
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing bacterial affinity and cytocompatibility with four-arm star glycopolymers and antimicrobial α-polylysine†
Dicky
Pranantyo
a,
Li Qun
Xu
a,
Zheng
Hou
b,
En-Tang
Kang
 *a and
Mary B.
Chan-Park
*b
*a and
Mary B.
Chan-Park
*b
aDepartment of Chemical & Biomolecular Engineering, National University of Singapore, Kent Ridge, Singapore 119260. E-mail: cheket@nus.edu.sg
bCentre of Antimicrobial Bioengineering, School of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637459. E-mail: mbechan@ntu.edu.sg
First published on 2nd May 2017
Abstract
A series of four-arm star copolymers, incorporating glycopolymer and antimicrobial polypeptide domains, was developed in the design of forthcoming anti-infective agents. Mannose, glucose, and galactose-based glycopolymers with a variety of well-defined chain lengths were prepared via atom transfer radical polymerization, whereas linear α-polylysine was prepared via ring-opening polymerization of N-carboxyanhydride monomers. Copper-catalyzed azide–alkyne cycloaddition was employed for ‘click’ conjugation of the glycopolymer arms and the polypeptide chains. The glycopolymer–polypeptide conjugates were non-hemolytic and exhibited higher cytocompatibility than the linear α-polylysine. The conjugates with shorter chains of mannose-based glycopolymer arms showed an enhanced bactericidal efficacy against Gram-negative and Gram-positive bacteria, with a therapeutic selectivity half of that of the linear α-polylysine. The pendant mannose moieties of the conjugates increased microbial targeting due to their specific affinity for bacterial surfaces, and binding competition with free mannopyranoside was demonstrated. Therefore, the molecular combination of glycopolymers and polypeptides without loss of their respective activities provides an interesting concept in the design of antimicrobial agents to combat infectious disease.
1. Introduction
Disease induced by bacterial infections is a common occurrence in our daily life, and often accounts for high medical cost, chronic illness or even death of patients. Traditional antibiotics that fight these infections need to penetrate the target microorganisms and react with the metabolic activity of the cells, such as interfering with critical bacterial enzymes, disrupting certain protein syntheses, inhibiting gyrase and DNA replication, or sequestering essential nutrients.1–3 These mechanisms of action only target specific intracellular functions and leave the morphology of the bacteria intact, allowing them the opportunity to evolve and develop drug resistance.4 With excessive consumption of these metabolic-based antibiotics, the number of resistant bacteria and superbugs rapidly grows, causing a worldwide concern which requires immediate attention.5 It is thus of utmost interest to design a new type of anti-infective agent with a more straight-forward killing mechanism to minimize the risk of bacteria developing resistant traits.The use of antimicrobial peptides (AMPs) is an emerging concept for the next-generation treatment of pathogenic infections.6 These AMPs generally contain positively charged ions along their amphipathic α-helical or β-sheet structures, which can interact with bacterial membranes.7 Upon interaction, the cationic AMPs destabilize or even penetrate through the lipophilic domain of the cell membranes, leading to membrane disintegration and cytoplasmic leakage.8,9 Under these conditions, bacteria eventually die without developing resistance. Nevertheless, the scarcity of natural peptides has placed a limitation to their potential application, as they are part of the natural immune system extractable only in infinitesimal amounts. Among the conventional well-established techniques to prepare synthetic peptides, solution-coupling and solid-phase syntheses provide precise control over chain composition, with the drawback of employing multistep procedures in a tedious, time-consuming and expensive manner.10–12 In comparison, the development of the N-carboxyanhydride (NCA) ring-opening polymerization (ROP) technique pioneered the translation of peptide designs into polymer synthesis, which offers a convenient means to produce polypeptides in a large quantity and at a low cost.13 This powerful NCA-ROP technique allows the preparation of polypeptides with a narrowly defined chain length and intrachain composition.14 Polypeptides consisting of a combination of lysine, alanine, phenylalanine, and leucine amino acids were prepared via the NCA-ROP technique, and the synthetic polypeptides showed microbiocidal activity against a wide range of pathogenic