
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring degree of esterification and branching of poly(glycerol sebacate) by energy efficient microwave irradiation†
Chi Ching
Lau
 a,
Mustafa Kemal
Bayazit
a,
Jonathan Campbell
Knowles
bc and
Junwang
Tang
*a
a,
Mustafa Kemal
Bayazit
a,
Jonathan Campbell
Knowles
bc and
Junwang
Tang
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK. E-mail: junwang.tang@ucl.ac.uk
bDivision of Biomaterials and Tissue Engineering, UCL Eastman Dental Institute, 256 Gray's Inn Road, London, WC1X 8LD, UK
cDepartment of Nanobiomedical Science & BK21 Plus NBM Global Research Center for Regenerative Medicine, Dankook University, Cheonan 330-714, Republic of Korea
First published on 12th June 2017
Abstract
Poly(glycerol sebacate) (PGS) is known as an exciting biomaterial owing to its tunable mechanical properties and controllable degradation rate. However, it is always challenging to control these properties. In this study, we have proposed a solvent-based system to provide a better control of reaction temperature in a microwave cavity, which can minimize evaporation of monomers, and water was collected to analyse the degree of esterification. Pre-PGSs with varied degrees of esterification were prepared using both single mode and multimode microwave cavity irradiation (MI) in this solvent-based reaction system. For a similar degree of esterification of pre-PGSs, the reaction time was almost halved with a better control on mechanical properties by single mode MI compared to multimode MI. Furthermore, the single mode MI approach was compared with the conventional heating (CH) approach. The mechanical properties and degradation rate of PGSs can be controlled readily by using the single mode MI approach compared to CH, which are crucial for their application as a biomaterial. It has been found that the single mode MI not only accelerates the pre-polymerisation process rate by six times, but also speeds up the curing time to the same extent. The Young's modulus of PGSs prepared by single mode MI is increased from 0.77 to 3.14 MPa when the degree of esterification is 66.82%, which is 50% higher than that reported in the literature. Furthermore, PGS using a highly branched pre-PGS prepared by the single mode MI method has a large degree of flexibility. It can achieve a much higher Young's modulus than that obtained by CH with a short curing time (<10 hours). In addition, the residual mass of PGSs prepared by single mode MI is varied from 78.54% to 92.96% compared to the CH method that ranges from 84.24% to 93.31%. Thus, these highly branched PGSs produced by single mode MI also show a wider degradation window (approximately 59% higher degree of flexibility than the CH method), which is found to be highly dependent on the degree of esterification and curing time of the pre-polymer, and controlled by branching.
Introduction
Poly(glycerol sebacate) (PGS) has been studied intensively after the first report by Langer's group in 2002 due to its unique properties.1 For instance, its rapid degradation kinetics is compatible with the healing rate of tissues (i.e. 6–12 weeks), a modulated degradation rate is favorable for a drug carrier, and tailored mechanical properties are suitable for a nerve guide material.2–4 The cytotoxicity of a biopolymer is always a concern before any clinical application, but both reactants of PGS are endogenous monomers that can be naturally found in human metabolites; glycerol is involved in the synthesis of phospholipids and sebacic acid is a metabolite in fatty acid oxidation.5,6 From the previous studies, PGS that showed less inflammation is favourable in tissue engineering than poly(lactide-co-glycolide), a currently widely utilized biomaterial.4 In addition, the cytotoxicity of PGS can be further reduced by adding a filler into the system. The percentage of dead cells decreased significantly as the crosslink level of PGS increased (e.g. by adding Bioglass).7,8 Furthermore, a highly toughened PGS showed excellent cytocompatibility compared to a soft PGS.9 This is probably due to the rapid degradation of the soft PGS which induces an acidic and cellular toxicity environment. In contrast, controllable degradation of a highly crosslinked PGS maintains a low concentration of potentially toxic degraded sebacate in the medium,2,9,10 which is highly preferable but very challenging.The bio-application of PGS is thus widely known to be determined by its physical properties (e.g. degree of crosslinking and degradation rate) that can be manipulated by a synthesis approach. Synthesis of PGS by a conventional approach is very time and energy consuming, which normally requires pre-polymerisation under inert gas conditions to prevent oxidation of reactants for 24 h and curing under vacuum conditions for 48 h.1 Microwave irradiation (MI) has recently shown a great potential to replace conventional heating (CH), either for organic or inorganic synthesis.11,12 It offers homogeneous and fast heating due to volumetric and selective interaction between the microwave and polar molecules, often leading to better crystallinity of the prepared materials. Furthermore, MI with remarkably increased reaction rates and high reproducibility can readily tune the product selectivity and yields.13–15 There are a few reports discussing the effect of microwave irradiation on the pre-polymerization of PGS,16,17 in which a household multimode microwave oven was used, which may result in an ineffective control of the temperature rise and reaction rate. An uncontrolled temperature rise could both trigger the evaporation of the monomer (e.g. glycerol due to a low boiling point of 290 °C) and change the stoichiometric ratio of the reactants that potentially altered the physical and chemical properties of the resulting PGSs.18 In addition, multimode MI provides a much weaker energy density than single mode MI,19 which can influence the degree of crosslinking of a polymer.
Given such attractive potentials of a single mode MI with a controllable reaction rate to biopolymer synthesis, to the best of our knowledge there is no report studying PGS synthesis using a single mode MI approach, benefiting from both well-controlled heating power and temperature rise. In this study, finely-tuned polyesterification of sebacic acid and glycerol to prepare PGS by single mode MI was carried out. To avoid change of the reactants’ ratio during MI synthesis, we carried out the reactions in a solvent (i.e. toluene with a lower boiling point of 110 °C) to protect the evaporation of glycerol (boiling point at 290 °C) and further set the maximum reaction temperature at 130 °C. All these have ensured that all precursors remained in the reaction medium throughout the microwave irradiation with a well-controlled reaction temperature. The degree of esterification (DE) was thoroughly investigated in order to control mechanical properties and degradation rates of PGS. For comparison, PGS synthesis was also conducted using CH. The mechanism underlying PGS formation by MI was also discussed.
