sol–gel transformation driven by the thermal disruption of the host–guest crosslink complexes.58
Finally, isothermally healable, and often functional (see for example ref. 59 and 60), polymer networks have been reported, which undergo healing processes without the need to increase the temperature significantly owing to the effective dynamic chemistries employed, such as disulfide exchange61/metathesis,62,63 imine-acylhydrazone64 or diarylbibenzofuranone65 type of crosslinking. These approaches are extremely useful in applications where a high increase in temperature could be deleterious such as in cell encapsulating materials or in functional microdevices.
3. Light-responsive polymer networks
Light is a clean, inexpensive and non-invasive stimulus that can be delivered in a precisely temporal and spatial manner by remote activation via lasers and other illuminating devices with numerous applications in biomedicine, diagnostics, surgery, biosensors and bioelectronics, and drug delivery systems.66 Commonly used photosensitive moieties that are used as synthons in photoresponsive materials include azobenzenes, spiropyrans (SP), oxetanes, coumarins, anthracene and cinnamoyl moieties, among others, and they have been successfully utilized to design optically controlled self-healing polymers by photochemical cycloaddition,67,68 photo-induced radical polymerization69 and reshuffling of (photo-reversible) covalent bonds,70–72 photoisomerizations73,74 or photothermal effects.75,76Kim and co-workers originally proposed the light-triggered healing of a polymer network via the photochemical cycloaddition of cinnamoyl groups to form cyclobutane crosslinks.67 Polymer films made of 1,1,1-tris-(cinnamoyloxymethyl) ethane and methacrylate-based monomers were broken into several pieces and subsequently exposed to ultraviolet (UV) light of λ > 280 nm for 10 minutes; the samples could recover only ≈14% of their initial flexural strength, although it could be further increased to 26% by simultaneously heating at 100 °C. They found that upon crack formation, cyclobutane would reverse to the original cinnamoyl groups which could undergo cycloaddition again when irradiated. Coumarin-type derivatives, which can also undergo photo-cycloadditions, have now been applied to self-repair PU networks when irradiated with UV light at room temperature,77,78 owing to the reversible photodimerization at ≈350 nm and photocleavage at 254 nm (Fig. 7). Although coumarin moieties have been extensively used in photochemistry due to their reversible photodimerization abilities, their use in the field of healable materials is only recently expanding and improving.
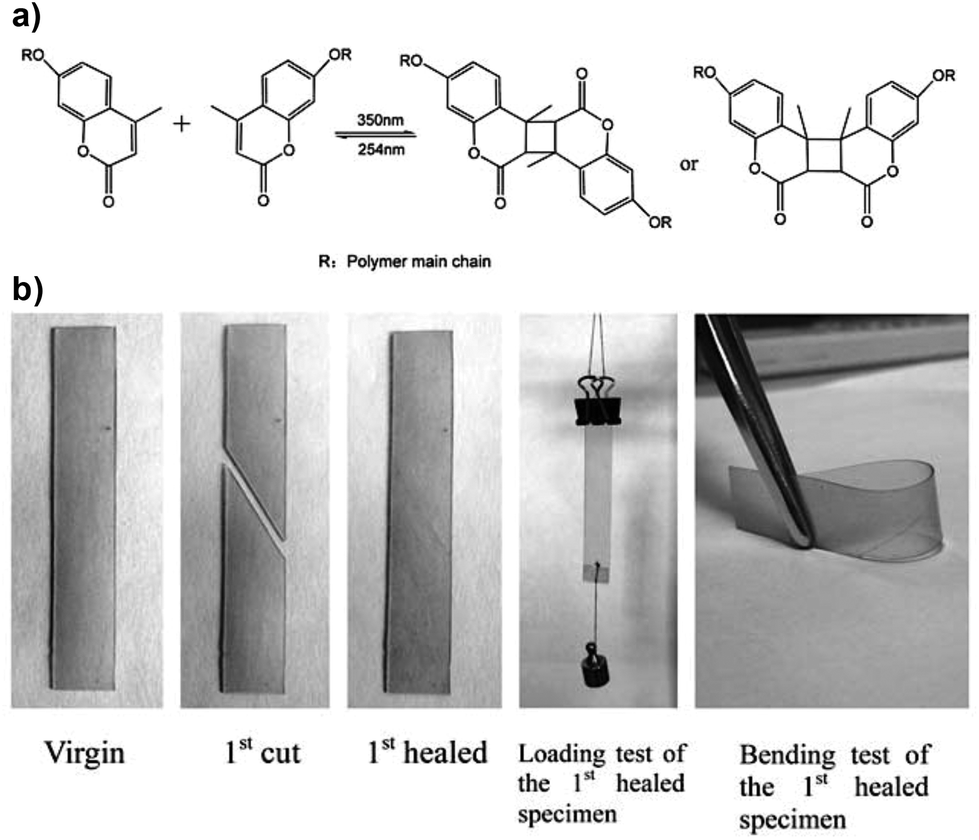 | ||
| Fig. 7 (a) Photodimerization and photocleavage of coumarin derivatives; (b) photos showing the remending of damaged PU networks by UV irradiation (the samples were exposed to 254 nm UV light for 1 min, brought in contact with each other and illuminated by 350 nm UV light for 90 min). Adapted from ref. 77 with permission from the Royal Society of Chemistry. | ||
In 2009, Ghosh and Urban proposed a polymer network based on the incorporation of an oxetane-substituted chitosan (OXE-CHI) precursor into a PU network that healed in 30 minutes when exposed to UV light (at room temperature), although the cut width was below 20 μm.69 They further demonstrated that OXE-CHI is preponderant for the self-repair mechanism, in addition to the stoichiometry of the reactive groups and pH variations in the damaged area.79 However, the mechanism based on the mechanical stress-sensitive oxetane opening would limit the repeated healing in the same area since its ring is not regenerated after reaction with chitosan chain radicals. Earlier, Bowman et al. had described a chemically crosslinked polymeric network based on pentaerythritol tetra(3-mercaptopropionate), triethyleneglycoldivinylether and 2-methyl-7-methylene-1,5-dithiacyclooctane monomers that experienced a photoreversible backbone cleavage and chain rearrangement without changing its mechanical properties. The polymer network, when irradiated (320 to 500 nm), exhibited rapid stress relief by sequential photocleavage and reformation of the crosslinks due to the diffusion of radicals in the backbone produced through addition–fragmentation chain transfer reaction, which conferred plasticity and equilibrium shape changes to the matrix.80
As previously discussed, the host–guest type of molecular interaction is another notable non-covalent strategy employed to fabricate reversible gel networks. Typically, host components, such as CDs, can be decorated with many functional groups, and owing to their hydrophobic internal cavity, can accommodate different guest molecules, typically including adamantane (Ad), ferrocene (Fc) and azobenzene (azo).81,82 Although the application of supramolecular chemistry in self-repairing materials has been extensively studied, the light-induced healing of supramolecular polymer networks has received greater consideration only very recently. Light-switchable supramolecular hydrogels based on host–guest interaction can be prepared by the complexation of β-CD with azo moieties;83 the latter can undergo a reversible and efficient trans–cis photoisomerization transformation upon UV and visible light irradiation that either favours or blocks the interaction with the CD cavity.84 The mechanism relies on the conversion of the trans isomer to the less stable cis isomer after UV irradiation, which will slowly relax to the trans form under dark conditions or is accelerated by using visible light.
For example, Jiang et al. explored the disassembly and reassembly of a pseudopolyrotaxane hydrogel, composed of PEG as the axis and α-CD as “threaded rings” (as reported by Harada and Kamachi),85 which reversible photoresponsive character was added by incorporating an azo moiety into the matrix (Fig. 8a).74 The complexation of CD with PEG formed microcrystal complexes that acted as physical crosslinkers to form the hydrogels within 4 hours; the addition of a competitive guest, 1-[p-(phenyl-azo)benzyl]pyridinium bromide, could dissociate the gel by replacing the PEG units in the complex. The alternation of UV (365 nm) and visible light (435 nm) allowed for a reversible sol-to-gel and gel-to-sol transition of the polymer network, respectively, for repeated cycles, due to the transition of the azo moiety from the trans to the cis form, which escapes from the α-CD cavity and enables PEG to move back and regenerate the hydrogel, and vice versa. Harada et al. further exploited this system86 by synthesizing polyacrylamide-based hydrogels functionalized with α-CD and azo compounds as the host and guest units, respectively (Fig. 8b).87 The assembly of both host and guest gels could be rapidly achieved by simple agitation under aqueous conditions. On irradiation with UV light, the gels dissociated due to the photoisomerization of the azo from trans to cis, which could then be reverted by visible light irradiation. The photoswitching character of the gel-assembly was also tested by placing a β-CD gel adjacent to the α-CD-azo gel followed by UV irradiation and agitation, which resulted in a pairing exchange reaction to form a β-CD-azo gel. The weakening of the adhesion forces (again driven by the trans–cis photoisomerization of azo) of the α-CD gel favoured the macroscopic assembly with the β-CD gel possibly because the association constant of the latter is higher, and forms a larger host binding cavity and an inclusion complex with both trans- and cis-azo guests. Inspired by these works, Tian and co-workers developed a supramolecular hydrogel based on the recognition between host β-CD and guest α-bromonaphthalene (α-BrNp) acrylamide-based copolymers.73 The hydrogels could be formed in about 15 minutes and could exert gel–sol phase transition by cooling and heating around 65 °C. Besides the thermoresponsive character, the gel also engendered a room temperature phosphorescence (RTP) signal. The addition of an azo-based polymer to the binary BrNp/CD system could adjust the RTP emission reversibly by competitive complexation with the β-CD under UV irradiation (by forming or deforming the pseudorotaxanes). The hydrogels prepared at a high concentration (2 × 10−2 M) showed a G′ around 3000 Pa (vs. ≈300 Pa at 1 × 10−2 M), and could heal within 1 minute after being cut in half and re-joined into one single piece, although only after 1 hour the gels could recover their original shape and mechanical properties in full. More recently, Liu et al. followed a similar approach towards the preparation of photoresponsive gels by copolymerizing AAm with a host–guest macro-crosslinker (poly-CD/azobenzeneacrylamide), which self-healing capability could be activated and deactivated with light.88 As described above, the photoisomerizable azo moiety could change its binding affinity with the β-CD upon UV irradiation. As a result, the crosslinking density of the network decreased as evidenced by the drop of G′ from 30 to 25 kPa at low strain values. Moreover, the self-healing ability of these gels after exposing to UV and visible light for 15 minutes each, or in darkness at 30 °C for 5 hours, was comparable with a healing efficiency close to 90%.
 | ||
| Fig. 8 (a) Representation of sol–gel transition of photoresponsive pseudopolyrotaxane hydrogels driven by competitive host–guest interactions. Adapted with permission from ref. 74. (b) Schematic depiction of photo-controlled interactions of CD/azo moieties upon irradiation with UV and visible light. Adapted with permission from ref. 86. Copyright 2010, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Light-controlled reversible change of rheological properties of fluids have also been reported,89 which may be applicable in microfluidics or as injectable formulations for cells and biologics or responsive fillers. For example, the Raghavan group reported photorheological fluids obtained by doping lecithin/sodium deoxycholate wormlike reverse micelles with an SP derivative.90 Under UV irradiation, SP compounds could be isomerized to the merocyanine (MC) form, which induced a 10-fold decrease in viscosity, which in turn resulted in the fluid flowing under gravitational force; when the UV light was switched off (or after visible light irradiation), the samples could recover to their initial viscosity and colour within 10 minutes, due to the photo-conversion of MC back to SP, and this cycle could be repeated 10 times without loss of response.