microorganisms, including clinically significant bacteria Serratia marcescens and fungi Candida albicans.15 Under a transmission electron microscopy study, micelles consisting of a poly(L-lactide) core and poly(phenylalanine-stat-lysine) shell were reported to readily pierce and destroy the membrane of Gram-negative and Gram-positive bacteria.16 However, it should be noted that synthetic cationic peptides are often toxic and detrimental to mammalian cells.17 While ε-polylysine produced via natural fermentation of the Streptomyces genus has been used as a food preservative due to its low cytotoxicity, α-polylysine prepared by NCA-ROP is highly cytotoxic and thus cannot be readily used in consumer-based products. In order to actuate their potential applicability, cationic peptides can be molecularly engineered to reduce their toxicity. For example, the preparation of cationic polymers with hydrophilic, non-hemolytic antimicrobial properties was achieved by controlling the alkyl backbone length of tetramethyldiamines and dibromoalkanes in a polyaddition reaction.18 Poly(2-methyloxazoline) with a quaternary ammonium terminal and a cleavable ester satellite terminal was also reported to exhibit bactericidal activity with low hemotoxicity.19
The glycopolymer is a class of synthetic macromolecules with pendant carbohydrate moieties. It possesses ideal biocompatibility with mammalian bodies and other living cells in general, and hence, would make a feasible candidate for moderating the toxicity of anti-infective agents.20,21 For example, glucosamine was employed to functionalize star polymers comprising of antimicrobial polylysine, which resulted in dramatic improvement of cytocompatibility toward human aortic smooth muscle cells.22 On the other hand, glycomaterials also display interesting characteristics in many biological recognition events, such as cell–cell adhesion and development of new tissues, and thus have potential applications in targeted drug delivery, tissue engineering, and synthesis of biocompatible materials.23,24 Micro-organisms exhibit preferential adhesion to specific substrates, and this characteristic is a plausible target in the design of new antimicrobial compounds. It was suggested that the phagocytic cells of certain nonopsonized Escherichia coli express type I fimbriae that recognize mannose.25 For example, gold nanoparticles with mannose-based pendant motifs showed strong binding capability to FimH lectin domains and induced the aggregation of E. coli ORN 178.26 However, the adhesion of bacteria is polyvalent and bacterial adhesins often recognize more than one type of cell-surface molecule.27,28 A library of galactosylated polymers with different statistical, gradient, and block microstructures were proven to have higher aptitude in clustering Ricinus communis (castor bean) agglutin RCA120 than other glycopolymers.29 Glycopeptide dendrimers based on galactose also showed high affinity to LecB and LecA lectins, and efficiently induced the biofilm dispersal of Pseudomonas aeruginosa.30 It is thus a promising concept to employ glycopolymers in the design of antimicrobial compounds for modulating toxicity and simultaneously targeting bacterial cells.
In this report, a library of mannose, glucose, and galactose-based glycopolymers was conjugated with antimicrobial polypeptide to form a four-arm star microstructure. The two α-polylysine arms would deliver the killing effect toward bacteria, whereas the other two glycopolymer arms would moderate the toxicity toward mammalian cells and enhance binding affinity for the target bacteria. The antimicrobial properties toward various Gram-positive and Gram-negative bacteria were determined by measuring the minimum inhibitory and bactericidal concentrations of the glycopolymer–polypeptide conjugates. In vitro hemolysis and cytotoxicity assays were carried out to investigate the biological characteristics of the conjugates.
2. Experimental section
2.1. Materials