Experimental
General procedure for synthesis of poly(glycerol sebacate)
Equimolar glycerol (99%, Sigma Aldrich) and sebacic acid (>99% Sigma Aldrich) were measured and pre-mixed in a round bottom flask before adding dry toluene (30 mL) into the mixture. Concentrated H2SO4 (3 μL) was then added into the mixture. The reactor was connected to a Drechsel bottle for water collection. This mixture was then ramped under nitrogen gas in a dynamic mode (150 W) in the CEM Discover SP system (single mode) for 3 minutes (one cycle) with a temperature limit of 130 °C. It was then cooled to room temperature. The cycle was repeated to get 12 and 27 minute samples. The abovementioned experimental procedures were repeated using a CEM MARS system (multimode microwave) under similar microwave conditions (150 W, 3 min per cycle). The cycle was repeated to get a total preparation time of 21 minutes. Condensed water was collected for calculating the degree of esterification. The pre-PGSs were purified by removing the unreacted glycerol and then were dried using the pump and left in the fumehood overnight at room temperature. The dried prepolymer was then cured in a vacuum oven at 120 °C for 2–48 h. PGS samples were then cooled to room temperature under vacuum conditions. For the control experiments, the pre-polymerisation step was carried out by the conventional heating method. The reactor was connected to Dean–Stark apparatus for water collection. Similar to the previous method, water was collected while toluene was used as a solvent. The curing procedure remained unchanged.Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dilution with a matrix (α-cyano-4-hydroxy-cinnamic acid) in water–acetonitrile (2:8, v/v) and 0.5% formic acid. 3 μL of the resulting sample was deposited onto the MALDI target plate and allowed to dry. Samples were analysed using a Waters MALDI micro MX (Waters, UK) with a nitrogen laser in the reflectron mode using delayed extraction (500 ns) and an accelerating voltage of 120 V, pulse 2500, and detector 2000. The simulated isotopic peak patterns were constructed using online software MoIE- Molecular Mass Calculator v2.02 (http://mods.rna.albany.edu/masspec/MoIE).
:1 dilution with a matrix (α-cyano-4-hydroxy-cinnamic acid) in water–acetonitrile (2:8, v/v) and 0.5% formic acid. 3 μL of the resulting sample was deposited onto the MALDI target plate and allowed to dry. Samples were analysed using a Waters MALDI micro MX (Waters, UK) with a nitrogen laser in the reflectron mode using delayed extraction (500 ns) and an accelerating voltage of 120 V, pulse 2500, and detector 2000. The simulated isotopic peak patterns were constructed using online software MoIE- Molecular Mass Calculator v2.02 (http://mods.rna.albany.edu/masspec/MoIE).
Degradation study
For each sample, five or more polymer specimens were cut into dimensions of 5 × 5 mm2 before storing in the standard phosphate buffer saline (PBS, 1×) at 37 °C for 28 days. The initial weight of the PGS samples was recorded before storing in the PBS solution. During the degradation test, the PBS solution was refreshed daily. At pre-set times (after 7, 14, 21, and 28 days), these degraded specimens were dried in a drying oven at 60 °C for 12 h and the residual mass of the specimens was recorded.Results and discussion
Synthesis of a poly(glycerol sebacate) pre-polymer (pre-PGS) using single mode and multimode microwave irradiation (MI)
Pre-PGSs have been prepared by using a multimode domestic microwave,16,17 thus the influence of single mode and multimode microwave irradiation (MI) on pre-PGSs synthesized was first studied here. Single mode MI was found to be more energy efficient than multimode MI to synthesize a viscous pre-PGS with a similar degree of esterification. For instance, 12 min of microwave treatment is needed to synthesize the pre-PGS with ca. 70% DE by single mode MI, which was not achieved until 21 min of irradiation using multimode MI, while the pre-PGSs prepared by both methods showed a characteristic ester linkage (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond) at 1734 cm−1 and 1730 cm−1, respectively (see ESI Fig. S1†). The rapid reaction time is attributed to the high intensity in a single mode microwave.19,20 Other than the reaction time, the mechanical properties of the crosslinked PGSs, obtained by 8 h curing of pre-PGSs synthesized by both microwave methods, were also studied (see Fig. 1). Based on Fig. 1a, the Young's modulus of the PGSs synthesized by single mode MI (2.60 ± 0.34 MPa) is smaller than the PGSs synthesized by multimode MI (3.31 ± 0.30 MPa), while the ultimate tensile strength of the PGSs by single mode MI is somewhat higher than that by multimode MI (approximately 0.46 ± 0.06 MPa for single mode MI and 0.41 ± 0.15 MPa for multimode MI – see ESI, S2†). The elongation at break for PGSs is 27.73 ± 1.87% and 17.00 ± 7.77%, for single mode and multimode MI, respectively (Fig. 1b). In short, PGS synthesized by single mode MI has 20% smaller Young's modulus than that prepared by multimode MI. This can be readily increased by increasing microwave power or reaction time. Simultaneously elongation at break can also be tuned, therefore the degree of flexibility to tune PGS properties can be easily controlled by single mode MI. In addition, PGSs fabricated by single mode MI have a slightly higher ultimate tensile strength. Owing to the potential to tune PGS properties with a higher degree of flexibility, a faster reaction rate and higher energy efficiency, the experimental conditions were then optimised using single mode MI and further compared with CH.
O bond) at 1734 cm−1 and 1730 cm−1, respectively (see ESI Fig. S1†). The rapid reaction time is attributed to the high intensity in a single mode microwave.19,20 Other than the reaction time, the mechanical properties of the crosslinked PGSs, obtained by 8 h curing of pre-PGSs synthesized by both microwave methods, were also studied (see Fig. 1). Based on Fig. 1a, the Young's modulus of the PGSs synthesized by single mode MI (2.60 ± 0.34 MPa) is smaller than the PGSs synthesized by multimode MI (3.31 ± 0.30 MPa), while the ultimate tensile strength of the PGSs by single mode MI is somewhat higher than that by multimode MI (approximately 0.46 ± 0.06 MPa for single mode MI and 0.41 ± 0.15 MPa for multimode MI – see ESI, S2†). The elongation at break for PGSs is 27.73 ± 1.87% and 17.00 ± 7.77%, for single mode and multimode MI, respectively (Fig. 1b). In short, PGS synthesized by single mode MI has 20% smaller Young's modulus than that prepared by multimode MI. This can be readily increased by increasing microwave power or reaction time. Simultaneously elongation at break can also be tuned, therefore the degree of flexibility to tune PGS properties can be easily controlled by single mode MI. In addition, PGSs fabricated by single mode MI have a slightly higher ultimate tensile strength. Owing to the potential to tune PGS properties with a higher degree of flexibility, a faster reaction rate and higher energy efficiency, the experimental conditions were then optimised using single mode MI and further compared with CH.
 | ||
| Fig. 1 (a) Young's modulus and (b) elongation at break of PGSs prepared by 12 min single mode MI and 21 min multimode MI, followed by 8 h curing. | ||
Preparation of poly(glycerol sebacate) pre-polymer (pre-PGS) using single mode MI and CH
A two-step preparation procedure was applied to synthesise all PGSs. The first step was pre-polymerisation under MI or CH conditions and the second was curing of pre-PGSs in a vacuum oven. In a typical procedure, a mixture of sebacic acid and glycerol in dry toluene was heated to reflux temperature to prepare pre-PGSs and the condensed water was collected as a measure of degree of esterification (DE) – as illustrated in ESI Fig. S3.†21 In this mixture, glycerol interacts much stronger with the microwave than others. This is because the loss tangent of glycerol (tanσ = 0.651) is higher than toluene (tanσ = 0.040) due to the multi-hydroxide groups attached, and the long chain of polar molecules (i.e. sebacic acid) always has a weaker interaction than a short chain with a microwave.22,23 The pre-PGSs were then purified by only removing unreacted glycerol and further characterised.
