
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the CDSA of poly(lactide) containing triblock copolymers†
Wei
Yu
,
Maria
Inam
,
Joseph R.
Jones
,
Andrew P.
Dove
 * and
Rachel K.
O'Reilly
*
* and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: r.k.o-reilly@warwick.ac.uk; a.p.dove@warwick.ac.uk
First published on 16th August 2017
Abstract
Crystallisation-driven self-assembly (CDSA) has become an extremely valuable technique in the preparation of well-defined nanostructures using diblock copolymers. The use of triblock copolymers is considerably less well-known on account of more complex syntheses and assembly methods despite the functional advantages provided by a third block. Herein, we show the simple preparation of well-defined tuneable 1D and 2D structures based on poly(lactide) triblock copolymers of different block ratios synthesised by ring-opening polymerisation (ROP) and reversible addition–fragmentation chain transfer (RAFT) polymerisation, where a phase diagram based on a novel unimer solubility approach is proposed. Using a series of poly(L-lactide)-b-poly(N,N-dimethylacrylamide) (PLLA-b-PDMA) diblock copolymers and PDMA-b-PLLA-b-PDMA triblock copolymers with different core/corona ratios, single solvent CDSA processes revealed that comparatively hydrophilic polymers were liable to achieve 2D platelets, while the less hydrophilic counterparts yield ‘transition state’ wide cylinders and pure 1D cylinders. The length of crystalline core block is also shown to play an important role in fixed corona/core ratio systems, where a longer core block is prone to form cylindrical structures due to a lack of overall solubility, whereas a shorter block forms platelets. Importantly, this approach reveals contrary results to conventional theories, which state that longer solvophilic blocks relative to the core-forming block should favour more curved core/corona interfaces. Our morphological transitions are shown in both di- and tri-block copolymer systems, showing the generalisation of these assembly methods towards promising methodologies for the rational design of PLLA-based nanocarriers in the biomedical realm.
Introduction
Block copolymer self-assembly has attracted a great deal of attention in recent decades as a consequence of the ability to fabricate a variety of morphologies, from simple spherical, cylindrical, lamellae and vesicular architectures, to complex and sophisticated hierarchical nanostructures.1–8 Among these, cylindrical structures have exhibited extensive potential in various unique applications, such as additives for polymer resin toughening9,10 and templates for electronic materials.11 Particularly in the biomedical arena, cylindrical assemblies have been shown to display longer in vivo circulation times and also show a preference for altered cell-internalization pathways when compared to their spherical counterparts.12–14The use of semi-crystalline polymers has provided an unprecedented route to the facile fabrication of cylindrical micelles using crystallisation-driven self-assembly (CDSA), where the formation of cylindrical structures is driven by the crystallisation of the core block to form micelles with low interfacial curvature.15 This phenomenon has been studied extensively by Manners, Winnik and co-workers using polyferrocenyldimethylsilane (PFS)-containing organometallic–organic diblock copolymers in alkane solvents.15–18 The size and morphology of the assemblies can be precisely adjusted with the instruction of a seed-growth methodology.19–22 Various functional groups have also been incorporated, including fluorescent marking,23 metal nanoparticle incorporation24 and photo-responsibility,25 demonstrating a diverse range of potential uses within drug delivery processes.26
As a semi-crystalline polymer, PLLA is renowned for its outstanding biocompatibility and biodegradability, which allows use in bio-relevant applications. In our previous studies, we have shown that PLLA-containing block copolymers can be fabricated using a combination of reversible addition–fragmentation chain transfer (RAFT) and ring-opening polymerisation (ROP), where we have been able to achieve tuneable cylindrical micelles with a range of coronal blocks by CDSA.27–29 Further work was carried out to enrich the functionality of the PLLA-based cylinders30,31 and successfully realize stereocomplexation-triggered morphological transitions,32 indicating the promising future of these cylindrical particles in the biomedical realm.
Similar to conventional phase separation-driven assembly, it can be expected that triblock copolymers will lead to an alternate morphology when undergoing CDSA as a consequence of the extra coil phase. Wang et al. prepared a series of coil–crystalline–coil poly(ferrocenylphenylphosphine) (PFP)-b-PFS-b-poly(dimethylsiloxane) (PDMS) triblock copolymers and, upon increasing the degree of polymerisation of PFP from 6 to 11, the morphology transitioned from cylindrical to spherical micelles. It was proposed that the longer PFP block interfered with the crystallisation of the PFS block to a greater extent than the shorter blocks, thus leading to amorphous spheres.15 Similarly, Schmalz et al. found that polystyrene (PS)-b-polyethylene (PE)-b-poly(methyl methacrylate) (PMMA) (ABC) triblock terpolymers formed worm-like micelles, whereas the corresponding PE diblock copolymers (PE-b-PMMA) formed platelet-like structures. It was also found that, as the composition or molecular weight of the triblock terpolymer was changed, the morphology of the structures also changed, and, therefore, it was possible to control the type of structure formed, i.e. spherical, worm-like (cylindrical) or lamellar.33 Although these studies focused on the CDSA behaviour of ABC type coil–crystalline–coil triblock copolymers, seldom have carried out a systematic study of the assembly behaviour of a simplified triblock terpolymer model, i.e. ABA type copolymers. It is expected that these polymers will achieve different assembly morphologies compared to their corresponding coil–crystalline diblock copolymers as a consequence of the difference in solubility and crystallisation behaviour.
Herein, we report the preparation of well-defined amphiphilic diblock and triblock copolymers, PDMA-b-PLLA and PDMA-b-PLLA-b-PDMA, with a range of block lengths synthesised by ROP and RAFT polymerisation. Using a simple, single solvent assembly system, we show that the hydrophobicity of the polymer plays an important role in the assembly process for both di- and tri-block copolymer systems, where a clear transition from cylindrical to platelet structures can be observed on increasing polymer solubility.