Sodium azide (99.5%), 2,2-bis(bromomethyl)propane-1,3-diol (98%), 2-bromopropionyl bromide (97%), triethylamine (99%), D-(+)-mannose (99%), D-(+)-glucose (99.5%), D-(+)-galactose (98%), sulfuric acid (95%), sodium bicarbonate (99.7%), magnesium sulfate (99.5%), 2-hydroxyethyl methacrylate (99%), boron trifluoride diethyl etherate, N-ε-carbobenzoxy-L-lysine (99%), triphosgene (98%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), copper(I) bromide (CuBr, 99.9%), copper(II) bromide (CuBr2, 99%), sodium (99.9%), propargylamine (98%), trifluoroacetic acid (99%), hydrobromic acid (33 wt% in acetic acid), methyl α-D-mannopyranoside (MeMan, 99%), and fluorescein isothiocyanate (FITC, 97.5%) were purchased from Sigma-Aldrich Chem. Co. (St Louis, MO). Dimethyl sulfoxide, ethyl acetate, dichloromethane, hexane, acetic anhydride, tetrahydrofuran, toluene, ethanol, methanol, chloroform, dimethylformamide (DMF), and diethyl ether were of analytical or HPLC grade. Escherichia coli (E. coli, ATCC 25922), Pseudomonas aeruginosa (P. aeruginosa, ATCC 15692) and Staphylococcus aureus (S. aureus, ATCC 25923), and Staphylococcus epidermidis (S. epidermidis, ATCC 12228) were purchased from American Type Culture Collection (ATCC, Manassas, VA). Four-arm ‘clickable’ initiator (AzBr, 456.09 g mol−1),31N-ε-carbobenzoxy-L-lysine N-carboxyanhydride (CbzLys-NCA, 306.32 g mol−1) monomer, and the glycomonomers of mannose (AcManEMA, 460.43 g mol−1), glucose (AcGluEMA, 460.43 g mol−1) and galactose (AcGalEMA, 460.43 g mol−1)32,33 were synthesized by referring to the previous methods with some modifications (section 1, ESI†).2.2. Synthesis of the glycopolymers (P(GlyEMA))
AcManEMA polymer (P(AcManEMA)) was prepared via atom transfer radical polymerization. AzBr (124 mg, 0.27 mmol), AcManEMA (5 g, 10.86 mmol), PMDETA (94 mg, 0.54 mmol), and CuBr2 (11 mg, 0.05 mmol) were added into 10 mL of toluene. The mixture was stirred and degassed with argon for 30 min. CuBr (78 mg, 0.54 mmol) was added to the mixture and the flask was quickly sealed. The mixture was stirred at room temperature for 24 h. The mixture was passed through a column of aluminium oxide to remove the copper residues, precipitated in ethanol, and dried under reduced pressure to obtain AcManEMA polymer (P(AcManEMA)20, white solid, yield 67%). Using a similar method, another set of P(AcManEMA) with a different number of repeat units was prepared using [AcManEMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[AzBr]:[PMDETA]:[CuBr]:[CuBr2] molar feed ratios of 20:1:2:2:0.2 and 60:1:2:2:0.2, resulting in P(AcManEMA)10 and P(AcManEMA)30, respectively.
:[AzBr]:[PMDETA]:[CuBr]:[CuBr2] molar feed ratios of 20:1:2:2:0.2 and 60:1:2:2:0.2, resulting in P(AcManEMA)10 and P(AcManEMA)30, respectively.
P(AcManEMA) (3 g) was dissolved in 10 mL of methanol/chloroform (1/1 volumetric ratio). Sodium methoxide solution (1 M) was freshly prepared by dissolving sodium in methanol, and added dropwise into the P(AcManEMA) solution under stirring until pH 9. The reaction was carried out at room temperature for 4 h to allow the formation of a white precipitate. The precipitate was dialyzed against doubly-distilled water for 3 days and lyophilized to obtain 2-(D-mannosyloxy)ethyl methacrylate polymer (P(ManEMA), white solid, yield 50%). In addition, 2-(D-glucosyloxy)ethyl methacrylate polymer (P(GluEMA)) and 2-(D-galactosyloxy)ethyl methacrylate polymer (P(GalEMA)) were prepared using similar procedures as that for P(ManEMA) by replacing the AcManEMA starting materials with AcGluEMA and AcGalEMA, respectively.
2.3. Synthesis of N-ε-carbobenzoxy-L-lysine polymer (P(CbzLys))
Propargylamine (36 mg, 0.66 mmol) was dissolved in 5 mL of DMF and degassed with argon for 15 min. CbzLys-NCA (4 g, 13.1 mmol) was dissolved in 20 mL of DMF and transferred via syringe to the propargylamine solution under stirring. The mixture was degassed with argon for 30 min and sealed tightly. The reaction was allowed to proceed at room temperature for 24 h. The mixture was precipitated thrice in 200 mL of diethyl ether and dried under reduced pressure to obtain N-ε-carbobenzoxy-L-lysine polymer (P(CbzLys), pale yellowish solid, yield 90%).2.4. Preparation of the glycopolymer–polylysine conjugates (P(GlyEMA)–P(Lys))
P(ManEMA)20 (1.5 g, 0.12 mmol) and P(CbzLys) (1.3 g, 0.24 mmol) were dissolved in 20 mL of DMF and degassed with argon for 15 min. PMDETA (41.6 mg, 0.24 mmol) and CuBr (34.4 mg, 0.24 mmol) were added to the mixture under stirring. The mixture was degassed with argon for another 15 min and sealed tightly. The reaction was allowed to proceed under stirring at 50 °C for 24 h. The mixture was continuously reprecipitated in ethanol to remove the blue catalyst residue. The precipitate was dried under reduced pressure to obtain P(ManEMA)20–P(CbzLys) copolymer (white solid, yield 75%).P(ManEMA)20–P(CbzLys) (1 g, 0.04 mmol, ∼1.76 mmol of L-lysine repeat unit) was dissolved in 30 mL of trifluoroacetic acid. Hydrobromic acid (4-fold mol of L-lysine repeat unit) was added to the solution. The deprotection was allowed to proceed under stirring at room temperature for 4 h. The solution was precipitated in diethyl ether, dialyzed against doubly-distilled water for 3 days, and lyophilized to obtain P(ManEMA)20–P(Lys) copolymer (white cotton-like solid, yield 91%).