Pre-PGSs, prepared by both methods, with different DEs are listed in Table 1. One can see that MI provides at least a six-fold faster reaction rate than the CH method by comparing the amount of water collected. According to the water collection profile (see ESI Fig. S3†), MI almost starts the esterification process when it is turned on. However, it takes more than 30 minutes to collect water droplets by CH. MI (dielectric heating) speeds up the reaction rate significantly by generating the heat homogeneously in bulk solution via dipole rotation where the polar species (e.g. glycerol molecules) align themselves with a rapidly changing electrical field of the microwave such that the reactants could be activated selectively. More importantly, MI provides the heat internally and tends to eliminate the ‘thermal wall effect’.24,25 Hence, the condensed water molecules are evaporated faster in the microwave due to the large dielectric loss of water molecules, which further shifts the reaction towards polymer formation.26–28
| Methods | Heating time (min) | Volume of water (mL) | DE (%) |
|---|---|---|---|
| MI | 3 | 0.40 | 18.18 |
| 12 | 1.47 | 66.82 | |
| 27 | 1.95 | 88.64 | |
| CH | 50 | 0.90 | 40.91 |
| 77 | 1.50 | 68.18 | |
| 166 | 2.00 | 90.91 |
DE can be used as a measure of the polymerisation degree and more importantly for predicting the unreacted alcohol groups after the pre-polymerisation process. In this study, the DE is calculated using eqn (1) stated below:
 | (1) |
The results of DE are shown in Table 1. We also observed that pre-PGSs prepared by the MI method with 18.18% DE and 66.82% DE were in wax and viscous liquid forms, respectively, comparable with the previous reports.17 On the other hand, the DE is increased by a longer reaction time in both heating methods, however, this increment is drastic by MI. For instance, a minimum six-fold increase in the reaction rate can be obtained by MI compared to by CH (ca. at similar DE 70%).
ATR-FTIR was used to confirm the formation of ester functional groups (Fig. 2). A sharp peak is found at 1730 cm−1, which corresponds to the carbonyl (CO) stretching mode of the ester linkage, and the bands appearing at 2924, 2829 and 2824 cm−1 are attributed to the C–H stretching of the polymer backbone.16,17 The relative intensity of characteristic ester carbonyl increases as the reaction time increases, suggesting a high degree of polymerisation.17 The peak at 1689 cm−1 is referred to the carbonyl stretching of the remaining free sebacic acid after pre-polymer formation. The pre-PGSs prepared by CH and MI show a broad peak around 3448 cm−1 and 3400–3469 cm−1, respectively, attributed to hydroxyl (alcohol group) stretching of the pre-polymer. As the MI time increases, the right shift of the alcohol group is observed. The precursor glycerol contains two alcohol groups. One is the primary alcohol group and the other is the secondary alcohol group, which has a mixed peak around 3290 cm−1 (ESI Fig. S4†). The primary alcohol group has a higher frequency than the secondary alcohol group.29 The observed continuous right shift may indicate the pre-PGS prepared by MI contains more primary than secondary alcohol groups. In other words, the MI approach uses more secondary alcohol groups than the CH approach.
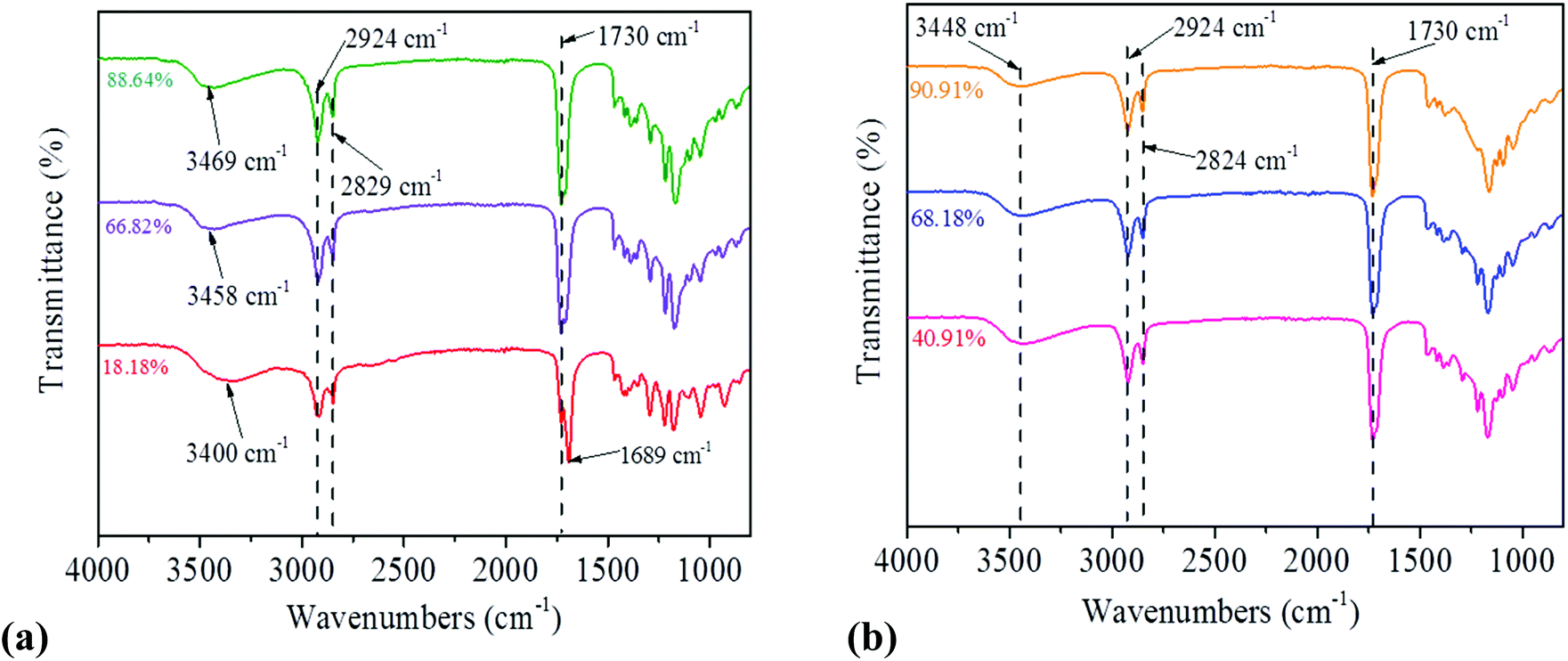 | ||
| Fig. 2 ATR-FTIR spectra of pre-PGSs prepared at different DEs by (a) MI and (b) CH methods. These pre-polymers showed a strong signal of an ester bond (CO) at 1730 cm−1 and an hydroxyl bond (–OH) around 3400–3469 cm−1 when using the MI method. Similarly, the pre-polymer that was prepared via CH also showed a CO signal at 1730 cm−1 and a –OH signal at 3448 cm−1. | ||