Experimental section
Materials
Chemicals and solvents were purchased from Sigma-Aldrich, Acros, Fluka, TCI, Fisher Chemical, Alfa Aesar or VWR. L-Lactide was purchased from Corbion-Purac and recrystallized once from dichloromethane and twice from toluene. The monomer was dried over 3 Å molecular sieves for 3 days and recrystallized from toluene. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) and (−)-sparteine were distilled over CaH2 before use. 1-(3,5-Bis(trifluoromethyl)phenyl)-3-cyclohexyl-thiourea was prepared and purified as reported.29 2,2′-Azobis(isobutyronitrile) (AIBN) was received from Molekula. After recrystallisation from methanol it was stored at 4 °C. Deuterated solvents were used as received from Apollo Scientific. Raft agent 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) was synthesised based on previous work.34Instrumentation
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz. All spectra were recorded in CDCl3 unless otherwise stated. The chemical shifts were reported as δ in parts per million and quoted downfield from the internal standard tetramethylsilane (δ = 0 ppm). Diffusion-ordered spectroscopy (DOSY) NMR spectroscopy was performed on a Bruker AV-500 AVANCE spectrometer equipped with a 5 mm broadband observe (BBO) z-axis gradient probe capable of generating nominal maximum field strengths of 53.5 G cm−1. The measurement was performed using stimulated echo and LED pulse sequences incorporating bipolar-gradient pulses for diffusion, using a diffusion time of 100 ms and a LED delay of 5 ms. For each experiment, pulsed field gradients with a duration of 2.5 ms followed by a recovery delay of 200 μs were applied with increases from 5% to 95% of the maximum strength in 32 equally spaced steps. Experiments were carried out on samples at a polymer concentration of 10 mg mL−1 in deuterated chloroform with active temperature regulation at 298 K. The DOSY spectrum was processed by the Bruker Topspin S3 software package (version 2.1).Size exclusion chromatography (SEC) was performed using an Agilent 1260 Infinity Multi-Detector GPC System fitted with a refractive index and UV detector, and equipped with a guard column (Varian PLGel) and two PLGel 5 μm mixed-D columns. The mobile phase was DMF and 5 mM NH4BF4, at a flow rate of 1 mL min−1 at 50 °C. All data was analysed using Cirrus v3.3 and Agilent GPC/SEC software v1 with calibration curves produced using Varian Polymer Laboratories linear PMMA standards. Mass spectra were obtained using a Bruker Ultraflex II matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometer. The MALDI-ToF samples were prepared using a trans-2-[3-(4-t-butyl-phenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) matrix and sodium trifluoroacetate (NaTFA) as a cationization agent. Samples were prepared as follows: DCTB (2 μL, 10 mg mL−1 in tetrahydrofuran), sample (2 μL, 1 mg mL−1 in tetrahydrofuran) and NaTFA (2 μL, 0.1 mg mL−1 in tetrahydrofuran) were added to the MALDI-ToF plate successively. The samples were measured in reflection ion mode and calibrated using SpheriCal (1200–8000 g mol−1) standards.
Transmission electron microscopy (TEM) was performed using a JEOL 2100FX at 200 kV. TEM samples were prepared on a formvar/carbon film TEM grid. In short, 2 μL of sample solution (1 mg mL−1) was deposited on a grid and left air dry. 5 μL of uranyl acetate (UA, 1%) solution was then dropped on the grid and left for 60 seconds before blotting. The sample was kept in a desiccator overnight before characterisation. TEM samples were also prepared by using graphene oxide (GO)-covered TEM grids which are almost electron transparent and give excellent contrast.35 Generally, one drop of the sample solution (2 μL) was added onto a GO grid, and, after 2 min, the solution was blotted away before drying. The GO grids were prepared as following: lacey carbon grids (400 mesh, Cu, Elektron Technology UK LTD) were cleaned by air plasma from a glow-discharge system (2 min, 20 mA) to improve the hydrophilicity of the lacey carbon. One drop of GO solution (0.10–0.15 mg mL−1) was deposited on the grid and left to air-dry.
Dynamic light scattering was conducted using a Malvern Zetasizer Nano instrument equipped with a 4 mW He–Ne 633 nm laser module at 25 °C, with data analysis using Malvern DTS 6.20 software. Measurements were carried out at a detection angle of 173° (backscattering). All determinations were made in triplicate unless otherwise stated (with 10 measurements recorded for each run).
Wide Angle X-ray Scattering (WAXS). WAXS was performed on a Panalytical X'Pert Pro MPD equipped with a Cu Kα1 hybrid monochromator as the incident beam optics. Typically, ca. 30 mg of freeze-dried particles was placed in a 10 mm sample holder, and standard “powder” 2θ–θ diffraction scans were carried out in the angular range from 10° to 30° 2θ at room temperature.
Synthesis of PLLA homopolymers