Polymers with metal centres possess striking optical properties, which make coordination chemistry particularly attractive for optically trackable healable polymers. The binding between a metal and macromers can be adjusted by reversible bonds of low to high dissociation energies, which offers tunability and controllable self-repairing properties to the macromolecules by incorporating different metal ions and ligands.91 The work by Weder et al. on photoresponsive metallosupramolecular polymers is a distinctive example due to their interesting healing behaviour based on the photothermal effect and broad application.75 The researchers used a rubbery poly(ethylene-co-butylene) core with 2,6-bis(1′-methylbenzimidazolyl)pyridine (Mebip) ligand end-groups coordinated with metal ions Zn2+ or La3+ (Fig. 9). The hydrophobic core and the polar metal–ligand motif resulted in a soft–hard phase separated lamellar structure. The exposure of UV light to the damaged site excites the metal–ligand complexes, which absorbed energy was converted into heat as they return to their ground state, causing the depolymerization of the metallopolymers and the decrease of the viscosity. The dissociation of the hard phase increases the mobility of the macromers, which promotes the recovery of the mechanical properties, thus allowing for a rapid repair of the network through formation of new bonds on cooling after switching off the light. The healing efficiency was determined to reach nearly 100 ± 36% by stress/strain experiments but it was inversely proportional to the metal/polymer ratio. The same group further improved the mechanical properties of this system by functionalizing the materials with hydrogen-bonding ureidopyrimidone derivatized cellulose nanocrystals.92 Similar hybrid multi-responsive soft actuators were recently reported by Mauro et al. who prepared metallosupramolecular polymers based on zinc(II)-terpyridine coordination nodes bearing photoisomerizable diazobenzene units or luminescent phenylene-ethylene moieties (acting as supragelators in organic solvents).93 The resultant soft organogels (G′ ca. 50 Pa) displayed light-triggered orientation-dependent volume reduction under UV light, photoluminescence and rapid self-healing ability owing to the dynamic metal–ligand coordination bonds; after placing two pieces in close contact for 15 minutes the gel could heal and recover its properties.
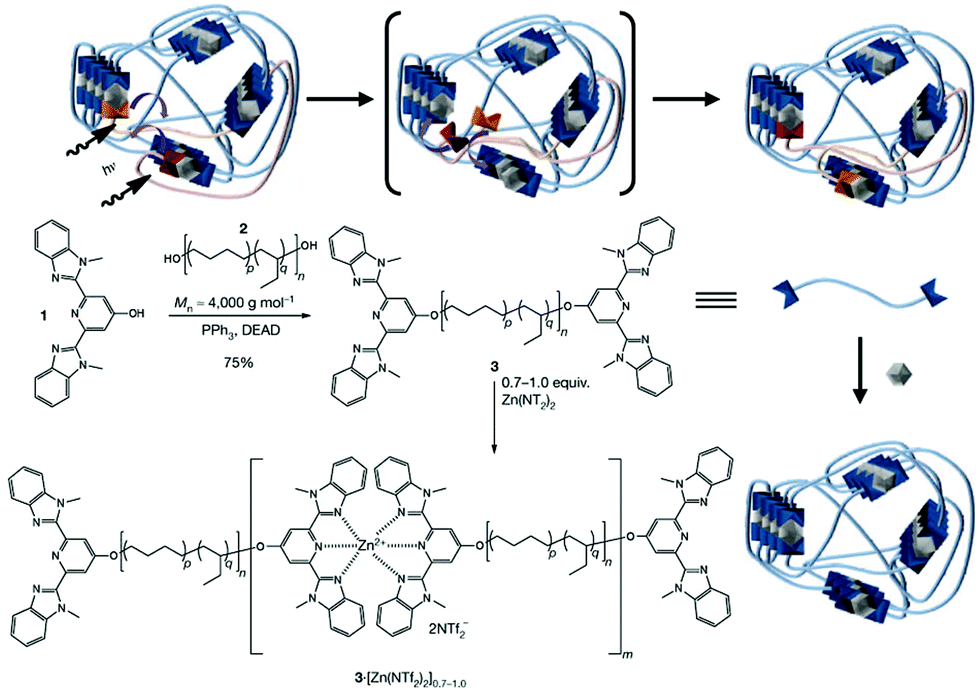 | ||
| Fig. 9 Synthesis of a metallopolymer network and its healing mechanism. Adapted with permission from ref. 75. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
It should be noted that UV light cannot penetrate deeply into tissues and may damage biological components, and has only a small fraction in sunlight, which can hamper the use of azo molecules in biomedical and solar-energy applications. Nevertheless, intrinsic visible and near infrared (NIR) light-responsive azo-based polymers have been recently developed, which can solve some problems.94
Dynamic exchange reactions between reversible covalent bonds under mild conditions constitute an alternative strategy to develop self-healing polymer networks triggered by light, heat or redox agents, which include disulfides, allyl sulfides and trithiocarbonates. Disulfide bonds usually undergo degenerative exchange reactions, in the presence of catalysts UV light or heat, in which they are disrupted and reformed via radical or ionic intermediates; they are also relevant in the biological field because of the cysteine aminoacids that are found in proteins and other biopolymers. For example, the Matyjaszewski group demonstrated the self-repairing of covalently crosslinked poly(n-butyl acrylate) networks, under UV irradiation and in an inert atmosphere, taking advantage of the dynamic reshuffling of trithiocarbonates, which can act as photoinitiators in radical polymerizations and react among themselves to re-crosslink the network.70 Thiuram disulfide (TDS) moieties have similar reaction exchange properties that can also be used to synthesize light-responsive self-healing polymers. Departing from previous work, the same group addressed the weaknesses of that approach by incorporating TDS units in the polymer backbone that could heal in air under visible light, without the need for solvent and under milder conditions (Fig. 10a).71 The exchangeable character of TDS, owing to the cleavage of disulfide that forms dithiocarbamate-based free radicals, is responsible for the dynamic nature of the matrix through light-triggered reorganization of the crosslinking units. The mechanical properties of the gels improved with light exposure time and recovered their original values within 24 hours (breaking stress >380 kPa and 175% elongation at break); in these systems it is important to allow enough time for the covalent bonds to reform during the network remodelling. Recently, Kloxin and co-workers incorporated a photoactive monomer comprising a dithiocarbamate group into a PU network, which could remend itself at the damaged site and simultaneously increase the network strength (i.e. photo-healing and photo-strengthening).72 It was shown that by introducing a labile crosslinker, followed by photostimulation with UV irradiation (365 nm), generated stable free radicals that initiated a secondary radical polymerization of ethylene glycol dimethacrylate (EGDMA) monomers impregnated into the network prior to the formation, which in turn, enhanced the moduli of the gels. After 2 hours of irradiation, the material could heal after damage and G′ could increase from 0.3 to 2 MPa, which could be controlled by the initial concentration of EGDMA. Additionally, this approach allowed for a relative spatial control of the mechanical properties as well as the potential shape change of the network at the damaged site.
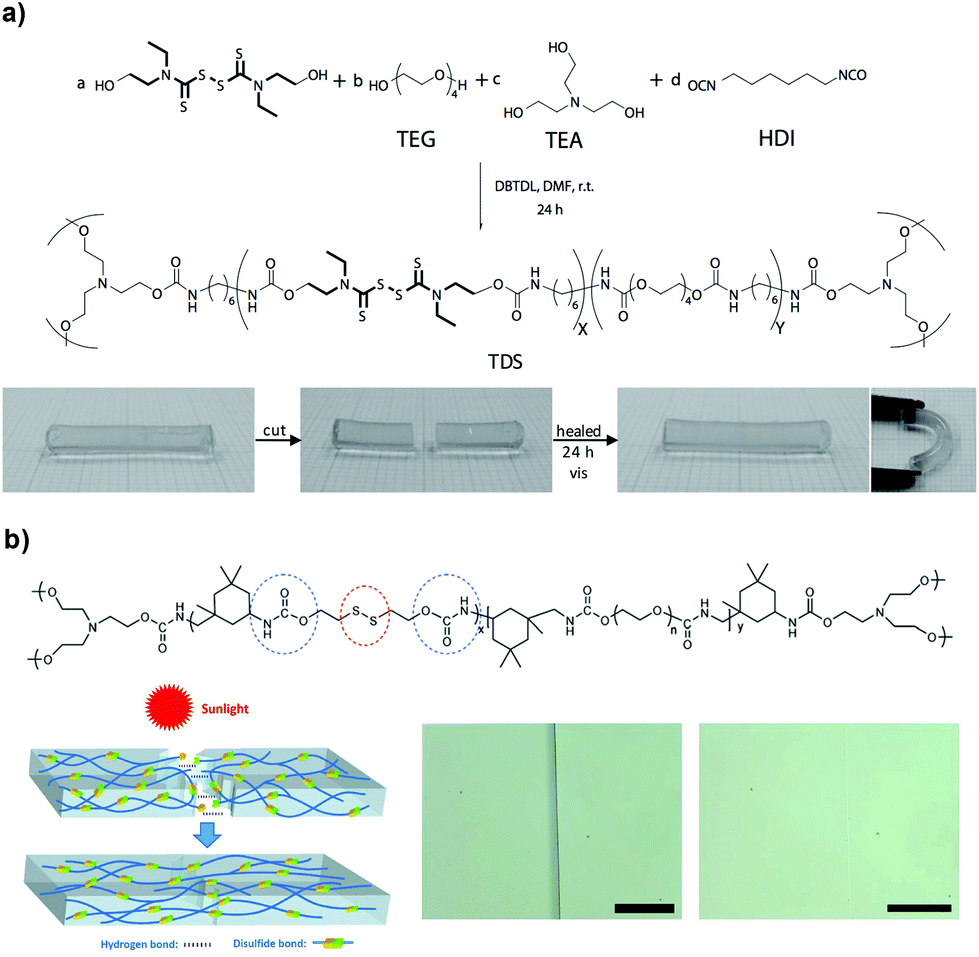 | ||
| Fig. 10 (a) Illustration of the self-healing reaction of TDS-based gels under visible light at room conditions. Adapted with permission from ref. 71. Copyright 2012, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (b) A PU-based gel exhibits self-healing behaviour when exposed to sunlight (intensity ≈ 90 mW cm−2). Adapted from ref. 98 with permission from the Royal Society of Chemistry. | ||
As mentioned above, it is pertinent to develop materials that can heal under mild conditions with visible light, as is the case with TDS and diselenide bonds-containing systems. In an effort to develop materials with healable properties under mild conditions with the use of visible light, Xu et al. studied the dynamic exchange character of diselenide bonding (Se–Se bond has a bond energy of 172 kJ mol−1, which is weaker in comparison with the S–S bond, 240 kJ mol−1)95 and demonstrated that the metathesis reaction can be induced with visible light but stops in the dark.96 In this sense, a series of diselenide-containing PU elastomers with different mechanical and healing properties were synthesized.97 After irradiating with visible light (table lamp, 2600 Lux), the materials could heal and virtually recover their initial Young's modulus within 48 hours. In addition, by using a directional blue laser (457 nm), the healing process could be faster and remotely triggered, and the integrity of the samples could be retained; after 30 minutes, the networks could recover almost 100% of their Young's modulus and the breaking stress could reach ≈80%.