Based on the obtained IR spectra, the highly branched pre-PGS structure by the MI approach is proposed as depicted in Fig. 3. MI interacts strongly with glycerol as mentioned above, leading to the activation of both alcohol groups in glycerol, which react more efficiently with sebacic acid than that in the CH approach. In order to justify this hypothesis, the obtained pre-polymers were further characterized by MALDI-TOF and 1H NMR. 1H NMR spectra of the obtained pre-PGSs, synthesised using both heating methods, were recorded using acetone-d6 (δ 2.09 ppm) as the deuterated solvent. As depicted in Scheme 1, 5 different sets of protons (Ha–e) can be assigned to the repeating units of pre-PGSs.30
 | ||
| Fig. 3 Proposed scheme for the possible structure of the pre-PGS that was prepolymerised via MI and CH methods, and the crosslinked PGS after the curing process. The dotted line in the figure indicates the cross-linking. | ||
 | ||
| Scheme 1 Polycondensation of PGS using equimolar glycerol and sebacic acid where R refers to hydrogen or a branched chain. The red circled region is referred to the secondary alcohol group of glycerol. | ||
1H NMR spectra of the pre-PGSs, prepared by MI (66.82% DE) and CH (68.18% DE), display the chemical shifts at δ 1.32 ppm (Ha), δ 1.59 ppm (Hb), and δ 2.32 ppm (Hc), which are attributed to the –CO–CH2–CH2–CH2– group in the pre-polymer from the precursor sebacic chain (Fig. 4a and b).31,32 The additional peaks at δ 3.50–5.50 ppm identified in the spectrum are ascribed to the Hd and He in the PGS molecular chain from glycerol.33 All these assigned chemical shifts are also presented in the 1H NMR spectra of the pre-PGSs with a low DE (<41%) by MI and CH methods (ESI Fig. S5†). Consistently, 13C NMR analysis of these samples shows signals: 25–34 ppm for the sebacate methylene carbon, 60–75 ppm for the methylene and methine carbons of glycerol and glyceride units, and 172–174 ppm for the signals of carboxylic acids and esters (ESI Fig. S6†).34
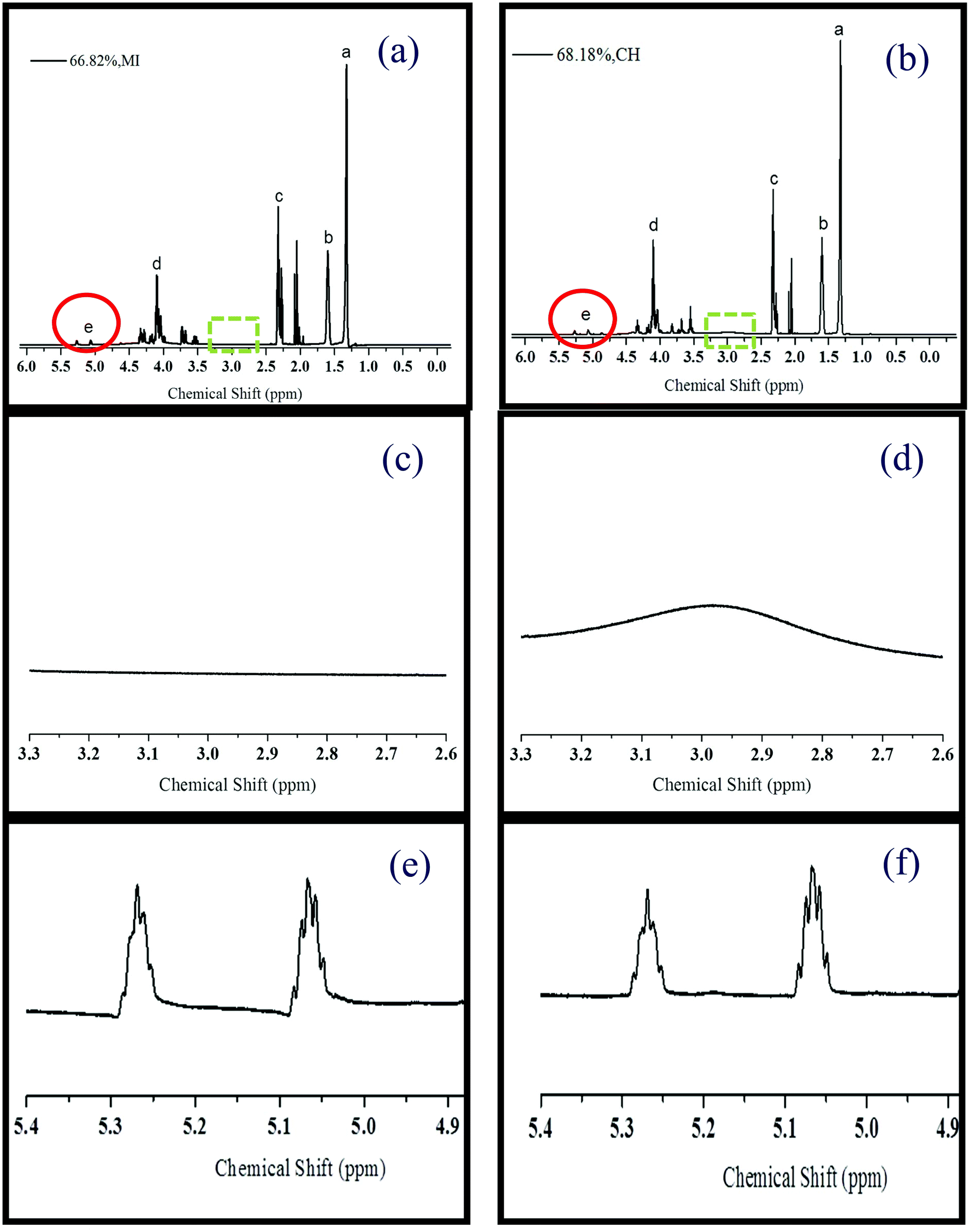 | ||
| Fig. 4 1H NMR spectra, in acetone-d6, of the pre-polymer with (a) 66.82% (MI) and (b) 68.18% (CH). The sebacic chain peak in the pre-PGS is illustrated at δ 1.32 ppm (Ha), δ 1.59 ppm (Hb), δ 2.32 ppm (Hc) while Hd and He attributed to the glycerol chain in the pre-polymer. The 1H NMR spectra demonstrate the typical molecular structure of pre-PGS where the additional peak at 2.09 ppm is the solvent acetone-d6 peak. The next two figures refer to the region of δ 2.65–3.30 ppm for (c) 66.82% and (d) 68.18% of the pre-PGS that was synthesized by MI and CH, respectively. The significant peak in (d) suggests the excess of the secondary hydroxyl group (He) of glycerol existing in the pre-polymer prepared by the CH method. The last two figures illustrate the region at δ 4.90–5.40 ppm for the pre-PGS with (e) 66.82% – MI and (f) 68.18% – CH. These two peaks are assigned to He, which corresponded to 1,2-diacylglyceride (δ 5.07 ppm) and 1,2,3-triacylglyceride (δ 5.27 ppm). | ||
More importantly, a significant broad signal is observed at δ 2.65–3.30 ppm in the 1H NMR spectrum of the pre-PGS (68.18% DE) prepared using the CH method (Fig. 4d). This signal is assigned to the secondary hydroxyl groups of glycerol (He in Scheme 1), which is in agreement with the previous reports.35 In contrast, no such signal is observed in the 1H NMR spectrum of the pre-PGS (66.82% DE) prepared using MI (Fig. 4c). The presence of excessive secondary alcohol groups in the pre-PGS prepared by CH suggests that the CH method produces linear polymer chains, whereas MI facilitates branching by selectively activating all alcohol groups as indicated in Fig. 3, which is crucial to control the properties of the final polymer.