Ring-opening polymerisation (ROP) of L-lactide was carried out using a dual-headed initiator, 1,3-propanediol, with an organic catalyst, DBU. Typically, for the synthesis of homopolymer PLLA32, L-lactide (2.5 g, 17.35 mmol) monomer, 1,3-propanediol (34.59 μl, 0.48 mmol), DBU (25.94 μl, 0.17 mmol) and dichloromethane (25 mL) were mixed in an ampoule in a glove box, under nitrogen. The solution was left stirring at room temperature for 5 minutes for 100% monomer conversion. The mixture was purified by precipitation twice in hexane and once in methanol and dried in vacuo to yield a white powder (80% yield). Mn, NMR = 4.6 kDa, DP = 32. Mn, SEC = 10.4 kDa, ĐM = 1.05. 1H NMR (CDCl3): δ (ppm) 5.15 (q, OCH(CH3)CO, 3JH–H = 7.1 Hz), 4.43–4.29 (m, 2H, HOCH(CH3)CO), 4.19 (t, 4H, OCH2CH2CH2O, 3JH–H = 5.7 Hz), 2.09–1.90 (m, 2H, OCH2CH2CH2O), 1.57 (d, OCH(CH3)CO, 3JH–H = 7.1 Hz).Synthesis of dual functionalised macro-CTA (CTA-PLLA-CTA)
RAFT agent 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT), synthesised according to a previous method,36 was coupled to the PLLA polymer backbone by esterification. In a typical coupling reaction, PLLA32 (1.4 g, 0.324 mmol), DDMAT (1.18 g, 3.24 mmol), 4-dimethylaminopyridine (DMAP) (0.0396 g, 0.324 mmol) and N,N′-dicyclohexylcarbodiimide (DCC) (0.668 g, 3.24 mmol) were mixed together in an ampoule with 12 mL chloroform. The solution was left stirring at room temperature for 3 days. The solution was filtered and the filtrate precipitated in diethyl ether three times and the resultant polymer was dried in vacuo (1.1 g, 70% yield). Mn, NMR = 5.2 kDa, Mn, SEC = 9.9 kDa, ĐM = 1.06. 1H NMR (CDCl3): δ (ppm) 5.15 (q, OCH(CH3)CO, 3JH–H = 7.1 Hz), 4.43–4.29 (m, 1H, HOCH(CH3)CO), 4.19 (t, 4H, OCH2CH2CH2O, 3JH–H = 5.7 Hz), 3.26 (t, 4H, SCH2CH2, 3JH–H = 7.5 Hz), 2.09–1.90 (m, 2H, OCH2CH2CH2O), 1.57 (d, OCH(CH3)CO, 3JH–H = 7.1 Hz). 1.24 (s, C10H21), 0.87 (t, 6H, CH3CH2, 3JH–H = 6.8 Hz).Synthesis of triblock copolymers poly(N,N-dimethylacrylamide)-b-poly(L-lactide)-b-poly(N,N-dimethylacrylamide) (PDMA-b-PLLA-b-PDMA)
Dual functionalised macro-CTA was chain-extended using RAFT polymerisation with N,N-dimethylacrylamide (DMA) as the monomer (Scheme 1). In a typical reaction of PDMA122-b-PLLA32-b-PDMA122, CTA-PLLA32-CTA (50 mg, 0.01 mmol), DMA (0.33 ml, 3.2 mmol) and 10 mg ml−1 AIBN solution (16.42 μl, 0.001 mmol) were mixed in 1 mL 1,4-dioxane in an ampoule. The solution was degassed via three freeze–pump–thaw cycles and refilled with argon. The solution was placed in an oil bath at 70 °C for 5 h. The product was precipitated in diethyl ether three times and dried in vacuo (90% conversion, 0.18 g, 60% yield). Mn, NMR = 28.9 kDa, DP = 245. Mn, SEC = 38.4 kDa, ĐM = 1.08. 1H NMR (CDCl3): δ (ppm) 5.15 (q, OCH(CH3)CO, 3JH–H = 7.1 Hz), 4.19 (t, 4H, OCH2CH2CH2O, 3JH–H = 5.7 Hz), 2.88 (s, CON(CH3)2), 1.57 (d, OCH(CH3)CO, 3JH–H = 7.1 Hz), 1.24 (s, C10H21), 0.87 (t, 6H, CH3CH2, 3JH–H = 6.8 Hz).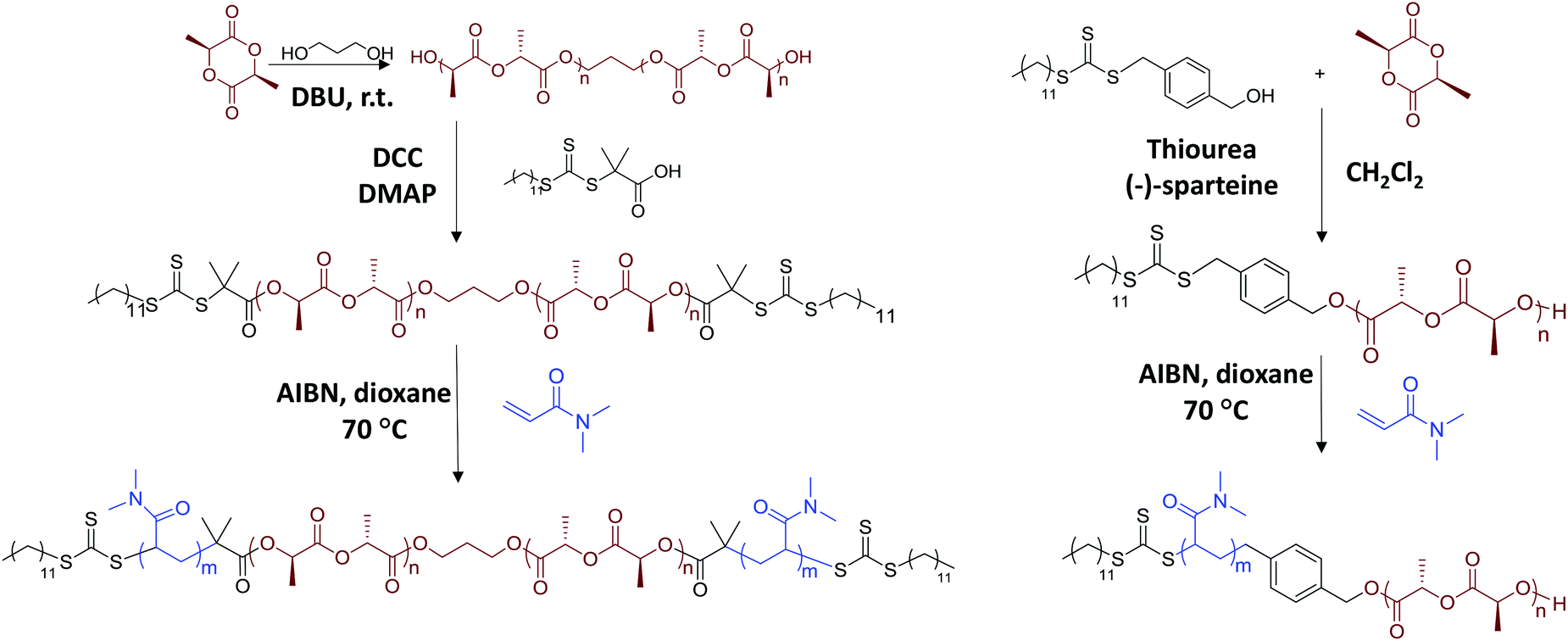 | ||
| Scheme 1 Synthesis of PLLA-b-PDMA diblock and PDMA-b-PLLA-b-PDMA triblock copolymers. | ||
Crystallisation-driven self-assembly of PDMA-b-PLLA-b-PDMA triblock copolymers and PDMA-b-PLLA diblock copolymers
All triblock copolymers were assembled in HPLC grade methanol. Typically, 5 mg polymer was totally dissolved in 1 mL methanol with vortexing and sonication at room temperature. The assembly solution was left to age in a sealed vial at room temperature for one day. Diblock copolymers were assembled in ethanol. Typically, 5 mg polymer was dissolved in 1 mL of ethanol and heated to 90 °C for 8 hours before cooling to room temperature and ageing overnight.37Results and discussion
Synthesis of PDMA-b-PLLA-b-PDMA triblock copolymers
In order to investigate how the crystalline block length affects the assembly morphology, a series of PLLA block lengths (PLLA25, PLLA32, PLLA50, PLLA68) were targeted (Scheme 1, Table S1†). Evidenced by SEC characterisation (Fig. S1†), all homopolymers were well-defined with monomodal distributions (ĐM < 1.1), with consistent molecular weights in comparison with theoretical calculations. Further analysis using MALDI-ToF MS and 1H NMR spectroscopy (Fig. S2 and S3†) suggested an absence of unwanted chain transesterification processes or initiation from water.The PLLA homopolymers bearing hydroxyl functional groups at both ends were reacted with RAFT agent, 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT), using N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) as activating reagents (Scheme 1). The coupling reaction was carried out in a concentrated solution for an extensive period of time (3 days) using an excess of the carboxylic group functionality to promote quantitative attachment of the RAFT agent to both ends of the homopolymer. As observed by 1H NMR spectroscopy of macro-initiator CTA-PLLA32-CTA (Fig. 1), the integral value of the triplet associated with the methylene proton of the thiocarbonyl group at δ = 3.24 ppm was equal to the corresponding value of the methylene protons in the initiator (δ = 4.18 ppm), confirming that both ends of the polymer had been successfully functionalised with DDMAT. Comparable results were also achieved with the rest of the series (Fig. S4†).