2.5. Characterization
Chemical structures of the synthesized products were characterized by 1H NMR spectroscopy on a Bruker ARX 300 MHz spectrometer, using deuterated chloroform, dimethyl sulfoxide, or water as a solvent. Fourier transform infrared (FT-IR) spectroscopy measurements were carried out on a BioRad FTS-135 spectrophotometer. Gel permeation chromatography (GPC) was performed on a Waters GPC system equipped with a Waters 1515 isocratic HPLC pump, a Waters 717 plus Autosampler injector, and a Waters 2414 refractive index detector at a flow rate of 1 mL min−1 at 35 °C, using an Agilent PL Mixed gel D 5 μm column and DMF with LiBr 0.05 M as eluent.2.6. Determination of minimum inhibitory and bactericidal concentration (MIC and MBC)
Bacterial cells were cultured overnight in Mueller-Hinton broth (MHB) medium at 37 °C to a mid-log phase and diluted to 2 × 105 colony forming units (CFU) per mL. Stock solutions of P(Lys) and P(GlyEMA)–P(Lys) were prepared in the MHB medium at a concentration of 2048 μg mL−1. The compound solutions were serially diluted 2-fold, and 100 μL of each dilution was placed in each well of a 96-well plate (Greiner Bio-one, Germany). Then, 100 μL of the bacterial suspension was added to the compound solution in each well. The plate was incubated at 37 °C overnight and observed by the naked eye. The growing bacteria suspension appeared cloudy, while the non-growing one appeared clear.34 MIC was recorded as the lowest concentration of the compound that inhibited the growth of the bacteria in the well. The optical density was measured at 600 nm wavelength using a UV microplate reader (Multiskan GO, Thermo Scientific). After the MIC assay, 100 μL of the medium in the wells with no visible growth was aspirated and spread on Mueller-Hinton agar. The agar plates were incubated at 37 °C overnight to check the viability of the bacteria. MBC was recorded as the lowest concentration of the compound that killed 99.9% of the initially inoculated bacteria.2.7. Inhibition of the interaction of the P(ManEMA)–P(Lys) with bacteria
P(ManEMA)10–P(Lys) (115 mg, 0.01 mmol) and FITC (4 mg, 0.01 mmol) were dissolved in deionized water and stirred in the dark for 24 h. The mixture was dialyzed against deionized water for over 3 days and lyophilized to obtain FITC-labelled P(ManEMA)10–P(Lys) (yellow cotton-like solid, yield 90%). The FITC-labelled P(ManEMA)10–P(Lys) was dissolved in phosphate-buffered saline (PBS), or in PBS solution containing MeMan 0.1–0.3 M, to a concentration of 50 μg mL−1. Bacterial cells were suspended in PBS to a concentration of 2 × 108 CFU mL−1, and 0.5 mL of this bacterial suspension was added to 0.5 mL of FITC-labelled copolymer solution. The mixture was agitated briefly using a vortex mixer and kept in a refrigerator at 4 °C for 3 h. The mixture was centrifuged at 2700 rpm for 10 min to remove the supernatant, washed and resuspended in 1 mL of PBS. The bacterial suspension was quantified using a flow cytometry analyzer (BD LSR Fortessa, NJ). To obtain qualitative images, one droplet of the bacterial suspension was spread on a glass slide, sandwiched with a cover slip, and observed under a fluorescence microscope (Nikon Eclipse Ti-U, Japan).2.8. Determination of hemolytic activity