A further study was carried out to understand MI induced branching by observing the 1H NMR between δ 4.9 and δ 5.3 ppm, which corresponded to He and more specifically referred to 1,2,3-triacylglyceride (δ 5.27 ppm) as illustrated in Fig. 4e and f.34 Relative signal intensity ratios can be used to understand the changes in the presence of secondary alcohol groups of glycerol. The relative signal intensity ratio of Hb or Hc to Ha is a constant (ca. 0.5). This is because these protons are not involved in the esterification process. When reaching ca. 70% DE, this relative ratio of He/Ha is 15% higher under MI (0.0132) than that under CH conditions (0.0115). More interestingly, the relative ratios of He/Ha corresponding to 1,2,3-triacylglyceride (δ 5.27 ppm) are 0.012 and 0.010 for MI (18.18% DE) and CH (40.91% DE), respectively. Higher the relative He/Ha ratio at δ 5.27 ppm, higher the esterification of secondary alcohol groups of glycerol. This indicates that a higher branched pre-polymer was achieved by the MI method.
Other than 1H NMR spectra, the MALDI-TOF mass spectra, recorded from 500 to 5000 m/z, of the pre-PGSs synthesized by both methods were also used to verify the formation of oligomeric structures (ESI Fig. S7†). The profile of experimental spectra of the samples prepared by either MI or CH is consistent with the theoretical spectra (Fig. 5), suggesting the validity of modelling. However, the pre-PGS prepared by MI (DE = 18.18% or DE = 66.82%) shows a monoisotopic mass at ca. 2360 m/z (Fig. 5a and ESI Fig. S8†), appearing to be shifted by 4 m/z when compared with the theoretical monoisotopic mass at 2364 m/z ([C117H200O46Na]+) which is calculated using a likely linear PGS with 9 repeating units. The shift (4 m/z) is thought to be most likely due to the esterification reaction of secondary alcohol groups of glycerol to produce branched PGSs, which is in agreement with the NMR spectra. However, the pre-PGSs prepared by CH (DE = 68.18%, Fig. 5b) show a monoisotopic mass at ca. 1405 m/z ([C68H118O28Na]+, consisting of a linear PGS with 6 glycerol and 5 sebacic acid units) which is very close to the theoretical monoisotopic mass at ca. 1406 m/z ([C68H118O28Na]+) (Fig. 5b – both theoretical and experimental monoisotopic patterns). This comparable monoisotopic mass also suggests that MI produces different prepolymer structures than CH. Similar results can be obtained when DE is equal to 40.91% (ESI Fig. S9†).
 | ||
| Fig. 5 (a) MALDI-TOF spectra of the pre-PGS (18.18% DE-MI) show the maximum detected oligomer mass at 2360 m/z. These spectra are slightly left shifted when comparing to the theoretical MALDI spectra, GSGSGSGSGSGSGSGSGS [C117H200O46Na]+.(b) MALDI-TOF spectra of pre-PGS (68.18% DE-CH) depict the maximum oligomer mass at 1405 m/z, which is well-fitted with the theoretical spectra created by the software, i.e. GSGSGSGSGSG, [C68H118O28Na]+. | ||
Previous studies reported that a range of possible oligomers could only be produced from the reactants, glycerol and sebacic acid, when tuning their ratios.36 Surprisingly, different PGS structures (e.g. branched/heavily crosslinked or linear) can be obtained by only changing the heating method while maintaining a constant stoichiometric ratio of glycerol to sebacic acid (1:1). Overall results obtained from NMR and MALDI-TOF spectra suggest that the microwave promotes the formation of branching oligomers in pre-PGS (a detailed structure is presented in Fig. 3). Naturally, the primary alcohol group is likely to react with the carboxylic acid group to generate polymer linear chains, while the secondary alcohol group facilitates the branching. The microwave can produce a polymer with a higher molecular weight,37 thus, it very likely activated both secondary and primary alcohol groups simultaneously due to their similar polarity which couples well with MI. Therefore, at low DE, MI prefers branching in different directions while CH is likely to form a shorter chain prior to the curing process.