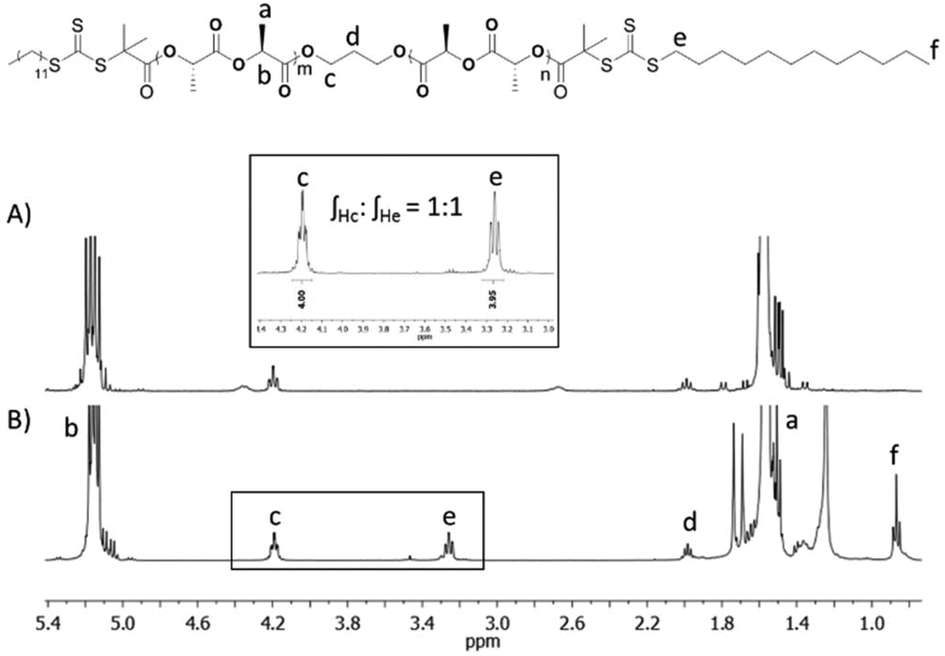 | ||
| Fig. 1 1H NMR spectra (400 MHz, CDCl3) of PLLA32 (A) and CTA-PLLA32-CTA (B) macro-initiator. | ||
SEC characterisation (Fig. S5†) showed a clear molecular weight shift (RI trace) and overlapping of the RI and UV traces (λ = 309 nm), which indicated successful attachment of the trithiocarbonate onto the polymer backbone. Furthermore, MALDI-ToF MS analysis of PLLA32 and CTA-PLLA32-CTA (Fig. 2 and Fig. S6†) also revealed a shift in mass distribution (Δm/z 692.25), which was calculated as exactly twice that of the molecular weight of DDMAT, with no mass peaks existing between the main distribution, thus confirming that attachment occurred at both ends of the polymer.
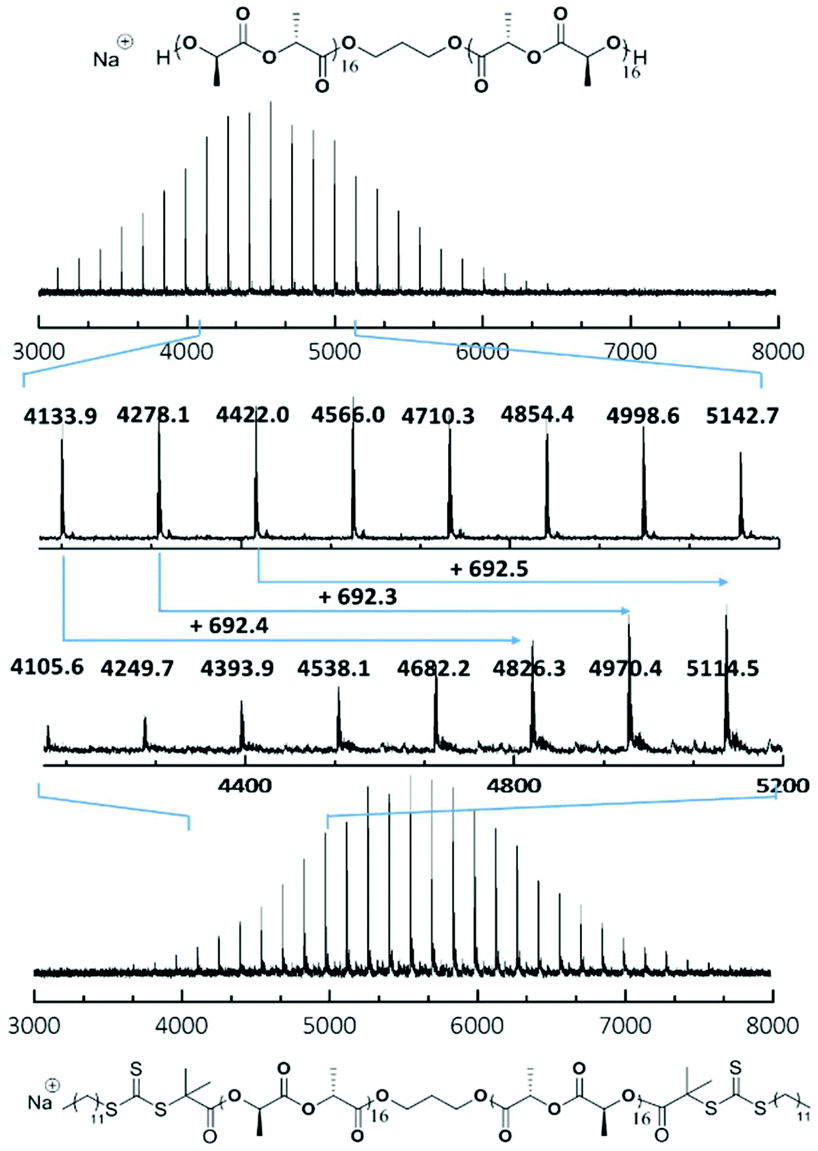 | ||
| Fig. 2 MALDI-ToF mass spectra of polymer PLLA32 and CTA-PLLA32-CTA showing a shift in molecular weight of 692.4, corresponding to the molecular weight of two DDMAT molecules. | ||
The bifunctional macro-CTA was used to grow hydrophilic blocks of poly(N,N-dimethylacrylamide) (PDMA) to produce ABA sequence, PDMA-b-PLLA-b-PDMA, triblock copolymers via RAFT polymerisation. Typically, the reaction was executed at 70 °C for 5 h to give a monomer conversion of 90% as determined by 1H NMR spectroscopy. Further 1H NMR spectroscopic analysis of the purified polymer (Fig. S7†) was used to confirm the block ratio and block length of the polymer blocks based on characteristic proton signals (δ = 2.90 and 5.14 ppm). The reaction conditions of the chain-growth of PDMA were adjusted, with respect to the core and corona lengths, to attain a range of well-defined triblock terpolymers T1–T13 (Table 1) with different hydrophobic ratios (11, 15, 25 and 32%). SEC analysis confirmed narrow molecular weight distributions for all polymers, ĐM < 1.2 (Fig. S8 and S9†).