Whole rabbit blood was purchased from InVivos Pte Ltd (Singapore), and the hemolysis assay was carried out under the protocol abiding by the Institutional Animal Care and Use Committee (IACUC), National University of Singapore (NUS). Stock solutions of P(Lys) and P(GlyEMA)–P(Lys) were prepared in PBS at a concentration of 8192 μg mL−1. The compound solutions were serially diluted 2-fold, and 100 μL of each dilution was placed in each well of a 96-well plate. Fresh whole rabbit blood was diluted in PBS at a concentration of 8% (v/v), and 100 μL of the blood suspension was added to the compound solution in each well. The blood suspension was also added to blank PBS as the negative control, and to PBS containing 0.1% Triton-X as the positive control. The plate was incubated at 37 °C for 1 h to allow the interaction between the compounds and the blood cells. Each mixture was centrifuged at 1500 rpm for 10 min, and the optical absorbance of the supernatant was measured at 560 nm under a microplate reader (Multiskan GO, Thermo Scientific). The percentage of hemoglobin release (HC) was calculated as [(AS − AN)/(AS − AP)] × 100%, where AS, AN, and AP are the optical absorbance of the supernatant from the incubated sample, negative control, and positive control, respectively.2.9. Cytotoxicity assay
Standard 3T3 mouse fibroblast cells were purchased from ATCC (Manassas, VA), and the cytotoxicity assay was carried out under a protocol abiding by the IACUC, NUS. The fibroblast cells were cultured in a 75 cm2 tissue culture flask containing 10 mL of DMEM medium supplemented with 10% fetal bovine serum, 1 mM L-glutamine, and 100 IU mL−1 penicillin. The flask was incubated under a humidified atmosphere of 95% air and 5% CO2 at 37 °C. The medium was refreshed every 2 days until the cells reached ∼80% confluence. The cells were detached from the flask using 12 mL trypsin-EDTA solution (5.3 mM) and collected by centrifugation at 1000 rpm for 10 min. The cells were resuspended in fresh culture medium, and 100 μL of the suspension containing 5000 cells was seeded in each well of a 96-well plate. The plate was incubated under a humidified atmosphere of 95% air and 5% CO2 at 37 °C for 24 h. Sample solutions were prepared by dissolving the P(Lys) and P(GlyEMA)–P(Lys) in the individual growth medium at two-fold serial dilutions. The medium in the wells was replaced with 100 μL of the prepared sample solutions, and the plate was incubated at 37 °C for 24 h. A nontoxic control experiment was carried out using the growth medium without the sample. After incubation, the medium in each well was replaced with 100 μL of MTT solution (0.5 mg mL−1 in the growth medium). After additional incubation for 4 h, the culture supernatant was aspirated and 100 μL of DMSO was added to dissolve the internalized purple formazan crystal. After 15 min, the optical absorbance was measured at 595 nm using a microplate reader (TECAN GENios). The results were expressed as percentages relative to the absorbance obtained in the control experiment.3. Results and discussion
3.1. Preparation of the four-arm star glycopolymer–polypeptide conjugates