Morphology of crosslinked PGS samples
After curing the pre-PGS in a vacuum oven, a significant reduction in the intensity of the hydroxyl band is observed as shown in ESI Fig. S10.† In order to investigate the effect of the heating method on the surface morphology, SEM images of both appearance and cross section were obtained (ESI Fig. S11†). For the internal structure analysis, the specimens were fractured in liquid nitrogen instead of cutting with a blade to prevent smearing. The SEM images show a smooth topography for the surface of either PGS samples and MI does not alter the morphology of PGS since all the crosslinked specimens show similar morphology. This morphology also suggests that the pre-PGS samples are free of the solvent (toluene) due to a complete removal of solvent before the curing process, which is very crucial prior to their use in bio-applications (also justified by the 1H NMR results, Fig. 4).Optimization of curing time and its mechanical properties
To further discover the effect of the branched pre-PGS on curing time and mechanical properties, we used a similar DE (ca. 70% DE) of the pre-PGS and further cured in a vacuum oven for 2–48 h. By reducing the curing time to 2 h, PGS specimens prepared by MI (DE = 66.82%) show faster toughening compared with CH samples (DE = 68.18%) – can be seen in ESI Fig. S12.† The PGS samples cured with a shorter time were also characterised using IR spectra where the ester linkage is found with a reduction of the hydroxyl bond from time to time – as seen in ESI Fig. S12.† This may be due to the branched pre-PGS samples that were produced by MI requiring shorter curing time to form a stiff polymer. It can be concluded that higher branching of the pre-PGS achieved by MI facilitates the formation of a crosslinked PGS in a very short curing time.Surprisingly, the Young's modulus of the specimens (after 48 h curing) fabricated in this study is at least 50% higher without changing the molar ratio of glycerol and sebacic acid as compared to the previous results.16,17,38,39 This is probably due to our modified synthetic procedure in which the unreacted glycerol was removed from the bulk pre-PGS prior to the curing process. Consequently, the remaining sebacic acid probably only functions as a crosslinking agent and thus produces a stiffer polymer. This novel preparation approach proposes that the PGS with a broad range of stiffness can be produced without an additional crosslinking agent, but by simply adjusting the degree of esterification and curing time. This highly toughened PGS may also improve its cytocompatibility.9
The mechanical properties of the crosslinked PGS (Fig. 6) prepared by both methods are clearly different. As the curing time increases, the Young's modulus of these specimens are increased but at a different step. The Young's modulus of PGSs by MI after 8 h curing is equivalent to that achieved in 48 h by CH, thus MI reduces the curing time by a factor of six, similar to results observed in the pre-polymerisation process. The Young's modulus of PGSs by MI after 8 h curing is increased from 0.77 ± 0.19 to 2.60 ± 0.34 MPa (a window of 1.83 MPa) and elongation at break is reduced from 53.41 ± 7.14 to 27.73 ± 1.87%. On the other hand, PGSs synthesised by CH at a similar curing time have smaller Young's moduli, ranging from 0.57 ± 0.06 MPa to 1.01 ± 0.21 MPa (a window of 0.44 MPa) and elongation at break is from 64.07 ± 10.71 to 48.72 ± 6.51%. The ultimate tensile strength of these PGS samples fabricated is depicted in ESI Fig. S13† from 0.26 ± 0.07 to 0.46 ± 0.06 MPa for MI samples and from 0.22 ± 0.03 to 0.35 ± 0.08 MPa for CH samples. The Young's modulus of these PGS specimens after 48 h of curing time is comparable (i.e. 3.14 ± 0.28 MPa and 2.87± 0.17 MPa for MI and CH, respectively).
 | ||
| Fig. 6 Young's modulus (MPa) and elongation at break of the crosslinked PGS (ca. 70% DE) prepared by (a, b) MI and (c, d) CH methods, respectively. The pre-PGS samples were pre-polymerised at a predetermined degree of esterification before curing for 2–48 h in a vacuum oven. | ||
As stated in previous studies, the extent of crosslinking affects the properties of a polymer significantly.40–42 A weak and soft polymer is produced at a low crosslink density. In contrast, a stronger and stiffer polymer can be formed at a higher crosslink density. These polymers would have different bio-applications. In this study, MI speeds up the curing process significantly and the CH method can produce PGSs with similar mechanical properties only if the curing time is long enough. Therefore, the Young's modulus of PGSs prepared by MI can be tuned easily, where a larger range of Young's moduli can be obtained (i.e. three times wider) with a short curing time compared to CH. This is because a higher degree of branching pre-PGS is synthesised by MI rather than a linear pre-PGS by CH.
Degradation of PGS
Degradation of PGS is a hydrolytic process where the ester bonds in the polymer chains react with water molecules, resulting in shorter chains via the surface erosion process. The degradation occurs within a region near the surface and provides a linear change in mechanical performance, because the surface erosion occurs much earlier than in the bulk of the sample.43 According to Wu et al., the initial porosity and pore size affected the degradation rate due to a variation of diffusion rate.44 However, PGS samples produced herein have similar morphology (see SEM images, ESI Fig. S11†), so it is not a dominating factor for the degradation rate of PGSs in our study.The surface erosion mechanism undergone by PGS is ideal for many drug delivery applications due to a slow water permeation rate and water-vulnerable drugs can be protected.45 Thus, the degradation properties of PGS specimens were tested in phosphate buffered saline (PBS) solution. Firstly, the PGS specimens with a different degree of esterification (ca. 18–90%) but with the same curing time of 24 h were investigated (Fig. 7). The degradation rate of PGS samples prepared by both methods decreases fast initially and then reaches a linear mass loss. With an increment in DE, the degradation rates of all samples are decreased. The residual mass of the PGS specimens (ca. 18.18% DE) after 28 days is 83.88 ± 2.02% (16.12% degraded) and the residual mass of PGS with 66.82% DE is 90.96 ± 0.96% (9.04% degraded). For the CH method, the residual masses of PGS samples are 91.89 ± 0.32% for 40.81% DE and 92.56 ± 0.24% for 68.18% DE, where these samples degrade less than 10%. When reaching ca. 90% DE, the PGS samples degrade slowest (only 6.20–7.42% degraded) where the residual masses are 92.58 ± 0.40% and 93.80 ± 0.40% for MI and CH, respectively.