| T-triblock | Block copolymers |
M
n, NMR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kDa) a (kDa) |
M
n, SECb (kDa) |
Đ
Mb |
Hydrophobic weightc (wt%) |
|---|---|---|---|---|---|
| D-diblock | |||||
| a Measured by 1H NMR spectroscopy in CDCl3. b Measured by DMF SEC using RI detection. c PLLA weight fraction in the block copolymer. | |||||
| T1 | PDMA42-b-PLLA25-b-PDMA42 | 11.3 | 17.8 | 1.05 | 32.1 |
| T2 | PDMA60-b-PLLA25-b-PDMA60 | 14.4 | 18.7 | 1.12 | 25.2 |
| T3 | PDMA105-b-PLLA25-b-PDMA105 | 24 | 32.5 | 1.10 | 15.1 |
| T4 | PDMA160-b-PLLA25-b-PDMA160 | 32.8 | 39.7 | 1.13 | 11.0 |
| T5 | PDMA45-b-PLLA32-b-PDMA45 | 13.5 | 19.5 | 1.04 | 32.2 |
| T6 | PDMA66-b-PLLA32-b-PDMA66 | 18.5 | 22.7 | 1.07 | 25.1 |
| T7 | PDMA122-b-PLLA32-b-PDMA122 | 28.9 | 38.4 | 1.08 | 15.3 |
| T8 | PDMA188-b-PLLA32-b-PDMA188 | 41.9 | 46.7 | 1.13 | 11.1 |
| T9 | PDMA75-b-PLLA50-b-PDMA75 | 22.1 | 30.9 | 1.04 | 32.2 |
| T10 | PDMA225-b-PLLA50-b-PDMA225 | 51.8 | 58.2 | 1.14 | 15.1 |
| T11 | PDMA295-b-PLLA50-b-PDMA295 | 65.5 | 69.6 | 1.17 | 11.0 |
| T12 | PDMA115-b-PLLA68-b-PDMA115 | 32.6 | 47.4 | 1.03 | 32.0 |
| T13 | PDMA315-b-PLLA68-b-PDMA315 | 72.3 | 73.5 | 1.14 | 15.0 |
| D1 | PLLA25-b-PDMA120 | 15.9 | 25 | 1.10 | 23.7 |
| D2 | PLLA25-b-PDMA225 | 26.3 | 36.3 | 1.17 | 13.9 |
| D3 | PLLA48-b-PDMA145 | 21.7 | 30.8 | 1.05 | 33.0 |
| D4 | PLLA48-b-PDMA240 | 31.1 | 41.5 | 1.05 | 22.8 |
| D5 | PLLA48-b-PDMA570 | 63.8 | 74.1 | 1.06 | 11.0 |
| D6 | PLLA48-b-PDMA950 | 101.5 | 122.2 | 1.10 | 6.9 |
Diffusion-ordered spectroscopy (DOSY) NMR was also carried out to confirm the absence of homopolymer to avoid any contamination which has the potential to significantly influence the CDSA results.38 The results obtained (Fig. 3) clearly showed the same diffusion coefficient value for protons corresponding to the PLLA core and the triblock terpolymer T3. This was distinguishably higher when compared to the PLLA32 homopolymer and macro RAFT agent CTA-PLLA32-CTA proton resonances, thus providing evidence for a pure triblock copolymer system without the presence of homopolymer. All of the diblock copolymers were prepared as previously reported (Fig. S10,†Table 1).37
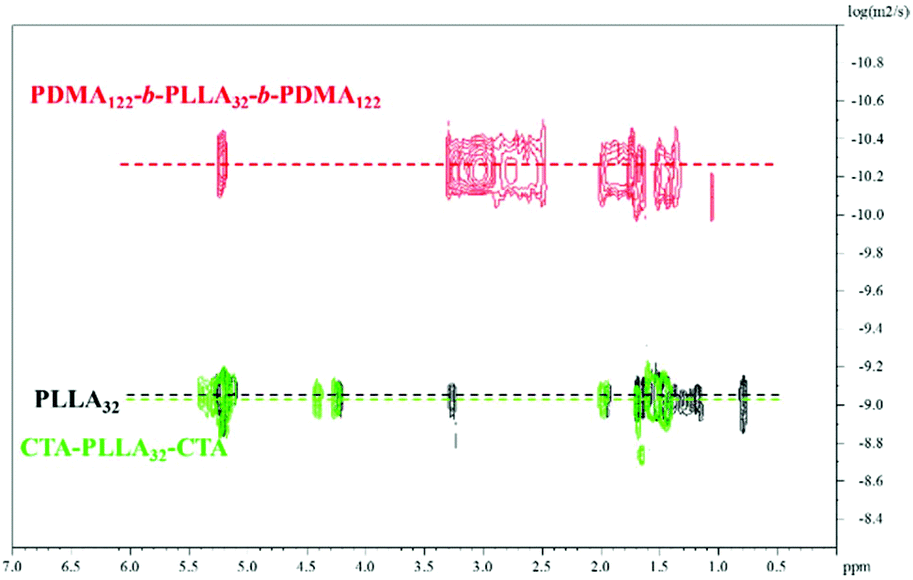 | ||
| Fig. 3 1H-DOSY NMR spectra (500 MHz) of homopolymer PLLA32, macro-initiator CTA-PLLA32-CTA and triblock copolymer PDMA-b-PLLA-b-PDMA T7 in CDCl3 at 298 K. | ||
Crystallisation-driven self-assembly of PDMA-b-PLLA-b-PDMA triblock copolymers
Previously, PLLA assemblies were prepared using a solvent switch and evaporation method which was time-consuming and laborious for further application.27,29,32 Recently, we reported that a single alcoholic solvent could be utilized to achieve CDSA for PLLA based diblock copolymers.37 Upon examination, triblock terpolymers were found to completely dissolve in methanol at 5 mg mL−1 at room temperature, where no Tyndall phenomenon was observed in solution for the first few hours, which indicates a lack of assembly into larger structures. After ageing at room temperature for 24 h, a strong Tyndall light path could be detected, which suggested the existence of large assemblies. Subsequent analysis of the sample using dynamic light scattering (DLS) confirmed the formation of monomodal assemblies (Fig. S11†).Tuning the size and morphology of the assembly by changing the hydrophobic and hydrophilic block length