The four-arm ‘clickable’ initiator (AzBr) was prepared via two-step nucleophilic substitutions on 2,2-bis(bromomethyl)propane-1,3-diol, by reacting the bromide and hydroxyl terminals with sodium azide and 2-bromopropionyl bromide, respectively (Scheme 1). In the 1H NMR spectra of the intermediate and final products (Fig. S1, ESI†), all the chemical shifts were accounted for properly, depicting successful synthesis of the AzBr. The glycomonomers were prepared via acetylation to protect the hydroxyl groups of the saccharides, followed by glycosidic reaction of the hemiacetal group with 2-hydroxyethyl methacrylate. In the 1H NMR spectrum of 2-(2′,3′,4′,6′-tetra-O-acetyl-D-mannosyloxy)ethyl methacrylate (AcManEMA) monomer (Fig. S2†), the chemical shifts characteristic of acetyl protons at 2.18–1.95 ppm and vinyl protons at 6.13 and 5.60 ppm indicate the presence of acetyl protecting groups and polymerizable methacrylate groups on the mannopyranoside, respectively.32 Similarly, these chemical shift characteristics are also present in the 1H NMR spectrum of 2-(2′,3′,4′,6′-tetra-O-acetyl-D-glucosyloxy)ethyl methacrylate (AcGluEMA) and 2-(2′,3′,4′,6′-tetra-O-acetyl-D-galactosyloxy)ethyl methacrylate (AcGalEMA) monomers (Fig. S3 and S4†).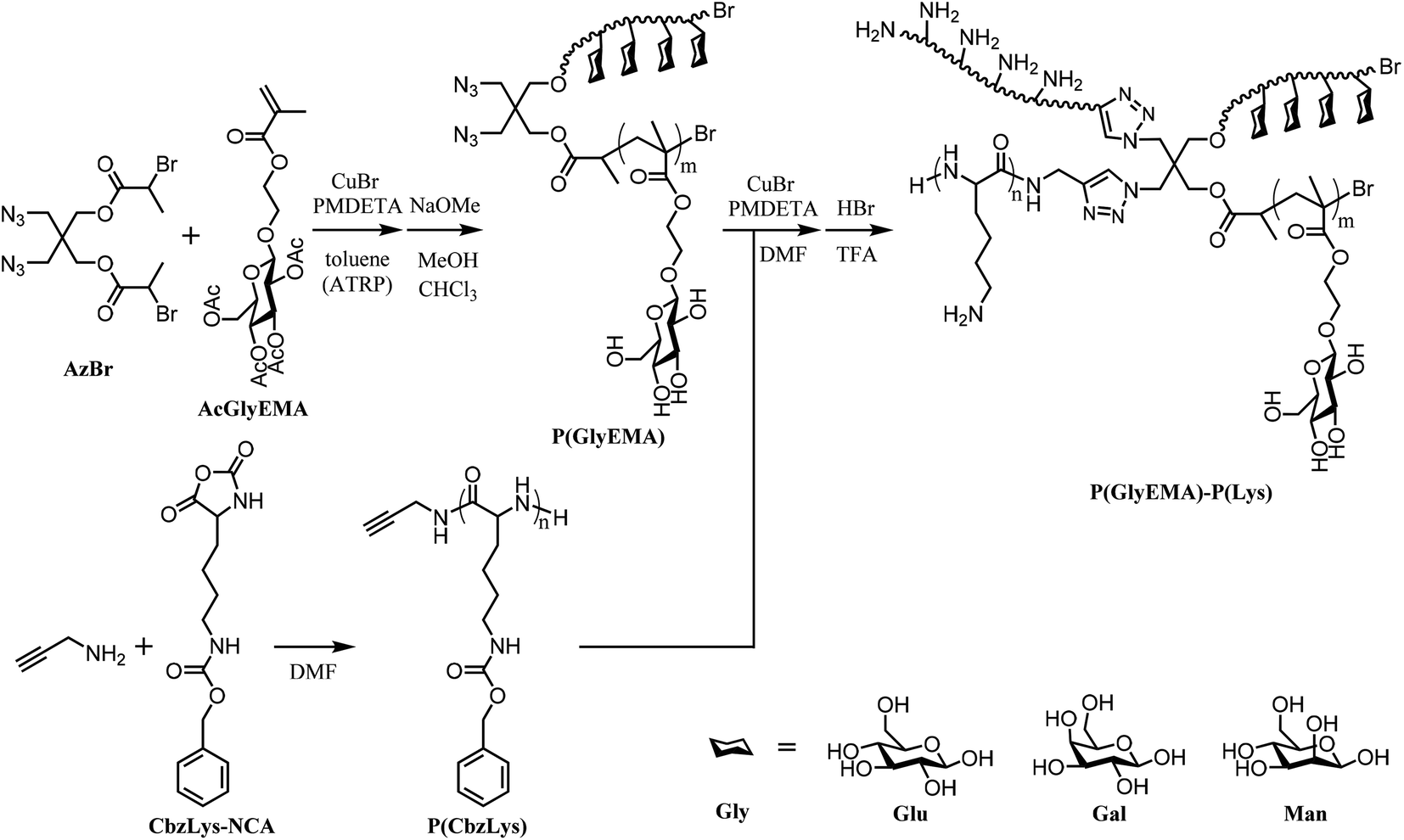 | ||
| Scheme 1 Preparation of the four-arm star glycopolymer–polypeptide conjugates via controlled radical polymerization, ring-opening polymerization, and ‘click’ chemistry. | ||
The two propionyl bromide arms of AzBr were used as the initiation sites for atom transfer radical polymerization (ATRP) of the glycomonomers (AcGlyEMA). In the 1H NMR spectra of the AcGlyEMA polymers (P(AcGlyEMA), Fig. S5†), two chemical shifts of ethylene protons from the polymer backbone emerge at 1.10–0.70 ppm. The chemical shift at 3.50 ppm is attributable to the ethylene protons adjacent to the azide groups of the initiator. The chemical shifts of the anomeric proton for P(AcManEMA), P(AcGluEMA), and P(AcGalEMA) appear at 4.87, 4.65, and 4.60, respectively.32,35,36 The degree of polymerization (Table 1) was estimated based on the peak integration ratio between these two chemical shifts, corresponding to the initiator protons and the anomeric proton. For the P(AcManEMA) polymers, the number of repeat units was calculated to be closely similar to the predetermined value, with moderately narrow polydispersity. Polymer chains of near theoretical length were also observed for the P(AcGluEMA) and P(AcGalEMA) polymers (not shown). A controlled manner of polymerization was accomplished using the ATRP technique to produce well-defined two-arm glycopolymers. After removal of the acetyl protecting groups, the four distinct chemical shifts at 2.18–1.95 ppm disappeared from the 1H NMR spectrum of P(ManEMA) (Fig. 1b). Disappearance of these acetyl shifts was also observed in the P(GluEMA) and P(GalEMA) spectra (Fig. S6†), indicating successful deprotection of the hydroxyl groups. In the FTIR spectra of all three glycopolymers (Fig. 2a), the presence of an azide stretching band at 2150 cm−1 confirms that the ‘clickable’ azide terminal remained intact during polymerization and deprotection.