 | ||
| Fig. 7 Degradation profile of the PGS [pre-polymerised by (a) MI and (b) CH methods and then cured for 24 h] in phosphate buffered solution. | ||
Secondly, the PGS samples with a similar DE (ca. 70%) but different curing times were also tested. As illustrated in Fig. 8, PGS specimens prepared by MI show a broader range of degradation profiles compared to CH samples. After a long curing time (≥8 h), the MI samples show a similar degradation profile to the CH samples where both degrade to approximately 87% (ca. 13% mass loss). Interestingly, when reducing the curing time to 4 and 6 h, the degradation rate of the sample prepared by MI changes significantly while only a small effect is observed on the CH samples. For instance, after 4 h of curing time, the residual mass of the MI sample is 78.54 ± 4.76% (21.46% degraded) compared to 85.37 ± 0.58% of the CH sample (14.63% degraded). On the other hand, by further increasing the curing time to 24–48 h, the PGS samples prepared by both methods show a slow and comparable degradation rate while the residual mass is above 90% – refer to ESI Fig. S14.†
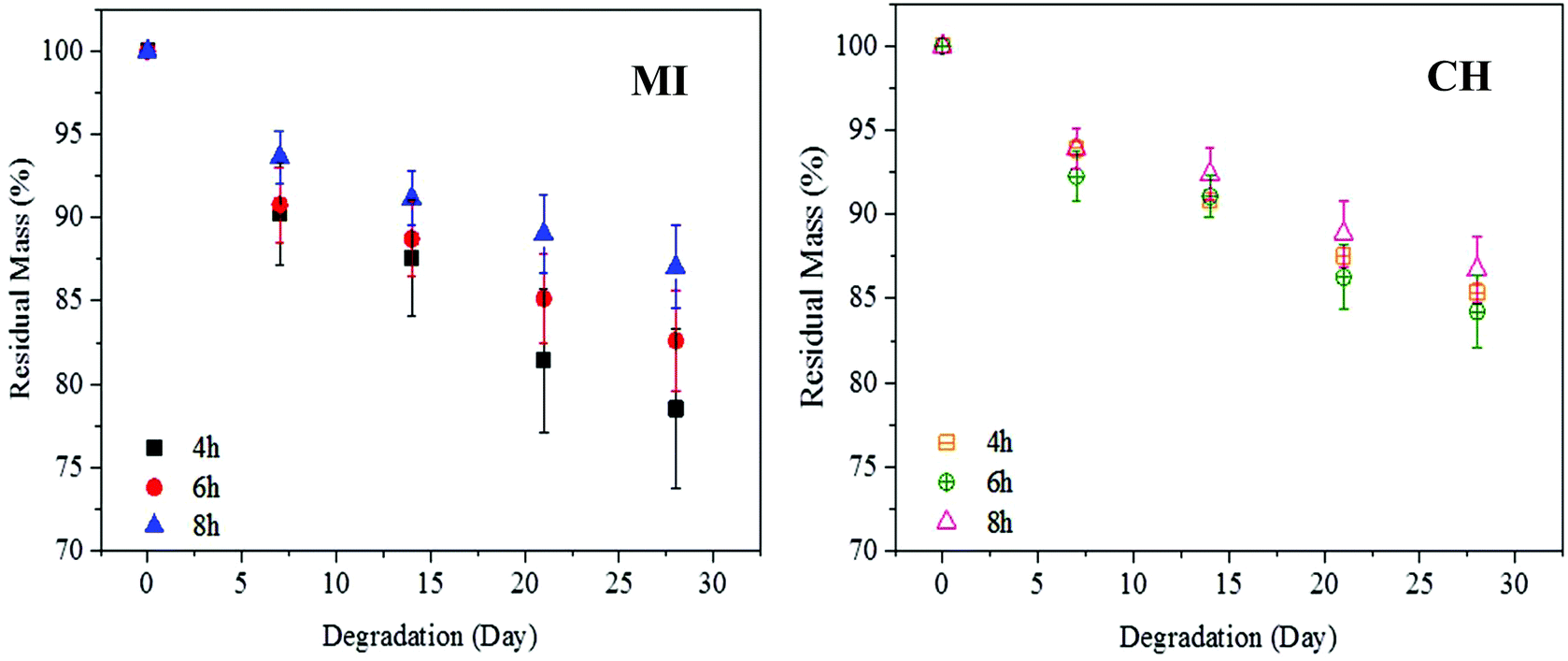 | ||
| Fig. 8 Degradation rate of the crosslinked PGS at 70% DE (pre-polymerised by MI and CH methods and then cured for 4–8 h) in phosphate buffered solution. | ||
Based on these degradation results, it can be found that the branched structure of PGS samples prepared by the MI method provides a wider degradation rate window compared to the PGS samples prepared by CH with similar DE. In other words, the polymers prepared by MI show a degradation rate ranging from 78.54% to 92.96% (a window of 14.42%), but those by CH ranging from 84.24% to 93.31% (a window of 9.07%). Thus, MI provides an approximately 59% wider degradation window compared to CH. The large and controllable degradation rates enable a more flexible drug delivery. A branched structure can give rise to a rapid hydrolytic cleavage due to more terminals which are attacked by water during degradation.46 In both methods, the increment in curing time or DE generally decreases the mass loss of PGS samples. This is because pre-polymerisation and the curing process both increase the number of ester bonds needed to be broken via surface erosion. Therefore, PGS with lower DE or shorter curing time degrades faster due to less number of ester bonds to be cleaved. On the other hand, the PGS prepared by both methods at a similar DE but a longer curing time (>8 h) shows a similar degradation rate (residual mass >90%). This is due to the fact that DE could reach a saturation point. As a result, the number of ester bonds to be broken is comparable. Therefore, the degradation rate does not alter much at this stage.
Conclusions
The pre-PGSs have been prepared by single mode MI in comparison with multimode MI and CH approaches. The reaction temperature has also been monitored and controlled with a solvent with a low boiling point to improve reproducibility and reduce the evaporation of monomers which is neglected in previous studies using multimode MI.16,17 It also offers good control over temperature, homogeneous temperature medium and an ease of manipulating the degree of esterification. In addition, the degree of esterification of pre-PGS was finely tailored by collecting the condensed water, which is crucial to control the mechanical properties and degradation rate.The single mode MI has been proven to be more energy efficient than multimode MI with a high degree of flexibility to tune PGSs Young's modulus and elongation at break. When comparing single mode MI with the CH method, single mode MI dramatically speeds up both pre-polymerisation and curing processes, e.g. by a factor of six, due to a fast and selective heating mechanism. The degree of esterification of pre-PGS can also be easily controlled by single mode MI which strongly affects the polymer properties (e.g. degradation rate and mechanical strength). Besides, the highly branched pre-PGSs are fabricated (proven by NMR and MALDI-TOF spectra) using the single mode MI method without changing the molar ratio of glycerol and sebacic acid. In addition, by using this proposed strategy, the Young's modulus of PGS (from 0.77 ± 0.19 to 3.14 ± 0.28 MPa) increases by 50% when compared to the reported literature.16,17,38,39 Moreover, the PGS samples prepared by single mode MI also reduce the curing time where a stiffer polymer is produced after 2 h curing in a vacuum oven. In addition, pre-polymer samples (DE = 66.82%) of the single mode MI method cured for only 8 h show a Young's modulus comparable to the one prepared by the CH method and cured for 48 h. For the degradation test, the degree of esterification and curing time affect the degradation rate significantly. This is because these two factors strongly control the degradation rate of PGSs due to the increment of ester linkage that needs to be cleaved. The highly branched pre-PGS fabricated by single mode MI provides a 59% wider window for degradation compared to the linear chain yielded by CH. In total, the new microwave approach provides a higher degree of freedom to tune mechanical properties (threefold) and degradation rate (59%) in order to meet the demands of various applications such as drug delivery vectors, which highly depend on the degradation rate of the polymer.
Acknowledgements
M. K. Bayazit and J. Tang acknowledge the Leverhulme Trust for financial support (RPG-2012-582). C. C. Lau is supported by a postgraduate scholarship from the Public Service Department of Malaysia.References
- Y. Wang, G. Ameer, B. Sheppard and R. Langer, Nat. Biotechnol., 2002, 20, 602 CrossRef CAS PubMed.
- Q. Chen, S. Liang and G. A. Thouas, Prog. Polym. Sci., 2013, 38, 584 CrossRef CAS.
- Z. J. Sun, C. Chen, M. Z. Sun, C. H. Ai, X. L. Lu, Y. F. Zheng, B. F. Yang and D. L. Dong, Biomaterials, 2009, 30, 5209 CrossRef CAS PubMed.