The morphologies and sizes of all the assemblies were further characterised by transmission electron microscopy (TEM). Successful CDSA results were observed for all triblock terpolymers using this simple methanol dissolution methodology, where unique morphologies of low dispersity confirmed the results attained by DLS analysis. A clear transition from diamond-shaped lamellae to cylinders can be observed on changing the block lengths of the triblock copolymers towards less soluble unimers (Fig. 4, where the white objects in Fig. 4b are artefacts of staining). The morphological transition observed by changing copolymer composition is in agreement with our previous research,37 and has also been observed by others.39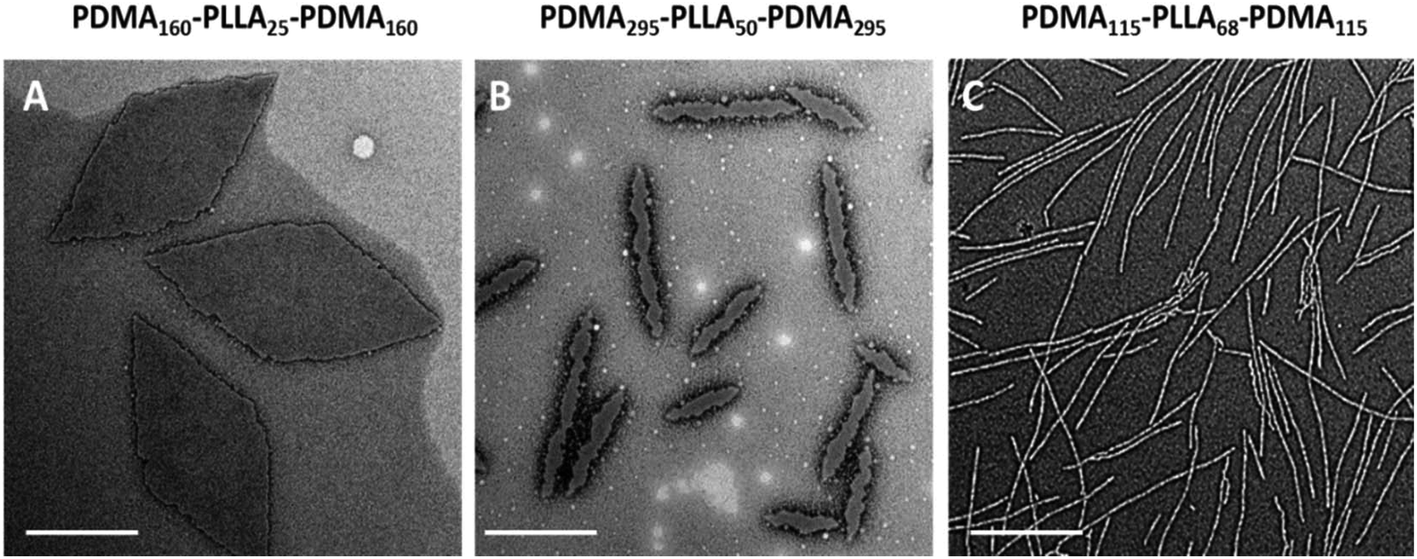 | ||
| Fig. 4 TEM analysis of micelles obtained from CDSA of the series of PDMA-b-PLLA-b-PDMA triblock copolymers T4 (A), T11 (B), T12 (C). Samples were negatively stained using uranyl acetate. Scale bar = 1 μm. | ||
Specifically, with shorter hydrophobic crystalline PLLA blocks (low degree of polymerisation (DP), 32) and a sufficiently long hydrophilic coronal blocks (hydrophilic weight 11% for example), diamond-shaped lamellae were achieved (Fig. 5E). Notably, these 2D sheets exhibited a regular diamond shape, where the average width and length is 1.0 ± 0.1 μm and 2.3 ± 0.2 μm respectively, which is in agreement with previous work.37 As the hydrophobic weight increased from 11% to 15% (Fig. 5F), sharp platelet assemblies were attained with slightly different average lengths and widths of 2.9 ± 0.2 and 0.9 ± 0.1 μm respectively. However, on decreasing the corona length, with a hydrophobic weight of 25%, elongated lamella or cylinders (mean width 55 ± 6 nm) were obtained (Fig. 5G). Samples were further characterised by AFM and on graphene oxide covered grids by TEM, confirming that the staining method had no impact on the results (Fig. S12 and S13†). It should also be noted that all of these assemblies were left to age over a month to ensure analysis of thermodynamically stable morphologies as opposed to kinetically trapped structures (Fig. S14†).
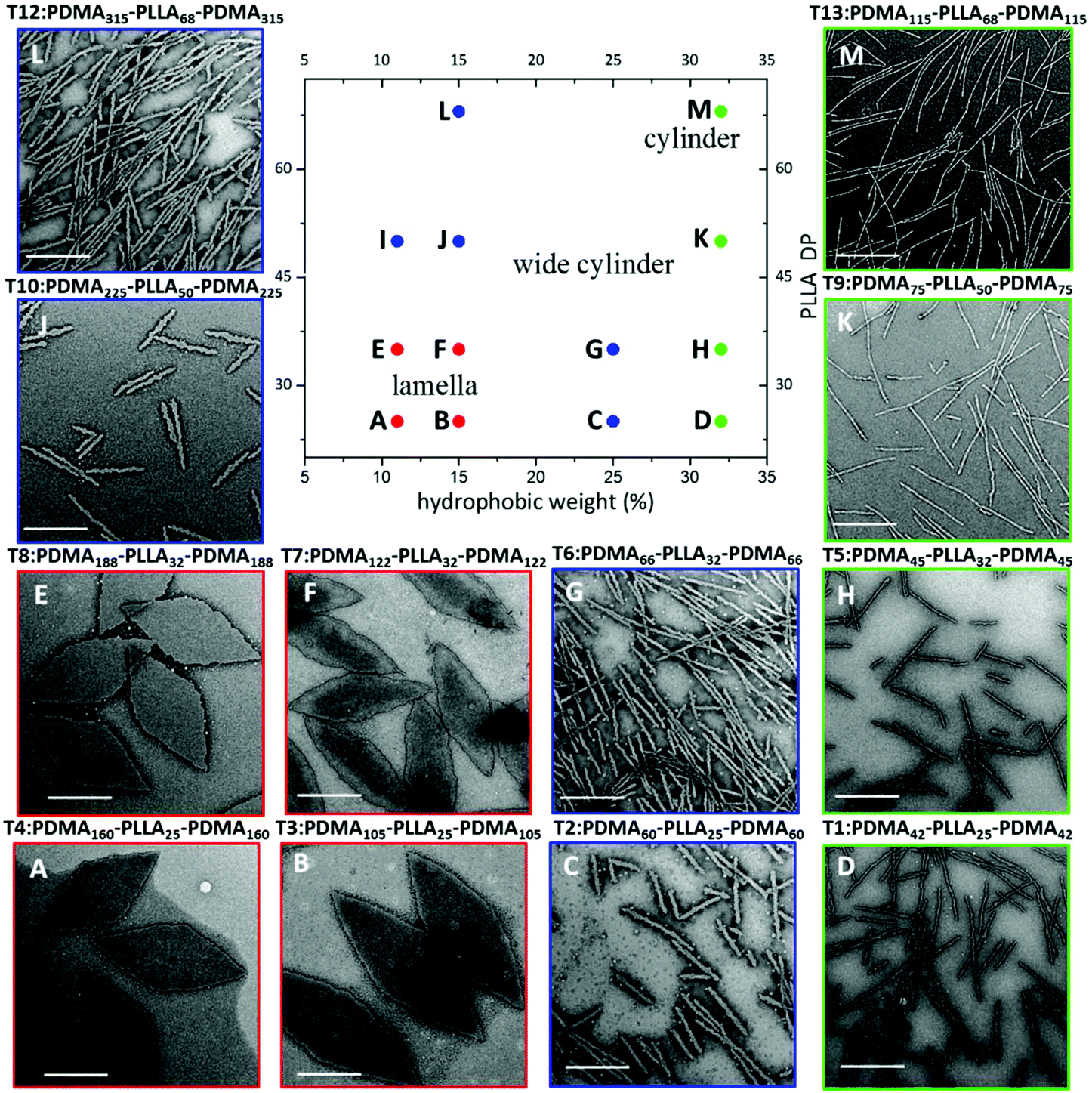 | ||
| Fig. 5 Phase diagram constructed for PDMA-b-PLLA-b-PDMA triblock terpolymers (T1–T13), T1 (D), T2 (C), T3 (B), T4 (A), T5 (H), T6 (G), T7 (F), T8 (E), T9 (K), T10 (J), T11 (I), T12 (L), T13 (M). TEM characterisation of T11 (I) is shown in Fig. 4(B). As the target PLLA DP and the hydrophobic weight were systematically varied, the achieved morphology changed from lamellae (red) to wide cylinders (blue) and pure cylinders (green). Samples were negatively stained using uranyl acetate for TEM characterisation. Scale bar = 1 μm. | ||
Based on these observations and in line with our previous results,37 we determined that the solubility of the polymers were of great significance in the CDSA process of these polymers. Block copolymers with a longer corona are more soluble, and thus remain as unimers undergoing a slower crystallisation process in forming large intact crystals (platelets), whereas, on the other hand, polymers with a shorter corona are less soluble in methanol and thus may form aggregates before crystallisation, leading to a less-crystalline cylindrical morphology. This was further confirmed by wide angle X-ray scattering (WAXS) characterisation (Fig. S15†). Accordingly, our results (Fig. 5H) showed thinner cylindrical assemblies (25.1 ± 3 nm in width) on increasing the hydrophobic ratio, which was consistent with our assumption.