 | ||
| Fig. 1 1H NMR spectra of the (a) P(AcManEMA)20 in CDCl3, (b) P(ManEMA)20 in D2O, (c) P(CbzLys) in DMSO-d6, and (d) P(ManEMA)20–P(Lys) conjugate in D2O. | ||
 | ||
| Fig. 2 FTIR spectra (a) and GPC chromatograms (b) of the ‘clickable’ glycopolymers, α-polylysine, and glycopolymer–polylysine conjugates. | ||
| Sample | Number of repeat units | Molecular weight, Mn (g mol−1) |
M
w/Mnc |
||
|---|---|---|---|---|---|
| Theoreticala | Experimentalb | Theoreticala | Experimentalb | ||
| a Theoretical repeat unit and molecular weight were derived from molar feed ratio of monomer and initiator. b Experimentally determined repeat unit and molecular weight were derived from integrated peak areas in the 1H NMR spectra. c Polydispersity index was derived from the GPC elution curves in DMF. | |||||
| P(AcManEMA)10 | 10 | 9.5 | 6302 | 6010 | 1.46 |
| P(AcManEMA)20 | 20 | 20.7 | 12148 |
12557 |
1.52 |
| P(AcManEMA)30 | 30 | 29.3 | 17994 |
17584 |
1.77 |
| P(CbzLys) | 20 | 19.6 | 5183 | 5081 | 1.31 |
The N-ε-carbobenzoxy-L-lysine polymer (P(CbzLys)) was prepared via ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydride (NCA), employing propargylamine as a ‘clickable’ initiator. All chemicals shifts in the 1H NMR spectrum are properly attributed to the respective protons of P(CbzLys) (Fig. S7†). The chemical shifts at 4.08 and 2.94 ppm are associated with the ethylene protons of the propargyl initiator and the ethylene protons adjacent to the pendant amino side groups, respectively.37 From the integrated peak area ratio between these two chemical shifts, a near-theoretical chain length with monomodal distribution was estimated for the P(CbzLys) (Table 1). In the FTIR spectrum of P(CbzLys) (Fig. 2a), a weak alkyne stretch band appears at 2125 cm−1, suggesting that the presence of the ‘clickable’ alkyne terminal has been preserved after polymerization.38
‘Click’ conjugation of the glycopolymers and α-polylysine was carried out via copper-catalyzed azide–alkyne cycloaddition, followed by removal of the carbobenzoxyl groups to reveal the peptide functional amines. All the chemical shifts characteristic of P(ManEMA) and α-polylysine appear in the 1H NMR spectrum of the P(ManEMA)–P(Lys) conjugate (Fig. 1d), marking the successful conjugation of the glycopolymer and polypeptide. In addition, all the chemical shifts of carbobenzoxyl protons at 7.42–7.08 and 5.00 ppm disappeared after the deprotection of the pendant amino moieties. All chemical shifts of the P(GluEMA)–P(Lys) and P(GalEMA)–P(Lys) conjugates can also be assigned according to their respective protons, demonstrating successful synthesis of the four-arm star glycopolymer–polypeptide conjugates. The chemical shift at 3.00 ppm is attributable to the ethylene protons adjacent to the amine moieties of the α-polylysine arms. The chemical shifts of anomeric carbon from the P(ManEMA), P(GluEMA), and P(GalEMA) arms are identified at 4.93, 4.53, and 4.46 ppm, respectively. The integrated peak area ratio of these two chemical shifts, representing the respective α-polylysine and glycopolymer arms, were used for calculating the mass fraction of L-lysine units in the conjugates (Table 3). The GPC chromatograms (Fig. 2b) illustrate the conversion of linear α-polylysine and two-arm glycopolymers into four-arm star copolymers of higher molecular weight. The linear α-polylysine shows a bimodal distribution as it assumes a coil-to-helix transition at around 15 repeat units.39 The controlled radical polymerization produced two-arm glycopolymers with unimodal chain length. After ‘click’ conjugation, the shift in molecular weight distribution of the conjugates shows the presence of a minor component of lower molecular weight residues, indicating the incomplete conversion of the linear and two-arm polymers into a four-arm star copolymer by the azide–alkyne cycloaddition.