An interesting approach was also reported by Zhang and co-workers who used sunlight to stimulate the self-healing of a PU network comprising disulfide bonds in the main chain (see Fig. 10b).98 By taking advantage of the photo-triggered reversible exchange reaction of disulfide bonds simply under illumination of the UV region of sunlight, and with the assistance of hydrogen bonding (which favoured initial and intimate contact across the interface), it was possible to restore the tensile strength of the damaged polymers up to 92% within 24 hours for at least 3 cycles.
Nanoparticles (NPs), nanorods and carbon nanotubes (CNTs), as well as clay and graphite nanosheets, are of particular interest in the design of polymer composites to not only enhance the mechanical or thermal properties but also to introduce photohealing attributes by acting as efficient photothermal antennae.6 Generally, this concept involves the use of remote sources of energy and its conversion in localized heat gradients at the damaged areas. Metallic nanoparticles, such as AuNPs, present surface plasmon resonance (SPR) at specific wavelengths (in the visible and NIR region), which introduces new opportunities for the use of tunable energy sources capable of localized efficient conversion of electromagnetic radiation into heat.76,99–101 Nanocomposites with light-triggered healing and shape memory properties combined were reported.102 Zhang and Zhao developed a chemically crosslinked PEO network loaded with AuNPs that displayed both optical healing and light-controlled shape memory, which could be activated individually in a sequential manner, based on the localized heating arising from the SPR absorption of AuNPs.103 The photothermal effect activated the rapid damage repair through crystal melting under laser exposure (532 nm, 3 s) followed by recrystallization upon cooling (that is, when the light was turned off), as shown in Fig. 11a. These polymer composites exhibited acceptable healing capabilities especially for thin and transparent specimens.
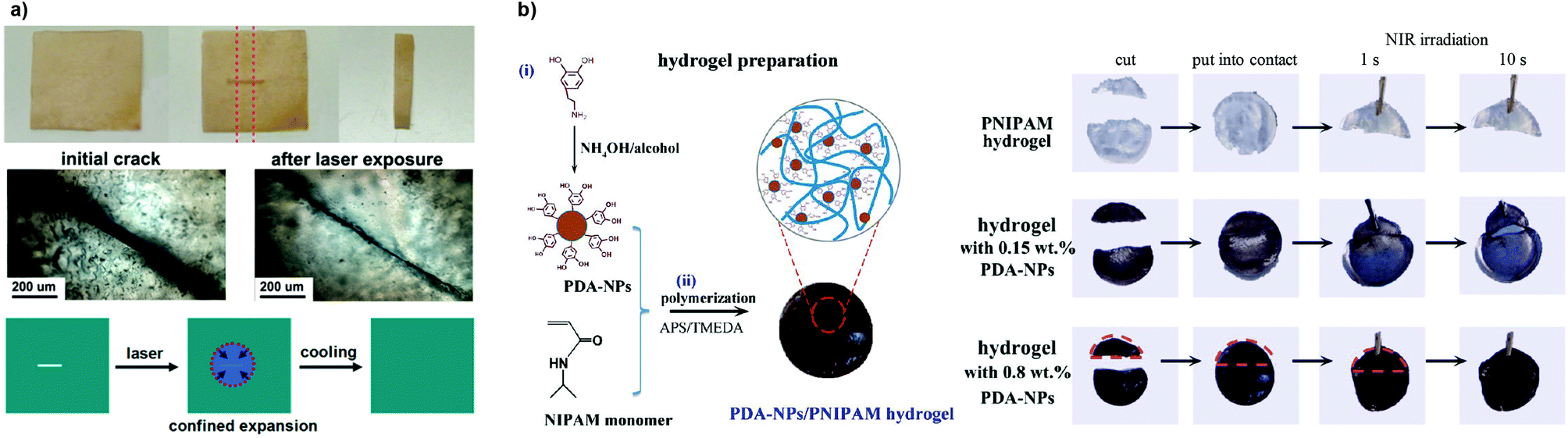 | ||
| Fig. 11 (a) Optical healing of a crosslinked PEO/AuNP polymeric film after exposing the crack to a laser (532 nm, 13 W cm−2) due to confined thermal expansion that helps bringing the surfaces together. Adapted with permission from ref. 103. Copyright, 2013 American Chemical Society. (b) PDA-NP/PNIPAM hydrogel preparation and its NIR-assisted healing for different conditions. Adapted with permission from ref. 105. Copyright, 2016 American Chemical Society. | ||
Alternative approaches have been recently reported using synthetically accessible polydopamine (PDA) NPs as photothermal agents.104 The incorporation of PDA-NPs into a PNIPAM-based network drove these gels NIR light-responsive with self-healing properties, volume change for pulsatile drug release and cell/tissue adhesiveness.105 If two halves of the gel (with 0.8 wt% of PDA-NPs) were brought in contact and irradiated for 10 s, they tended to heal and recover hydrogel tensile strength (Fig. 11b). In fact, the PDA-NPs also served as functional nanofillers and reinforced the network, increasing the elastic modulus. As shown by these works, PDA is an interesting photothermal filler as it shows relatively good biocompatibility, promotes cell adhesion in a wide range of substrates and proliferation by its inert and hydrophobic nature, and it is synthetically easy to prepare by dopamine monomer precursors.106,107
4. pH- and redox-responsive polymer networks
Chemoresponsive materials have attracted particular interest due to the easy control and integration of pH gradients in both laboratorial and industrial applications as well as in biological processes at the tissue/organ or cellular level. There are now thousands of reports on pH-responsive materials and more recently, their conceptualization in self-healable polymers has been reported by several groups, for example, polymer gels based on acylhydrazones as pH-switchable networks;108–110 Chen et al. reported on a dynamic covalent gel with acylhydrazone crosslinks synthesized by the condensation reaction of bis-acylhydrazine functionalized PEO with tris[(4-formylphenoxy) methyl]ethane (Fig. 12).111 The gels exhibited reversible gel–sol phase transition by adjusting the solution acidity (pH < 4) to hydrolyse the network within 1.5 hours, and regenerated their original structure in ≈20 s by restoring the pH. This process was repeated up to 8 consecutive cycles, where it was observed that the time of decomposing and reforming of the network became longer, and the mechanical properties deteriorated since G′ decreased from 70 to 30 kPa. Nevertheless, the gel possessed an autonomous self-healing behaviour once two pieces could merge into a single one (without any external intervention) by keeping them in contact for 7 hours. In another study, acylhydrazone bonds were integrated with the DA reaction for the design of double crosslinked hyaluronic acid (HA) based hydrogels.112 The DA crosslinking enhanced the structural integrity and mechanical strength of the network, whereas the acylhydrazone contributed with the pH-responsive behaviour and self-healing properties. At the same time, the aldehyde groups introduced could also react with local cartilage tissue amines (through Schiff base reaction), promoting good integration of the hydrogel with the surrounding tissue. Similarly, the gels could reversibly switch between gel and sol states within 20 minutes by adjusting the pH around 4, and nearly recovered their initial properties (G′ around 18 kPa and 55% elongation at break). When compared with control DA crosslinked hydrogels, these materials showed improved adhesive properties (10 kPa vs. 1.5 kPa adhesive stress).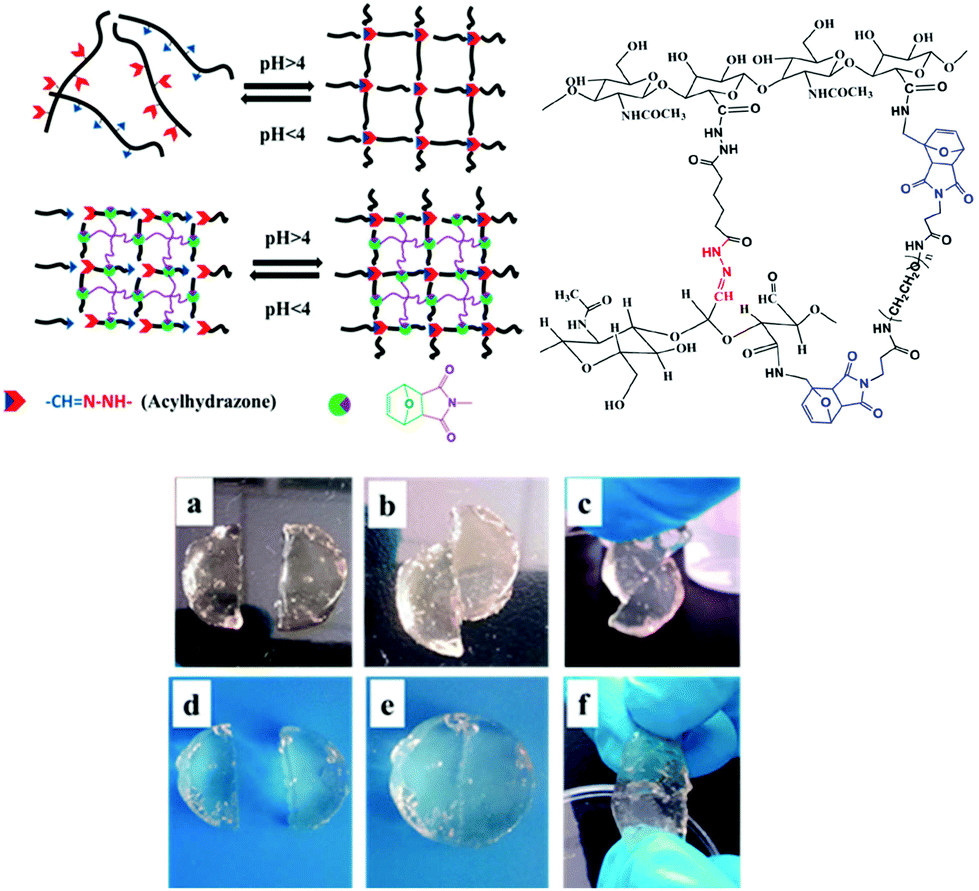 | ||
| Fig. 12 Illustration of pH-responsive HA hydrogels formed by the combination of the DA reaction and acylhydrazone bonds. The gel can heal within 3 h even without any external intervention (d–f), which contrasts with a control DA-only crosslinked network (a–c). Adapted with permission from ref. 111. Copyright, 2015 American Chemical Society. | ||
Wei et al. developed a simple method to prepare self-healing hydrogels by mixing chitosan and dibenzaldehyde-terminated PEG (within 1 minute at ambient temperature).113 The rapid formation of imine bonds (or Schiff base linkages) between aldehyde groups and the amino groups of the chitosan generated dynamic gels sensitive to pH and other biological stimuli (e.g. amino acids and vitamin B6 derivatives), which could recover their original mechanical properties after 2 hours. Likewise, other biomacromolecules containing amines can be used to fabricate analogous networks for biomedical applications.114,115 Another example was recently explored by Faust and co-workers, which combined polyisobutylene (PIB) dialdehyde functionalized polymers and side-chain amino acid-containing polymers to form a pH-sensitive dynamic covalent crosslinked network.116,117 These gels displayed poor swelling ability but good mechanical properties (G′ ≈ 17 kPa) and thixotropic behaviour, although a substantial decrease of G′ was observed with the decreasing of the [NH2]/[CHO] ratio (G′ ≈ 2 kPa), or the increasing number of regeneration cycles (G′ ≈ 1 kPa after 6 cycles).