Based on our success in utilising corona block lengths to tune CDSA nanostructures, we then investigated the impact of altering core block lengths. Maintaining the same hydrophobic ratio, we decreased the core block length from PLLA32 to PLLA25 to probe the influence of a shorter core block. TEM imaging revealed assemblies similar to those achieved with the same hydrophobic weight (with PLLA32), further confirming our solubility hypothesis (Fig. 5A–D). Similarly, when the core block was extended further (PLLA50 and PLLA68), the CDSA results of the corresponding triblock copolymers at 15% hydrophobic weight, PDMA225-b-PLLA50-b-PDMA225 (Fig. 5J) and PDMA315-b-PLLA68-b-PDMA315 (Fig. 5L), revealed cylinders of greater width as opposed to 2D platelets, where the widths were measured at 55 ± 4 nm for both cylinders. Consistently, a similar phenomenon was obtained from a series with 11% hydrophobic ratio (Fig. S16†). This morphological transition can also be explained using a solubility approach. Although the hydrophilic block was similar for each series of polymers, the comparatively longer core length resulted in a difficulty to solubilize as unimers. Therefore, shorter core length triblock copolymers led to 2D platelets, while a longer core length yielded cylindrical assemblies. As the corona block length of PLLA50 and PLLA68 was extended (Fig. 5K and M), thinner, conventional cylinders, of widths 18.1 ± 2 nm, 17.3 ± 2 nm respectively, were achieved, similar to the morphological transition from G to H. According to the summarized phase diagram (Fig. 5), as a consequence of the gradual decrease of the solubility from left bottom to top right, the assembly morphology undergoes a gradual transition from 2D diamond platelets to 1D cylinders, where a 1D wide cylinders can be considered as a ‘transition state’ morphology.
Comparison of the diblock system to the triblock system
In order to draw comparisons between di- and tri-block systems, we investigated the behaviour of the diblock copolymers PDMA-b-PLLA as a comparison to the terpolymers discussed above. Assemblies in methanol at room temperature (using the same approach as triblock copolymer system) were unsuccessful (Fig. S17†) as clear solutions could not be obtained, presumably because of the lack of the third PDMA block solubilising the polymer. However, it was previously shown that the solubility of the polymer was required to match that of the solvent for optimum nanostructure formation.37 To apply this to triblock and diblock copolymers, we initially observed the Tyndall effect using a series of alcoholic solvents.In the triblock copolymer system, as expected, the strongest Tyndall effect could be observed in methanol (Fig. 6A), indicating a much larger morphology (or a greater number of assemblies), whereas, for the diblock system, the most outstanding Tyndall effect was observed in ethanol. This observation was further supported by the results from static light scattering (SLS) (Fig. S18†). For triblock copolymers, TEM analysis also revealed that a blend of spheres, cylinders and incomplete platelets were obtained in ethanol (Fig. 6E). This can be attributed to the much greater solubilisation of the triblock copolymer, leading to limited crystallisation and hence mixed morphologies. The corresponding assemblies in methanol were pure intact diamonds (Fig. 6C), which supported the assumption that CDSA of triblock copolymers is easier to achieve in methanol. Similarly, for diblock copolymers, the cylindrical assemblies (Fig. 6D) achieved in methanol were less ordered than 2D platelets (Fig. 6F), indicating ethanol was the optimal alcoholic solvent.
 | ||
| Fig. 6 Triblock copolymer T3 and diblock copolymer D2 (with similar block lengths) were dissolved in different alcoholic solvents, i.e. methanol, ethanol, n-propanol, n-butanol, at a concentration of 5 mg mL−1. The assembly solution was heated to 90 °C before cooling to room temperature and aged for one day. A laser pen was used to monitor the Tyndall effect of the triblock copolymer (A) and the diblock copolymer (B). TEM characterisation of the assemblies of polymers T3 (C) and D2 (D) in methanol and polymers T3 (E) and D2 (F) in ethanol. Samples were negatively stained using uranyl acetate for TEM characterisation. | ||
To further confirm this assumption, d4-methanol and d6-ethanol were used to detect the assembly results of triblock copolymer T3 on a smaller scale. After ageing, the d4-methanol solution gradually became turbid and exhibited a stronger Tyndall effect than the corresponding d6-ethanol solution (Fig. S19†). 1H NMR spectroscopic analysis revealed a considerable suppression of the L-lactide proton signals in d4-methanol (Fig. S20†) after the assembly process (35% reduction compared to the original integral). The assemblies in d6-ethanol revealed a much lower suppression such that, after the assembly process (Fig. S21†), a reduction of only 8% was observed. These results indicate that more unimers participate in the assembly process in methanol, consolidating our initial speculations from the Tyndall effect.
Similar to the triblock system, a phase diagram can be produced for our series of diblock copolymers assembled in ethanol (Fig. S22†). For PLLA48-PDMAx diblock copolymers, as the corona lengths declined gradually (from 950 to 145), the assembly evolved from 2D platelets to a transition state (mixtures of diamond platelets and cylinders) and finally to a pure cylindrical phase. The same trend was also observed in the PLLA25-PDMAx diblock copolymers on the basis of TEM characterisation. Using the same theory, the enhanced solubility, as a consequence of the longer corona, contributes to the formation of well-ordered crystalline 2D platelets, while, conversely, the less soluble polymers (shorter corona blocks) lead to cylindrical assemblies.
As such, we propose a more generalised phase diagram encompassing both PLLA-containing di- and tri-block copolymer systems (Fig. 7), which consolidates our findings, i.e. for the CDSA process of PLLA based copolymers in a single alcoholic solvent, the solubility of the whole polymer plays an important role in determining the morphology of the assembly. The polymers that are more soluble in the assembly solvent achieve 2D platelets (red dots on the phase diagram), while the less soluble counterparts lead to 1D cylinders (green dots on the phase diagram) with a wide cylinder ‘transition state’ morphology in the middle (blue dots on the phase diagram).