- C. A. Sundback, J. Y. Shyu, Y. Wang, W. C. Faquin, R. S. Langer, J. P. Vacanti and T. A. Hadlock, Biomaterials, 2005, 26, 5454 CrossRef CAS PubMed.
- L. Sestoft, Acta Anaesthesiol. Scand., 1985, 29, 19 CrossRef.
- J. P. Bruggeman, B. J. de Bruin, C. J. Bettinger and R. Langer, Biomaterials, 2008, 29, 4726 CrossRef CAS PubMed.
- S. L. Liang, W. D. Cook, G. A. Thouas and Q. Z. Chen, Biomaterials, 2010, 31, 8516 CrossRef CAS PubMed.
- Q. Chen, L. Jin, W. D. Cook, D. Mohn, E. L. Lagerqvist, D. A. Elliott, J. M. Haynes, N. Boyd, W. J. Stark, C. W. Pouton, E. G. Stanley and A. G. Elefanty, Soft Matter, 2010, 6, 4715 RSC.
- Q. Z. Chen, H. Ishii, G. A. Thouas, A. R. Lyon, J. S. Wright, J. J. Blaker, W. Chrzanowski, A. R. Boccaccini, N. N. Ali, J. C. Knowles and S. E. Harding, Biomaterials, 2010, 31, 3885 CrossRef CAS PubMed.
- R. Shi, D. Chen, Q. Liu, Y. Wu, X. Xu, L. Zhang and W. Tian, Int. J. Mol. Sci., 2009, 10, 4223 CrossRef CAS PubMed.
- C. C. Lau, P. J. T. Reardon, J. C. Knowles and J. Tang, ACS Biomater. Sci. Eng., 2015, 1, 947 CrossRef.
- F. Wiesbrock, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2004, 25, 1739 CrossRef CAS.
- P. J. T. Reardon, J. Huang and J. Tang, Adv. Healthcare Mater., 2013, 2, 682 CrossRef CAS PubMed.
- P. J. T. Reardon, A. D. Handoko, L. Li, J. Huang and J. Tang, J. Mater. Chem. B, 2013, 1, 6170 RSC.
- M. K. Bayazit, J. Yue, E. Cao, A. Gavriilidis and J. Tang, ACS Sustainable Chem. Eng., 2016, 4, 6435 CrossRef CAS.
- H. M. Aydin, K. Salimi, Z. M. O. Rzayev and E. Pişkin, Biomater. Sci., 2013, 1, 503 RSC.
- X. Li, A. T. L. Hong, N. Naskar and H. J. Chung, Biomacromolecules, 2015, 16, 1525 CrossRef CAS PubMed.
- X. J. Loh, A. Abdul Karim and C. Owh, J. Mater. Chem. B, 2015, 3, 7641 RSC.
- S. Barlow and S. R. Marder, Adv. Funct. Mater., 2003, 13, 517 CrossRef CAS.
- N. Kuhnert, Angew. Chem., Int. Ed. Engl., 2002, 41, 1863 CrossRef CAS PubMed.
- I. Pomerantseva, N. Krebs, A. Hart, C. M. Neville, A. Y. Huang and C. A. Sundback, J. Biomed. Mater. Res., Part A, 2009, 91, 1038 CrossRef PubMed.
- D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563 CrossRef CAS PubMed.
- C. Gabriel, S. Gabriel, E. H. Grant, B. S. J. Halstead, D. Michael and P. Mingos, Chem. Soc. Rev., 1998, 27, 213 RSC.
- D. Dallinger, M. Irfan, A. Suljanovic and C. O. Kappe, J. Org. Chem., 2010, 75, 5278 CrossRef CAS PubMed.
- D. Adam, Nature, 2003, 421, 571 CrossRef CAS PubMed.
- R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2007, 28, 368 CrossRef CAS.
- B. T. Ergan, M. Bayramoğlu and S. Özcan, Eur. Polym. J., 2015, 69, 374 CrossRef CAS.
- S. Velmathi, R. Nagahata, J. Sugiyama and K. Takeuchi, Macromol. Rapid Commun., 2005, 26, 1163 CrossRef CAS.
- J. Coates, Interpretation of Infrared Spectra, A Practical Approach, Encyclopedia of Analytical Chemistry, 2006 Search PubMed.
- C. L. E. Nijst, J. P. Bruggeman, J. M. Karp, L. Ferreira, A. Zumbuehl, C. J. Bettinger and R. Langer, Biomacromolecules, 2007, 8, 3067 CrossRef CAS PubMed.
- S. Bodakhe, S. Verma, K. Garkhal, S. K. Samal, S. S. Sharma and N. Kumar, Nanomedicine, 2013, 8, 1777 CrossRef CAS PubMed.
- Y. Jia, W. Wang, X. Zhou, W. Nie, L. Chen and C. He, Polym. Chem., 2016, 7, 2553 RSC.
- H. Shi, Q. Gan, X. Liu, Y. Ma, J. Hu, Y. Yuan and C. Liu, RSC Adv., 2015, 5, 79703 RSC.
- Y. Li, W. D. Cook, C. Moorhoff, W. C. Huang and Q. Z. Chen, Polym. Int., 2013, 62, 534 CrossRef CAS.
- Q. Liu, M. Tian, T. Ding, R. Shi, Y. Feng, L. Zhang, D. Chen and W. Tian, J. Appl. Polym. Sci., 2007, 103, 1412 CrossRef CAS.
- D. Kafouris, F. Kossivas, C. Constantinides, N. Q. Nguyen, C. Wesdemiotis and C. S. Patrickios, Macromolecules, 2013, 46, 622 CrossRef CAS.
- R. Nagahata, D. Sano, H. Suzuki and K. Takeuchi, Macromol. Rapid Commun., 2007, 28, 437 CrossRef CAS.
- X. L. Guo, X. L. Lu, D. L. Dong and Z. J. Sun, J. Biomed. Mater. Res., Part A, 2014, 102, 3903 CrossRef PubMed.
- Q. Z. Chen, A. Bismarck, U. Hansen, S. Junaid, M. Q. Tran, S. E. Harding, N. N. Ali and A. R. Boccaccini, Biomaterials, 2008, 29, 47 CrossRef CAS PubMed.
- M. S. Soh and A. U. J. Yap, J. Dent., 2004, 32, 321 CrossRef CAS PubMed.
- J. K. Gillham, Polym. Eng. Sci., 1979, 19, 676 CAS.
- D. L. Safranski and K. Gall, Polymer, 2008, 49, 4446 CrossRef CAS.
- S. Lyu and D. Untereker, Int. J. Mol. Sci., 2009, 10, 4033 CrossRef CAS PubMed.
- L. Wu and J. Ding, J. Biomed. Mater. Res., Part A, 2005, 75, 767 CrossRef PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602 CrossRef CAS PubMed.
- X. Zhu, Y. Zhou and D. Yan, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py00862g |
| This journal is © The Royal Society of Chemistry 2017 |