Huang and co-workers described responsive gels on the basis of (host–guest) crown ether recognition through the synthesis of a dibenzo[24]crown-8 (DB24C8) terminated four-arm star poly(ε-caprolactone) (PCL) and dibenzylammonium salt (DBAS) terminated two-arm PCL polymer.118 The mixture of the polymers at relatively high concentrations, in chloroform, produced supramolecular gels as a result of pseudorotaxane formation between the terminal DB24C8 and DBAS moieties.119 By varying the pH or temperature, the resultant gels demonstrated reversible gel–sol transition; the addition of a base resulted in the disruption of the polymer network driven by the deprotonation of DBAS, which could be reverted with the addition of acid. If a long and flexible alkyl linker was used between the host and guest moieties, it prevented the formation of [c2]daisy chains and acted as a physical junction in the network.120 The same group further developed this system using poly(methyl methacrylate) (PMMA) with pendant DB24C8 groups and two bisammonium crosslinkers with different end-group sizes,121 owing to the host–guest interaction of DB24C8 and DBAS moieties. These soft gels could act as degradable materials triggered by the pH stimulus (addition of triethylamine/trifluoroacetic acid), and showed fast healing capacity (<30 s) over several cycles driven by the reversible host–guest interaction, or the reformation of electrostatic and hydrogen-bonding interactions between bisammonium salt and DB24C8 units of the polymer. This concept was then vastly explored by other researchers during the past few years. Gu et al. presented a pH-sensitive soft gel composed of poly(methyl vinyl ether-alt-maleic acid) grafted with β-CD or Ad.122 The complexation between host β-CD and guest Ad units conferred a reversible nature to the gel, which could recover after damage without any external stimulus, although it possessed a pH-dependent (weak) mechanical strength (G′ ranging from 170 to 500 Pa). Recent studies from the Burdick group demonstrated the in vivo applications of HA hydrogels based on β-CD/Ad interactions as injectable therapeutics.123
Varghese and co-workers proposed an interesting and simple self-healing hydrogel made from acryloyl-6-aminocaproic acid (A6ACA) precursors containing terminal carboxyl groups and amide groups as dangling side chains.124,125 Photo-crosslinked A6ACA hydrogels, Fig. 13, through hydrogen bonding could fast heal in a few seconds under acidic conditions (pH < 3) and reversibly disassembled when exposed to basic pH for at least 12 cycles with minimum hysteresis. A detailed analysis of the pH-mediated self-healing mechanism showed that side chains with terminal carboxyl groups are mostly protonated at low pH and mediated the formation of hydrogen bonds with other carboxyl or amide groups across the interface; above pH 9, these carboxyl groups are deprotonated and induce electrostatic repulsion, preventing the formation of hydrogen bonds. The healed materials were strong enough to sustain large deformations, but their healing efficiency was inversely proportional to the crosslinker content and depended non-monotonically on the side-chain length. To achieve an efficient healing, these chains should be long enough and flexible to make the functional groups accessible across the interface to create H-bridges, and sufficiently short to minimize steric hindrance of the interacting groups and to prevent hydrophobic collapsing. This system inspired Jiang et al. who developed a zwitterionic hydrogel physically crosslinked via electrostatic interactions by changing the side chains of PA6ACA to poly(carboxybetaine acrylamide), which could undergo self-healing at pH 7 without compromising cell viability.126 More recently, hydrogels based on cytosine- and guanosine-modified HA were prepared using 1,6-hexamethylenediamine as a crosslinker under physiological conditions.127 The assembly was established based on the principle of nucleobase complementarity pairing via hydrogen bonding. These biodegradable gels (4 < G′ < 99 kPa after 20 minutes) exhibited a pH-triggered gel-to-sol transition below 6 and above 8, and revealed good healing capacity after 24 hours.
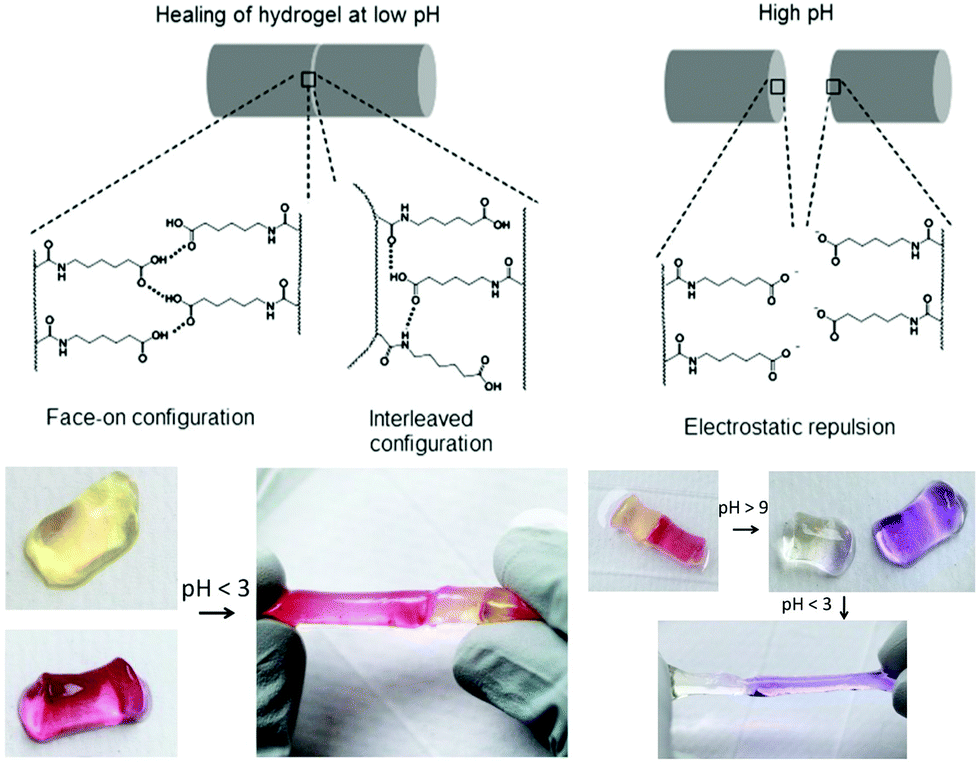 | ||
| Fig. 13 Self-healing mechanism of A6ACA hydrogels. At low pH the polymer network forms H-bonds and heals the damage, while at high pH the carboxyl groups in the polymer side chains become deprotonated, leading to strong electrostatic repulsion and preventing healing. Adapted from ref. 125. Copyright (2012) National Academy of Sciences. | ||
Metal–ligand coordination is thermodynamic stable, and depending on the metal ion and ligand used, can also be kinetically labile. The integration of different metal ions into polymer networks can impart the functional properties of these metals to the supramolecular materials prepared via this dynamic bonding, which may include light-emitting, conducting, mechano-responsiveness, etc.128 The pH variation experienced by mussel adhesive proteins during byssus secretion have inspired researchers to design pH-sensitive supramolecular hydrogels based on similar pH-mediated metal–ligand coordination. Natural mussel adhesion is mediated by byssus threads with a high concentration of 3,4-dihydroxyphenylalanine (DOPA), where catechol groups are known to form strong but reversible interactions with Fe3+ ions.129 Hydrogels were formulated from DOPA-modified PEG or polyallylamine networks with catechol-Fe3+ complexes as crosslinking points.130,131 The pH tunable behaviour of the system offered an easy control over its viscoelastic properties and healing behaviour due to its ability to switch from mono- to bis- and tris-catecholate complexes that form at different pH (from ≈5 to 9), without Fe3+ precipitation. The resultant gels could recover stiffness and cohesiveness within a few minutes after failure through the restoration of broken catechol-Fe3+ crosslinks.
The design of redox-responsive systems comprises the incorporation of species in the polymer matrix either as fillers or as monomeric synthons attached on the polymer backbone, that undergo oxidation–reduction reactions (i.e. gain or loss of electrons) triggered by an electrical potential or chemical agent, which modulate the material properties. Typically, these networks include Fc, conductive polymers, transition metal ions or redox-sensitive bonds (e.g. disulfides).132–135 Due to the reversibility and easy control of the redox process, these polymers and gels find useful applications as biosensors, batteries and biofuel cells, microactuators and drug delivery systems.