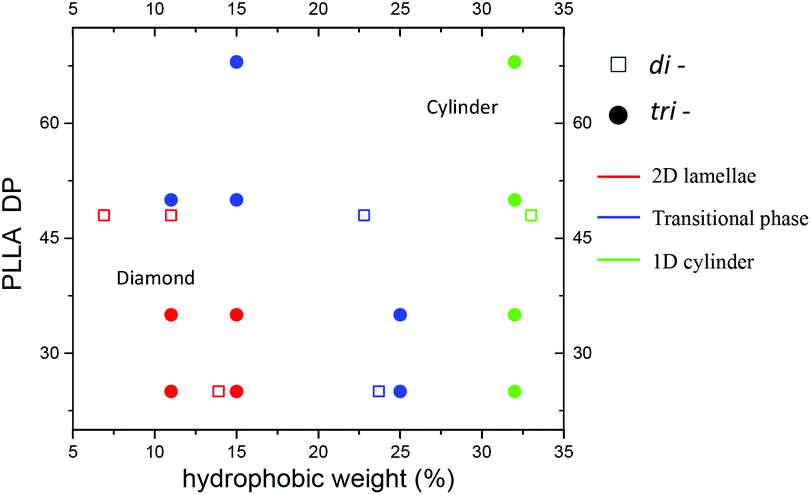 | ||
| Fig. 7 A combined phase diagram of PDMA-b-PLLA-b-PDMA triblock and PDMA-b-PLLA diblock copolymers. Triblock copolymers were assembled in methanol at room temperature whereas diblock copolymers were assembled in ethanol after elevating the temperature to 90 °C for 8 hours. The solid circles represent the triblock copolymers while the hollow squares represent the diblock copolymers. As the target PLLA DP and the hydrophobic weight were systematically varied, the achieved morphology ranged from 2D lamellae (red) to ‘transition state’ wide cylinders (blue) and pure cylinders (green). | ||
Conclusion
We have prepared a series of well-defined PDMA-b-PLLA-b-PDMA triblock copolymers through a combination of RAFT polymerisation and ROP. We have used a simple single component solution-phase methodology37 where we have been able to show shape-controlled CDSA for triblock copolymers through simple dissolution without any heating/cooling cycles.A morphological transition from 2D platelets to 1D cylinders was revealed through detailed investigation of the CDSA process by altering the core/corona block lengths. When the solvophilic corona length is increased, unimers are more soluble, leading to a slower crystallisation process and fully-formed 2D crystalline platelets. Similarly, shorter corona block copolymers are less soluble and thus aggregate before crystallisation, limiting the extent to which the crystalline block can adopt a preferred crystal conformation, forming cylindrical assemblies with few crystal defects. While the trends observed are consistent across both di- and tri-block copolymer morphologies, we propose that the increased hydrophilicity of the triblock copolymer system requires a more hydrophilic solvent to realise assemblies with controlled morphology.
Access to these uniform triblock copolymer organic nanomaterials with different morphologies, without the need of organic solvents and temperature control, greatly simplifies access to the sub-micro biocompatible assemblies which may show promise as a new class of drug carrier for further biomedical application.40,41
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Warwick and China Scholarship Council (W. Y.) for studentship funding. EPSRC are thanked for the award of a DTP studentship (M. I.) and ERC are acknowledged for support to A. P. D. (grant number: 681559) and R. O. R. (grant number: 615142).Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- P. Yang, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Macromolecules, 2016, 49, 6731–6742 CrossRef CAS PubMed.
- B. Fang, A. Walther, A. Wolf, Y. Xu, J. Yuan and A. H. Muller, Angew. Chem., Int. Ed., 2009, 48, 2877–2880 CrossRef CAS PubMed.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS PubMed.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- T. H. Epps III and R. K. O'Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC.
- E. G. Kelley, R. P. Murphy, J. E. Seppala, T. P. Smart, S. D. Hann, M. O. Sullivan and T. H. Epps III, Nat. Commun., 2014, 5, 3599 Search PubMed.
- J. Liu, Z. J. Thompson, H.-J. Sue, F. S. Bates, M. A. Hillmyer, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2010, 43, 7238–7243 CrossRef CAS.
- Y. S. Thio, J. Wu and F. S. Bates, Macromolecules, 2006, 39, 7187–7189 CrossRef CAS.
- H. Wang, X. Wang, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2008, 130, 12921–12930 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- S. E. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- A. Albanese, P. S. Tang and W. C. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- X. S. Wang, M. A. Winnik and I. Manners, Macromolecules, 2002, 35, 9146–9150 CrossRef CAS.
- J. Massey, K. N. Power, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 1998, 120, 9533–9540 CrossRef CAS.
- J. A. Massey, K. Temple, L. Cao, Y. Rharbi, J. Raez, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2000, 122, 11577–11584 CrossRef CAS.
- J. Raez, R. Barjovanu, J. A. Massey, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2000, 39, 3862–3865 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- T. Gadt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef PubMed.
- A. P. Soto, J. B. Gilroy, M. A. Winnik and I. Manners, Angew. Chem., Int. Ed., 2010, 49, 8220–8223 CrossRef PubMed.
- F. He, T. Gadt, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2011, 133, 9095–9103 CrossRef CAS PubMed.
- Z. M. Hudson, D. J. Lunn, M. A. Winnik and I. Manners, Nat. Commun., 2014, 5, 3372–3380 Search PubMed.
- J. Schöbel, M. Karg, D. Rosenbach, G. Krauss, A. Greiner and H. Schmalz, Macromolecules, 2016, 49, 2761–2771 CrossRef.
- H. Zhou, Y. Lu, H. Qiu, G. Guerin, I. Manners and M. A. Winnik, Macromolecules, 2015, 48, 2254–2262 CrossRef CAS.
- H. Qiu, V. A. Du, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2013, 135, 17739–17742 CrossRef CAS PubMed.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013, 46, 9074–9082 CrossRef CAS.
- N. Petzetakis, D. Walker, A. P. Dove and R. K. O'Reilly, Soft Matter, 2012, 8, 7408–7414 RSC.
- A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2014, 5, 1427–1436 RSC.
- L. Sun, A. Pitto-Barry, A. W. Thomas, M. Inam, K. Doncom, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2016, 7, 2337–2341 RSC.
- Z. Li, L. Sun, Y. Zhang, A. P. Dove, R. K. O'Reilly and G. Chen, ACS Macro Lett., 2016, 5, 1059–1064 CrossRef CAS PubMed.
- L. Sun, A. Pitto-Barry, N. Kirby, T. L. Schiller, A. M. Sanchez, M. A. Dyson, J. Sloan, N. R. Wilson, R. K. O'Reilly and A. P. Dove, Nat. Commun., 2014, 5, 5746–5755 CrossRef CAS PubMed.
- H. Schmalz, J. Schmelz, M. Drechsler, J. Yuan, A. Walther, K. Schweimer and A. M. Mihut, Macromolecules, 2008, 41, 3235–3242 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Chem. Commun., 2010, 46, 1091–1093 RSC.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- T. R. Wilks, A. Pitto-Barry, N. Kirby, E. Stulz and R. K. O'Reilly, Chem. Commun., 2014, 50, 1338–1340 RSC.
- M. Inam, G. Cambridge, A. Pitto-Barry, Z. P. L. Laker, N. R. Wilson, R. T. Mathers, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2017, 8, 4223–4230 RSC.
- G. Rizis, T. G. van de Ven and A. Eisenberg, Angew. Chem., Int. Ed., 2014, 53, 9000–9003 CrossRef CAS PubMed.
- M.-S. Hsiao, S. F. M. Yusoff, M. A. Winnik and I. Manners, Macromolecules, 2014, 47, 2361–2372 CrossRef CAS.
- M. Ding, J. Li, H. Tan and Q. Fu, Soft Matter, 2012, 8, 5414–5428 RSC.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01056g |
| This journal is © The Royal Society of Chemistry 2017 |