The versatility of supramolecular materials with redox-responsive self-healing properties based on host–guest interactions has attracted the interest of researchers. Harada et al. reported an elegant system using poly(acrylic acid) (PAA) modified with β-CD (as host polymer) and with Fc pendant groups (as guest polymer).136 By exploring the high affinity of β-CDs for Fc moieties in the reduced state, and their low affinity in their oxidized state, the authors fabricated a hydrogel at basic pH, which upon addition of redox reagents (sodium hypochlorite/glutathione) could induce a reversible gel–sol transition. Autonomous healing performance was tested for the soft gel (G′ ≈ 175 Pa) by mechanical rejoining of two cut pieces to form one after 24 hours at room temperature. Interestingly, when the interface of a gel was coated with sodium hypochlorite oxidant solution, healing was prevented, but by spreading a glutathione reducing agent, the cut surfaces re-adhered after 24 hours. The same group further described a similar but more complex approach based on the formation of a polymer network bearing two kinds of host–guest inclusion complexes – β-CD-Ad and β-CD-Fc – with different functional properties.137 Copolymers prepared with AAm or NIPAM as the main chain could form a supramolecular hydrogel through host–guest interactions, which exhibited self-healing, expansion–contraction and shape memory properties (Fig. 14). Self-healing of two separated pieces of a gel was observed with and without the presence of an oxidizing agent, and could be perceived within 2 or 3 hours, respectively, in wet conditions, although its recovery time increased to ca. 72 hours to achieve an adhesive strength ratio >68%. The redox responsiveness to ammonium cerium nitrate (CAN) (oxidizing agent) enabled the expansion and contraction of the gel because the Fc moiety in its oxidized form (i.e. hydrophilic) cannot be included in the cavity of β-CD that decreases the crosslinking density, whereas after reduction, the inclusion complex reformed and could release the water from the network. Analogously, Yuan et al. synthesized poly(N,N-dimethylacrylamide-r-glycidol methacrylate) and poly(N,N-dimethylacrylamide-r-2-hydroxyethyl-methacrylate) copolymers decorated with β-CD and Fc, respectively, as pendant groups, which can then assemble to form electrochemical- and redox-responsive hydrogels. These materials, whose reversible phase transition is perceptible after adding FeCl3/ascorbic acid solutions, or applying a potential of +0.5 V and −0.1 V (with saturated calomel electrode as reference), showed good healing behaviour and biocompatibility.138,139 Another example based on the incorporation of Fc groups in the polymer main chain was reported by Abe et al.:140 A redox-switchable network of entangled wormlike micelles could be formed by the addition of sodium salicylate to a ferrocenyl surfactant (11-ferrocenylundecyl)trimethylammonium bromide (FTMA). The oxidation of Fc groups triggered a phase transition of FTMA from the hydrophobic to the hydrophilic state with great influence in the aggregation degree. By simply tuning the applied electrical potential, it was possible to control the viscoelasticity of this electrorheological fluid due a change in the aggregation state of the micelles, which exhibited a zero shear viscosity around 15 Pa.
 | ||
| Fig. 14 (a) Redox-responsive supramolecular hydrogel exhibiting self-healing, expansion–contraction and shape memory properties. (b) Chemical structure of the host–guest inclusion complex used to fabricate (c) the polymer networks. (d) When two halves of a βCD-Ad-Fc PNIPAM gel were held together with or without CAN under wet conditions, the pieces adhere to (e) restore the original gel. No healing was observed if a competitive guest molecule was applied to one of the cut surfaces. Adapted with permission from ref. 137. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Indirectly related, but also relevant, organometallic polymers, such as poly(ferrocenylsilanes) (PFS), which contain alternating Fc and silane groups as repeating units, have also been used as redox-responsive polymeric networks.141 Vancso and co-workers reported PFS-based hydrogels by covalent crosslinking with PEG using a thiol-Michael addition click reaction.142 The reversible redox state of the PFS chains could be controlled, allowing for the tuning of the swelling/deswelling and mechanical properties of the gels.
Disulfide bonds as structural linkages are employed as well to form self-healing networks in response to relevant redox conditions.143 Matyjaszewski et al. presented the synthesis of poly(n-butyl acrylate) multiarm star polymers, by atom transfer radical polymerization (ATRP), containing thiol groups at the periphery.144 Self-healing organogel films crosslinked through disulfide bonds were formed by depositing these polymers on silicon wafers in the presence of an oxidizing agent. The atomic force microscopy (AFM) tests revealed the rapid healing behaviour of the films attributed to the regeneration of S–S bonds via a thiol–disulfide redox reversible exchange reaction; hence, the healing efficiency and time were dependent on the film thickness and width of the damage, which decreased from 2 hours to 20 minutes when the cut width was reduced from 5 to 2 μm, with a Young's modulus of 40 kPa (film thickness: 1.5 μm).
5. Magnetic-responsive polymer networks
The development of magnetic-responsive hydrogels generally involves the incorporation of ferromagnetic or superparamagnetic particles in a matrix, which trigger the self-healing process145 or shape memory effect146,147via electromagnetic induction. The heat generated during this process is driven by the oscillation of the NP magnetic moment (Néel relaxation) which is responsible for the temperature rise in situ, and facilitates increased polymer chain diffusivity to repair the damage.148,149 These systems find practical applications in turbines or automobile parts, batteries and electronics, as well as biomedical devices.Urban and co-workers described the self-repairing of a thermoplastic polymer network (p-methyl methacrylate/n-butyl acrylate/heptadecafluorodecyl methacrylate) containing superparamagnetic iron(III) oxide (γ-Fe2O3) NPs using a remotely oscillating magnetic field (Fig. 15a–c).150 Physically separated polymeric films could be repaired into a single piece, after 2 hours, for several cycles due to the oscillation of uniformly dispersed NPs (≈12 nm diameter, f = 300 kHz) at the frequency of the field, which resulted in a localized heat and subsequent interfacial diffusion to the damaged area, thus restoring the mechanical properties and dimensional stability of the network.
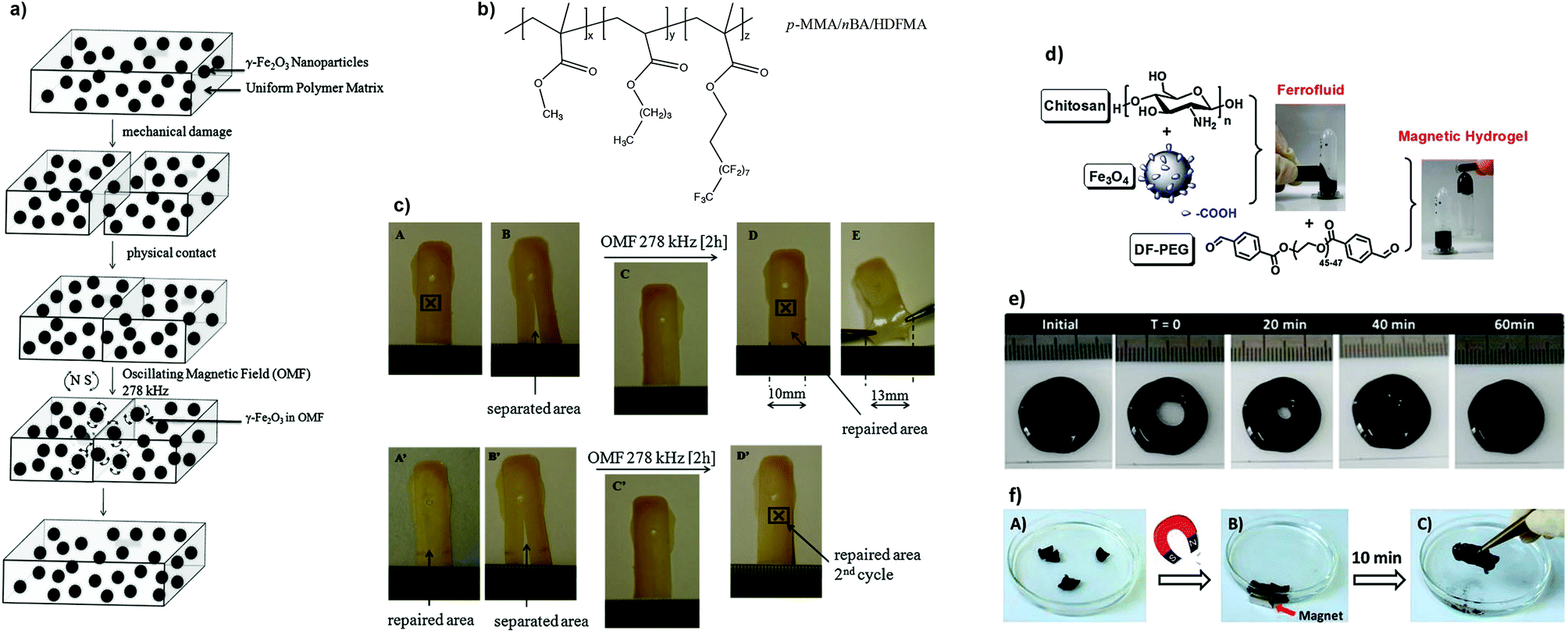 | ||
| Fig. 15 (a) Proposed mechanism of the repair process of the composite and (b) chemical structure of the polymer. (c) Photographs demonstrating the healing process of a p-MMA/nBA/HDFMA film containing 14% w/w of γ-Fe2O3 NPs in presence of an oscillating magnetic field. Adapted with permission from ref. 150. Copyright 2009, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (d) Preparation of chitosan-Fe3O4 ferrofluid and magnetic hydrogel, whose self-healing process can occur (e) without the application of a magnetic stimulus within 60 min, or (f) with an applied magnetic external field in ≈10 min. Adapted from ref. 152 with permission from the Royal Society of Chemistry. | ||
An alternative approach was proposed by Bowman et al. based on a healable retro-DA crosslinked network (1,1′-(methylene-di-4,1-phenylene)bismaleimide/pentaerythritol propoxylate tris(3-(furfurylthiol)propionate)) doped with chromium(IV) oxide particles.151 Hysteresis heating, driven by the alternating magnetic field, allowed the ferromagnetic polymer network to flow and heal fast for 10 cycles without significant loss of its mechanical properties; the self-limiting nature of the hysteresis heating controlled by the relatively low Curie temperature (TC) of the NPs (around 113 °C) protected the system from irreversible degradation.
Later, Wei et al. developed a simple method to prepare a magnetic rapidly self-healing gel (within 2 minutes) by embedding carboxy-modified magnetite (Fe3O4) NPs into a chitosan-PEG matrix.152 The composite (based on Schiff-base crosslinking between the pendant amine groups of chitosan and the benzaldehyde groups of PEG terminals) exhibited low cytotoxicity and could heal after 10 minutes when cut in different pieces and re-joined by the application of a magnetic stimulus, or by itself within 60 minutes without magnetic stimulation (Fig. 15d–f). This ferrofluid (G′ ≈ 400 Pa) could also be remotely operated by a magnet to pass through a narrow channel with an obstacle in the middle, showing the cooperation of magnetic and self-healing properties.
An interesting method was recently described by Chen and co-workers to synthesize a β-CD/N-vinylimidazole (host–guest) supramolecular hydrogel with embedded Fe3O4 particles through a magnetically induced frontal polymerization (MIFP), as shown in Fig. 16.153,154 This system took advantage of the magnetocaloric effect (in which an alternating magnetic field causes a thermal response in the material but without transfer of heat to its surroundings)155 to initiate the polymerization and create rapidly (<5 minutes) a self-healing gel with enhanced mechanical strength. The resulting materials could be repaired autonomously when damaged within 24 hours (up to 95% of healing efficiency), exhibited high tensile stress intensity (>50 kPa) and an elongation percentage at a break point of over 683%. In addition, when an external magnetic stimulus was applied, the healing time significantly decreased to 3 hours, without compromising the healing efficiency, owing to an amorphous flow of the gels in the joint region, as a result of the synergy between host–guest healing and the magnetocaloric effect.
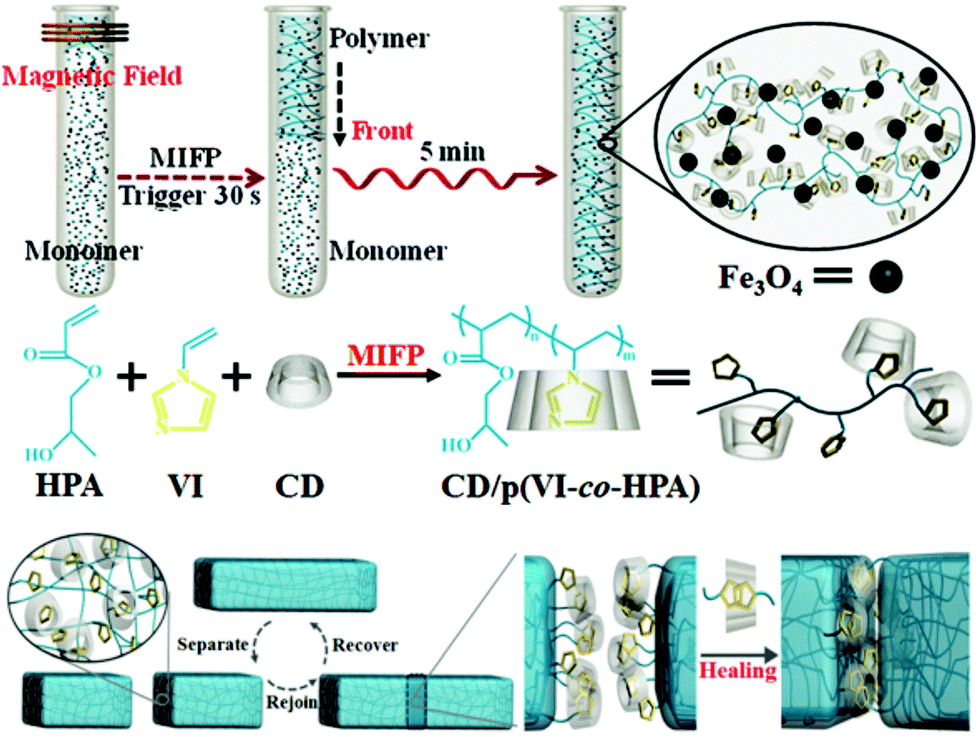 | ||
| Fig. 16 Schematics of the preparation of gels via MIFP and the self-healing host–guest concept. Reproduced with permission from ref. 153. Copyright 2013, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Nevertheless, the aforementioned examples rely mostly on systems that often require an external trigger to initiate the healing process, which can also be affected by ambient conditions. Alternative novel approaches have been explored, such as graphitic inks impregnated with magnetic Nd2Fe14B microparticles, which impart self-healing ability to printed films for electronic applications.156 The conductive nature and permanent magnetic properties of the particles allowed the films to recover rapidly (≈50 ms) and autonomously the mechanical and electrical contacts, even with a damage width up to 3 mm, for a large number of damage–heal cycles.
Yarn-based supercapacitors have attracted attention as energy-storage devices for healable wearable electronics.157 An elegant example from this front was reported by Zhi et al. where they fabricated yarn-based supercapacitors by wrapping magnetic electrodes around a healable carboxylated PU shell.158 Upon mechanical damage, the polymeric shell and the yarn electrodes (composed of multiple minuscule fibres) were brought in contact, and their configuration and their mechanical and electrochemical performances were restored, assisted by the magnetic field. They demonstrated that the yarn supercapacitor could recover up to 71% of its initial specific capacitance after four healing cycles.
It is therefore anticipated that novel composite healable polymer matrices will emerge in the near future with even more intricate magnetic fillers for tailor-made applications in the wearables field as well as in bioelectronics and microdevice fabrication.
6. Multi-responsive polymer networks
Although “mono”-responsive polymer networks have been extensively reported, reports on “dual” or “multi”-responsive healable networks by a single system are currently expanding.Rowan and co-workers took advantage of metal–ligand coordination to prepare diverse multi-responsive metallo-supramolecular hydrogels.159,160 These materials offer the possibility of designing multi-responsive self-repairing hybrid systems, whose properties can be tuned by changing the combination of polymers and metal ions used.161–163 For example, CNTs conjugated with terpyridine ligand-terminated PU and Zn2+ ions were used to design metallo-supramolecular nanocomposites that could self-heal upon exposure to NIR light, heat or solvent, showing high healing efficiency (>93%) and relatively short healing times (ranging from 30 minutes to 3 hours depending on the method used).164
Departing from previous works, Huang et al. reported quadruple stimuli-responsive self-healing polymer gels constructed by the complementary orthogonal reaction of heteroditopic monomers and addition of metal crosslinker PdCl2(PhCN)2, driven by metal–ligand interactions.165,166 Initially, a linear polymer was synthesized based on the host–guest interaction of a benzo-21-crown-7 (B21C7) unit and the dialkylammonium salt moiety of the monomer. The polymer gel was finally prepared by a coordination reaction between palladium(II) and the 1,2,3-triazole group in the backbone that acted as a ligand (Fig. 17). The transparent gels exhibited a reversible gel–sol type of transition in response to multiple distinct stimuli (pH-, thermo-, cation- and metallo-induced) with good mechanical properties, which G′ increased proportionally with the crosslinking density (from 10 Pa at 20% to 10 kPa at 100%). Particularly interesting, cation- and metallo-induced phase transitions could occur by adding K+ to disassemble the network, due to its affinity to the B21C7 unit, or with PPh3, which competes with Pd. The refinement of this system led to the development of a gel with faster self-healing behaviour (around 3 minutes) after rupture.166 These studies recently inspired other groups to employ crown ether-based recognition and metal–ligand coordination to assemble multiple-responsive self-repairing polymer gels.167
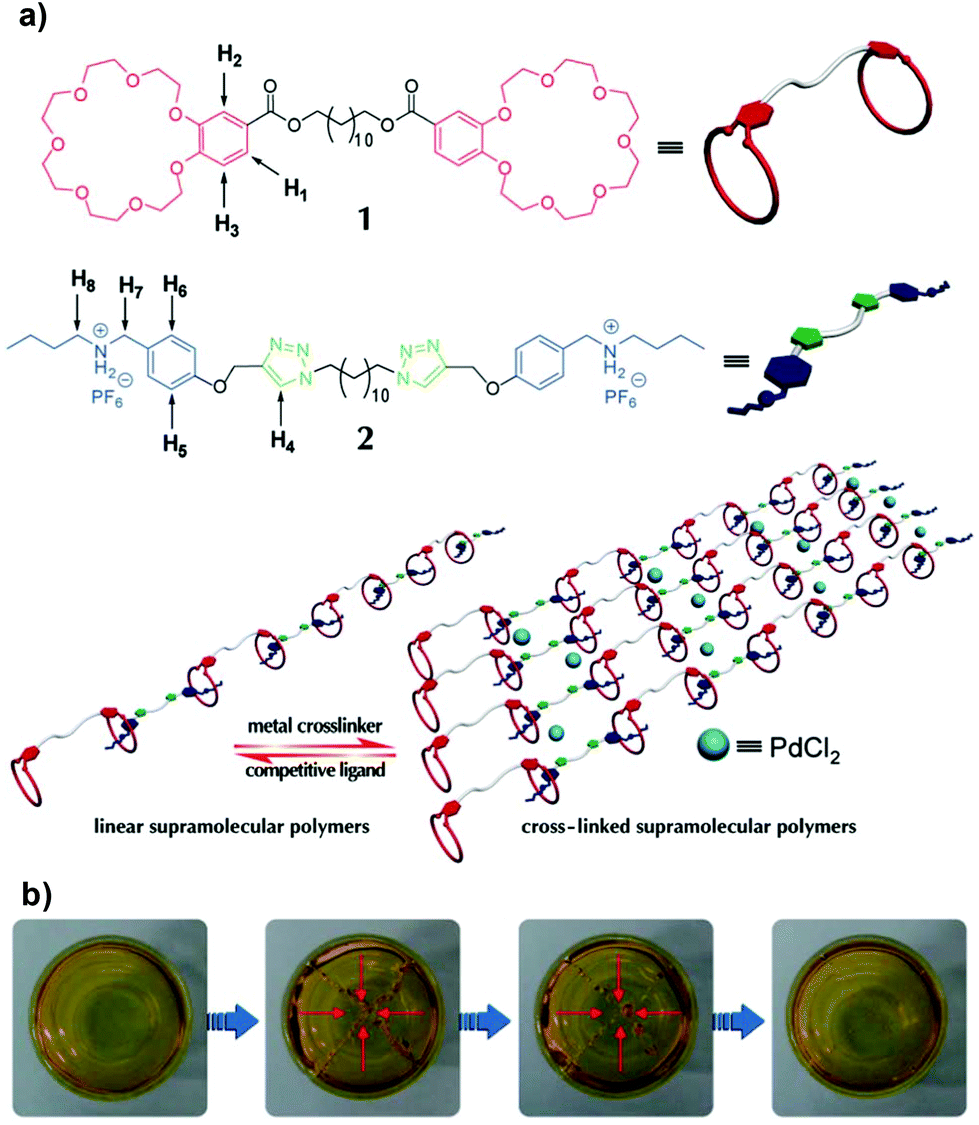 | ||
| Fig. 17 (a) Illustration of the formation of metallopolymer networks. (b) Photographs of a transparent supramolecular gel after damage and subsequent healing after 3 min. Adapted from ref. 166 with permission from the Royal Society of Chemistry. | ||
Previously, we described examples of β-CD/azo complexes to fabricate healable supramolecular gels. Wu et al. presented a variation of this approach based on the host–guest interaction between a CD, PEG and POSS copolymer, and an azo disulfide dimer.168 The hydrogels could respond reversibly to temperature, light and redox stimuli, and presented enhanced mechanical strength, due to the incorporation of POSS units, without an impact on their biocompatibility.
Dual responsive hydrogels with pH- and redox-triggered actuation could be prepared by combining acylhydrazone and disulfide bonds in the same system, which undergo several reversible gel–sol transition cycles.110,169 These bonds had proved to be compatible when combined in a single system and are independently operational upon pH changes.170 Hence, the opaque gels could heal themselves within 48 hours in both acidic and basic environments by selecting the appropriate repairing route: either they undergo disulfide exchange mediated self-healing under basic conditions or by acylhydrazone exchange in acidic and neutral conditions (the addition of aniline could accelerate the catalytic reaction and facilitate the healing process at neutral pH). It was demonstrated that the gelation time and the mechanical properties were dependent on the pH and the gelator concentration used (after 5 minutes, G′ around 4 kPa at pH 6; 10 wt%). The resulting materials presented acceptable healing efficiency after 48 hours, reaching >50% of the initial strength and elongation values at break. Following the aforementioned studies,113,152 Wei and co-workers synthesized pH- and temperature-responsive hydrogels by mixing a dibenzaldehyde end-functionalized poly(NIPAM-co-AA) copolymer with glycol chitosan at room temperature for approximately 20 minutes.171 The material possessed a G′max about 1000 Pa, and exhibited injectable and self-healing properties although its efficiency decreased after the second cycle. When heated above 37 °C and/or under acidic conditions, the gels were able to release the molecular dye rhodamine B and the anticancer drug cisplatin in a controlled manner. Besides, the gels were nontoxic and could be used to encapsulate fibroblasts during 24 hours, although further studies need to be performed to fully elucidate their ability as cell carriers.
Finally, a particularly interesting type of a dynamic covalent bond is the formation of reversible boronate ester bonds (in a pH dependent manner) between boronic acids and cis-diol-containing moieties, as is found in polyols, catechols, carbohydrates or glycoproteins. Electrochemical and optical sensors, pH-responsive hydrogels, insulin delivery systems, cell culture and capture, among others, are typical applications of these materials.172,173 In the context of self-healing materials, several works have emerged recently in the form of multi-responsive gels/polymer blends with numerous potential biomedical uses.174–183 These polymer networks are interesting in that they can undergo a gel–sol type of transition by pH fluctuations or the addition of cis-diol residues to trigger boronate ester de-complexation at the damaged site.
7. Conclusions and outlook
Self-healing polymer solids and gels are currently in a primitive phase and have not yet found many practical applications for various reasons that, as discussed in this article, researchers only very recently started to address. These are: (1) the incorporation of healable properties often compromise the overall mechanical properties (as shown in several examples developed by existing PU networks), (2) the chemistries employed are often (cyto-)toxic which prevent the use in the biomaterials field or in simpler applications that require the interaction with human skin or other organs (for example the furan synthons in DA networks fall in this category), (3) the repeatability of the healing capacity deteriorates with time, (4) healing times are very long, and (5) the chemistries involved: (i) do not survive/work in complex environments, i.e., in biological milieu or even in open air for prolonged times, which as a result can limit the potential applications of these materials as well as restrict their use as accessible building blocks with reasonable shelf life; and (ii) are not always compatible with other functional device components, for example, in the case of healable microelectronic devices/membranes, bottom-up fabrication approaches can be cumbersome.Several groups worldwide have dedicated some initial efforts to address these challenges, using double networks, nanocomposites, tough and stretchable gels, or by combining intricate manufacturing methodologies for cost-effective device fabrication.
It is still a challenge to develop intrinsically self-healing materials in a cost-effective way without changing their overall performance. Therefore, we anticipate that the constant refinement of the chemistries involved and the development of pragmatic solutions to the above-mentioned challenges may drive the field towards practical applications in the longer term. Conversely, in the near future it is more reasonable to expect that these materials will find applications as self-repairing materials of minimum damage (i.e. in the micro or milli-scale in the form of scratch/cut repairing) or in the form of transiently repairable materials for shorter-lived applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge UCL (UCL Excellence Fellowship), the Leverhulme Trust (ECF-2013-472) and the EPSRC (EP/M014649/1) for financial support.Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- P. Zhang and G. Li, Prog. Polym. Sci., 2016, 57, 32–63 CrossRef CAS.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- D. G. Shchukin, Polym. Chem., 2013, 4, 4871–4877 RSC.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Composites, Part B, 2016, 87, 92–119 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179–211 CrossRef CAS.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Acc. Chem. Res., 2008, 41, 1133–1142 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 CrossRef CAS PubMed.
- H. Eyring, J. Chem. Phys., 1936, 4, 283–291 CrossRef CAS.
- Y.-L. Liu and T.-W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- C.-H. Huang and Y.-L. Liu, RSC Adv., 2017, 7, 38360–38366 RSC.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802–1807 CrossRef CAS.
- E. B. Murphy, E. Bolanos, C. Schaffner-Hamann, F. Wudl, S. R. Nutt and M. L. Auad, Macromolecules, 2008, 41, 5203–5209 CrossRef CAS.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- G. B. Lyon, A. Baranek and C. N. Bowman, Adv. Funct. Mater., 2016, 26, 1477–1485 CrossRef CAS.
- N. Yoshie, S. Saito and N. Oya, Polymer, 2011, 52, 6074–6079 CrossRef CAS.
- Y.-L. Liu and C.-Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 905–913 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- P. J. Boul, P. Reutenauer and J.-M. Lehn, Org. Lett., 2005, 7, 15–18 CrossRef CAS PubMed.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J. M. Lehn, Chem. – Eur. J., 2009, 15, 1893–1900 CrossRef CAS PubMed.
- T.-W. Chuo and Y.-L. Liu, Compos. Sci. Technol., 2015, 118, 236–243 CrossRef CAS.
- Z. Xu, Y. Zhao, X. Wang and T. Lin, Chem. Commun., 2013, 49, 6755–6757 RSC.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS.
- H.-K. Lin and Y.-L. Liu, Macromol. Rapid Commun., 2017, 38, 1700051 CrossRef PubMed.
- N. Roy, E. Buhler and J.-M. Lehn, Polym. Int., 2014, 63, 1400–1405 CrossRef CAS.
- W. G. Skene and J.-M. P. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270–8275 CrossRef CAS PubMed.
- N. Kuhl, S. Bode, R. K. Bose, J. Vitz, A. Seifert, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 3295–3301 CrossRef CAS.
- R. Chang, X. Wang, X. Li, H. An and J. Qin, ACS Appl. Mater. Interfaces, 2016, 8, 25544–25551 CAS.
- Q. Wei, J. Wang, X. Shen, X. A. Zhang, J. Z. Sun, A. Qin and B. Z. Tang, Sci. Rep., 2013, 3, 1093–1098 CrossRef.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218–3227 Search PubMed.
- F. Wang, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2012, 22, 13076–13084 RSC.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- R. Vaiyapuri, B. W. Greenland, H. M. Colquhoun, J. M. Elliott and W. Hayes, Polym. Chem., 2013, 4, 4902–4909 RSC.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- Q. Wu, J. Wei, B. Xu, X. Liu, H. Wang, W. Wang, Q. Wang and W. Liu, Sci. Rep., 2017, 7, 41566–41577 CrossRef PubMed.
- J. Cui and A. D. Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- Y. Shao, H. Jia, T. Cao and D. Liu, Acc. Chem. Res., 2017, 50, 659–668 CrossRef CAS PubMed.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042–1048 CrossRef CAS PubMed.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS PubMed.
- Y.-G. Jia and X. X. Zhu, Chem. Mater., 2015, 27, 387–393 CrossRef CAS.
- T. Miao, S. L. Fenn, P. N. Charron and R. A. Oldinski, Biomacromolecules, 2015, 16, 3740–3750 CrossRef CAS PubMed.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- J. R. McKee, E. A. Appel, J. Seitsonen, E. Kontturi, O. A. Scherman and O. Ikkala, Adv. Funct. Mater., 2014, 24, 2706–2713 CrossRef CAS.
- F. Pitsch, F. F. Krull, F. Agel, P. Schulz, P. Wasserscheid, T. Melin and M. Wessling, Adv. Mater., 2012, 24, 4306–4310 CrossRef CAS PubMed.
- H. Wang, B. Zhu, W. Jiang, Y. Yang, W. R. Leow, H. Wang and X. Chen, Adv. Mater., 2014, 26, 3638–3643 CrossRef CAS PubMed.
- T.-P. Huynh and H. Haick, Adv. Mater., 2016, 28, 138–143 CrossRef CAS PubMed.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- R. Xu, I. Belharouak, J. C. M. Li, X. Zhang, I. Bloom and J. Bareño, Adv. Energy Mater., 2013, 3, 833–838 CrossRef CAS.
- Z. Wei, J. H. Yang, Z. Q. Liu, F. Xu, J. X. Zhou, M. Zrínyi, Y. Osada and Y. M. Chen, Adv. Funct. Mater., 2015, 25, 1352–1359 CrossRef CAS.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- C.-M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef CAS PubMed.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- M. B. Gordon, J. M. French, N. J. Wagner and C. J. Kloxin, Adv. Mater., 2015, 27, 8007–8010 CrossRef CAS PubMed.
- H. Chen, X. Ma, S. Wu and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 14149–14152 CrossRef CAS PubMed.
- X. Liao, G. Chen, X. Liu, W. Chen, F. Chen and M. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4409–4413 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- H. Zhang, D. Fortin, H. Xia and Y. Zhao, Macromol. Rapid Commun., 2013, 34, 1742–1746 CrossRef CAS PubMed.
- J. Ling, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2011, 21, 18373–18380 RSC.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- B. Ghosh, K. V. Chellappan and M. W. Urban, J. Mater. Chem., 2011, 21, 14473–14486 RSC.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- X. Chen, L. Hong, X. You, Y. Wang, G. Zou, W. Su and Q. Zhang, Chem. Commun., 2009, 1356–1358, 10.1039/B820894H.
- I. Tomatsu, A. Hashidzume and A. Harada, Macromolecules, 2005, 38, 5223–5227 CrossRef CAS.
- Y.-L. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef CAS PubMed.
- D. Wang and X. Wang, Prog. Polym. Sci., 2013, 38, 271–301 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- H. Yamaguchi, Y. Kobayashi, R. Kobayashi, Y. Takashima, A. Hashidzume and A. Harada, Nat. Commun., 2012, 3, 603 CrossRef PubMed.
- Q. Yang, P. Wang, C. Zhao, W. Wang, J. Yang and Q. Liu, Macromol. Rapid Commun., 2017, 38, 1600741 CrossRef PubMed.
- H. Sakai, Y. Orihara, H. Kodashima, A. Matsumura, T. Ohkubo, K. Tsuchiya and M. Abe, J. Am. Chem. Soc., 2005, 127, 13454–13455 CrossRef CAS PubMed.
- H.-Y. Lee, K. K. Diehn, K. Sun, T. Chen and S. R. Raghavan, J. Am. Chem. Soc., 2011, 133, 8461–8463 CrossRef CAS PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- M. V. Biyani, E. J. Foster and C. Weder, ACS Macro Lett., 2013, 2, 236–240 CrossRef CAS.
- E. Borré, J.-F. Stumbé, S. Bellemin-Laponnaz and M. Mauro, Angew. Chem., Int. Ed., 2016, 55, 1313–1317 CrossRef PubMed.
- P. Weis and S. Wu, Macromol. Rapid Commun., 2017 DOI:10.1002/marc.201700220.
- N. K. Kildahl, J. Chem. Educ., 1995, 72, 423 CrossRef CAS.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Adv. Mater., 2015, 27, 7740–7745 CrossRef CAS PubMed.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683–10690 CAS.
- M. B. Charati, I. Lee, K. C. Hribar and J. A. Burdick, Small, 2010, 6, 1608–1611 CrossRef CAS PubMed.
- Z. Cao, R. Wang, F. Yang, L. Hao, W. Jiao, W. Liu, Q. Wang and B. Zhang, RSC Adv., 2015, 5, 102167–102172 RSC.
- F. I. Altuna, J. Antonacci, G. F. Arenas, V. Pettarin, C. E. Hoppe and R. J. J. Williams, Mater. Res. Express, 2016, 3, 045003 CrossRef.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS.
- H. Zhang and Y. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 13069–13075 CAS.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353–1359 CrossRef CAS PubMed.
- L. Han, Y. Zhang, X. Lu, K. Wang, Z. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 29088–29100 CAS.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- D. E. Apostolides, C. S. Patrickios, E. Leontidis, M. Kushnir and C. Wesdemiotis, Polym. Int., 2014, 63, 1558–1565 CrossRef CAS.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- F. Yu, X. Cao, J. Du, G. Wang and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 24023–24031 CAS.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- F. Ding, S. Wu, S. Wang, Y. Xiong, Y. Li, B. Li, H. Deng, Y. Du, L. Xiao and X. Shi, Soft Matter, 2015, 11, 3971–3976 RSC.
- W. Yang, X. Wu, F. Liu, Y. Dou, Z. Hu and W. Hao, RSC Adv., 2016, 6, 34254–34260 RSC.
- K. Bauri, P. De, P. N. Shah, R. Li and R. Faust, Macromolecules, 2013, 46, 5861–5870 CrossRef CAS.
- U. Haldar, K. Bauri, R. Li, R. Faust and P. De, ACS Appl. Mater. Interfaces, 2015, 7, 8779–8788 CAS.
- Z. Ge, J. Hu, F. Huang and S. Liu, Angew. Chem., Int. Ed., 2009, 48, 1798–1802 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1905–1909 CrossRef CAS PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- X. Ma, N. Zhou, T. Zhang, W. Hu and N. Gu, Mater. Sci. Eng., C, 2017, 73, 357–365 CrossRef CAS PubMed.
- C. Loebel, C. B. Rodell, M. H. Chen and J. A. Burdick, Nat. Protoc., 2017, 12, 1521–1541 CrossRef CAS PubMed.
- S. Varghese, A. K. Lele, D. Srinivas, M. Sastry and R. A. Mashelkar, Adv. Mater., 2001, 13, 1544–1548 CrossRef CAS.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- T. Bai, S. Liu, F. Sun, A. Sinclair, L. Zhang, Q. Shao and S. Jiang, Biomaterials, 2014, 35, 3926–3933 CrossRef CAS PubMed.
- X. Ye, X. Li, Y. Shen, G. Chang, J. Yang and Z. Gu, Polymer, 2017, 108, 348–360 CrossRef CAS.
- R. Dobrawa and F. Würthner, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4981–4995 CrossRef CAS.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed.
- X. Sui, X. Feng, M. A. Hempenius and G. J. Vancso, J. Mater. Chem. B, 2013, 1, 1658–1672 RSC.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- T.-W. Chuo, T.-C. Wei and Y.-L. Liu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3395–3403 CrossRef CAS.
- T.-W. Chuo, J.-M. Yeh and Y.-L. Liu, RSC Adv., 2016, 6, 55593–55598 RSC.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- L. Peng, H. Zhang, A. Feng, M. Huo, Z. Wang, J. Hu, W. Gao and J. Yuan, Polym. Chem., 2015, 6, 3652–3659 RSC.
- Q. Yan, A. Feng, H. Zhang, Y. Yin and J. Yuan, Polym. Chem., 2013, 4, 1216–1220 RSC.
- K. Tsuchiya, Y. Orihara, Y. Kondo, N. Yoshino, T. Ohkubo, H. Sakai and M. Abe, J. Am. Chem. Soc., 2004, 126, 12282–12283 CrossRef CAS PubMed.
- B. Zoetebier, M. A. Hempenius and G. J. Vancso, Chem. Commun., 2015, 51, 636–639 RSC.
- X. Sui, L. van Ingen, M. A. Hempenius and G. J. Vancso, Macromol. Rapid Commun., 2010, 31, 2059–2063 CrossRef CAS PubMed.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- N. Hohlbein, A. Shaaban and A. M. Schmidt, Polymer, 2015, 69, 301–309 CrossRef CAS.
- S.-H. Lee, J.-H. Jung and I.-K. Oh, Small, 2014, 10, 3880–3886 CrossRef CAS PubMed.
- A. M. Schmidt, Macromol. Rapid Commun., 2006, 27, 1168–1172 CrossRef CAS.
- T. J. Ahmed, D. Stavrov, H. E. N. Bersee and A. Beukers, Composites, Part A, 2006, 37, 1638–1651 CrossRef.
- A. S. Ahmed and R. V. Ramanujan, J. Mater. Res., 2015, 30, 946–958 CrossRef CAS.
- C. C. Corten and M. W. Urban, Adv. Mater., 2009, 21, 5011–5015 CrossRef CAS PubMed.
- B. J. Adzima, C. J. Kloxin and C. N. Bowman, Adv. Mater., 2010, 22, 2784–2787 CrossRef CAS PubMed.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- C. Yu, C.-F. Wang and S. Chen, Adv. Funct. Mater., 2014, 24, 1235–1242 CrossRef CAS.
- X.-Y. Du, S.-S. Liu, C.-F. Wang, G. Wu and S. Chen, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2585–2593 CrossRef CAS.
- P. Nordblad, Nat. Mater., 2013, 12, 11–12 CrossRef CAS PubMed.
- A. J. Bandodkar, C. S. López, A. M. Vinu Mohan, L. Yin, R. Kumar and J. Wang, Sci. Adv., 2016, 2, e1601465 Search PubMed.
- H. Sun, X. You, Y. Jiang, G. Guan, X. Fang, J. Deng, P. Chen, Y. Luo and H. Peng, Angew. Chem., Int. Ed., 2014, 126, 9680–9685 CrossRef.
- Y. Huang, Y. Huang, M. Zhu, W. Meng, Z. Pei, C. Liu, H. Hu and C. Zhi, ACS Nano, 2015, 9, 6242–6251 CrossRef CAS PubMed.
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS PubMed.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS PubMed.
- J. Li, I. Cvrtila, M. Colomb-Delsuc, E. Otten and S. Otto, Chem. – Eur. J., 2014, 20, 15709–15714 CrossRef CAS PubMed.
- J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng, H. Xia and G. Chen, J. Mater. Chem., 2012, 22, 11515–11522 RSC.
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356–2359 RSC.
- Q. Zheng, Z. Ma and S. Gong, J. Mater. Chem. A, 2016, 4, 3324–3334 CAS.
- X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362–369 CrossRef CAS PubMed.
- X. Yan, D. Xu, J. Chen, M. Zhang, B. Hu, Y. Yu and F. Huang, Polym. Chem., 2013, 4, 3312–3322 RSC.
- J. Zhan, M. Zhang, M. Zhou, B. Liu, D. Chen, Y. Liu, Q. Chen, H. Qiu and S. Yin, Macromol. Rapid Commun., 2014, 35, 1424–1429 CrossRef CAS PubMed.
- X. Wang, J. Wang, Y. Yang, F. Yang and D. Wu, Polym. Chem., 2017, 8, 3901–3909 RSC.
- P. Zhang, F. Deng, Y. Peng, H. Chen, Y. Gao and H. Li, RSC Adv., 2014, 4, 47361–47367 RSC.
- Z. Rodriguez-Docampo and S. Otto, Chem. Commun., 2008, 5301–5303, 10.1039/B808725C.
- Y. Zhang, C. Fu, Y. Li, K. Wang, X. Wang, Y. Wei and L. Tao, Polym. Chem., 2017, 8, 537–544 RSC.
- Y. Kubo, R. Nishiyabu and T. D. James, Chem. Commun., 2015, 51, 2005–2020 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- M. C. Roberts, M. C. Hanson, A. P. Massey, E. A. Karren and P. F. Kiser, Adv. Mater., 2007, 19, 2503–2507 CrossRef CAS.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- M. Nakahata, S. Mori, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, ACS Macro Lett., 2014, 3, 337–340 CrossRef CAS.
- C. C. Deng, W. L. A. Brooks, K. A. Abboud and B. S. Sumerlin, ACS Macro Lett., 2015, 4, 220–224 CrossRef CAS.
- H. Meng, J. Zheng, X. Wen, Z. Cai, J. Zhang and T. Chen, Macromol. Rapid Commun., 2015, 36, 533–537 CrossRef CAS PubMed.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- M. Vatankhah-Varnoosfaderani, S. Hashmi, A. GhavamiNejad and F. J. Stadler, Polym. Chem., 2014, 5, 512–523 RSC.
- A. Pettignano, S. Grijalvo, M. Haring, R. Eritja, N. Tanchoux, F. Quignard and D. Diaz Diaz, Chem. Commun., 2017, 53, 3350–3353 RSC.
- J. Chen, Q. Su, R. Guo, J. Zhang, A. Dong, C. Lin and J. Zhang, Macromol. Chem. Phys., 2017, 1700166, DOI:10.1002/macp.201700166.
- M. Shan, C. Gong, B. Li and G. Wu, Polym. Chem., 2017, 8, 2997–3005 RSC.
| This journal is © The Royal Society of Chemistry 2017 |