3.2. Antimicrobial properties of the four-arm star glycopolymer–polypeptide conjugates
Gram-negative Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa), as well as Gram-positive Staphylococcus aureus and epidermidis (S. aureus and S. epidermidis), were employed to challenge the antibacterial activity of the four-arm star glycopolymer–polypeptide conjugates. The linear α-polylysine (P(Lys)) readily combat the bacterial growth with minimum inhibitory concentration (MIC) values of 10 and 32 μg mL−1 toward E. coli and P. aeruginosa, respectively, and 16 μg mL−1 toward both Staphylococci (Table 2). The trend is similar to that previously reported for the linear polylysine, albeit with lower MIC values overall.22 This discrepancy is probably due to the different bacterial strains used. In general, all of the glycopolymer–polypeptide conjugates exhibit higher MIC values than P(Lys). On the other hand, zeta potential (ζ) values of all the glycopolymer–polypeptide conjugates are lower than that of the linear α-polylysine (Table 2). Any additional increase in the glycopolymer length also lowered the ζ value of the conjugates, mainly because the glycopolymer arms are less cationic than the α-polylysine arms. It is hence postulated that an increase in glycopolymer length reduced the cationic density and electrostatic interactions with bacterial membranes, resulting in higher MIC values.| Sample | ζ, mV | MICa, μg mL−1 (μM) | MBCa, μg mL−1 (μM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | S. epidermidis | E. coli | P. aeruginosa | S. aureus | S. epidermidis | ||
| a Values in bracket were expressed as μM or μmol L−1. | |||||||||
| P(Lys) | 34.8 | 10 (3.8) | 32 (12.2) | 16 (6.1) | 16 (6.1) | 32 (12.2) | 64 (24.4) | 32 (12.2) | 32 (12.2) |
| P(ManEMA)10–P(Lys) | 28.9 | 18 (1.6) | 64 (5.5) | 32 (2.8) | 32 (2.8) | 64 (5.5) | 64 (5.5) | 64 (5.5) | 32 (2.8) |
| P(ManEMA)20–P(Lys) | 27.2 | 64 (3.7) | 128 (7.4) | 64 (3.7) | 64 (3.7) | 256 (14.7) | 256 (14.7) | 128 (7.4) | 128 (7.4) |
| P(ManEMA)30–P(Lys) | 12.2 | 256 (11.0) | 512 (22.0) | 128 (5.5) | 128 (5.5) | 512 (22.0) | 1024 (44.1) | 512 (22.0) | 256 (11.0) |
| P(GluEMA)10–P(Lys) | 28.1 | 64 (5.5) | 64 (5.5) | 32 (2.8) | 32 (2.8) | 64 (5.5) | 64 (5.5) | 64 (5.5) | 64 (5.5) |
| P(GluEMA)20–P(Lys) | 22.7 | 256 (14.7) | 256 (14.7) | 128 (7.4) | 128 (7.4) | 256 (14.7) | 512 (29.4) | 256 (14.7) | 256 (14.7) |
| P(GluEMA)30–P(Lys) | 10.9 | 256 (11.0) | 256 (11.0) | 128 (5.5) | 128 (5.5) | 1024 (44.1) | 1024 (44.1) | 512 (22.0) | 256 (11.0) |
| P(GalEMA)10–P(Lys) | 24.1 | 64 (5.5) | 128 (11.1) | 32 (2.8) | 64 (5.5) | 128 (11.1) | 256 (22.2) | 64 (5.5) | 64 (5.5) |
| P(GalEMA)20–P(Lys) | 18.7 | 128 (7.4) | 128 (7.4) | 128 (7.4) | 128 (7.4) | 256 (14.7) | 512 (29.4) | 256 (14.7) | 128 (7.4) |
| P(GalEMA)30–P(Lys) | 10.4 | 256 (11.0) | 256 (11.0) | 128 (5.5) | 256 (11.0) | 512 (22.0) | 1024 (44.1) | 512 (22.0) | 256 (11.0) |
The killing efficacy of all samples is recorded as minimum bactericidal concentration (MBC) values, which are similar to, or higher than, their respective MIC values. The antimicrobial efficacy also decreased with the increasing chain length of the glycopolymer arms. It should be noted that all the glycopolymers did not show any inhibitory effect when tested up to the highest concentration (2048 μg mL−1). Thus, the antimicrobial effect was mainly caused by the cationic α-polylysine arms, which might destabilize the bacterial membrane and induce cytoplasmic leakage. Among all of the glycopolymer–polypeptide conjugates, the lowest MIC value was observed for the P(ManEMA)10–P(Lys) conjugate, and is twice that of the linear P(Lys). However, on the basis of molarity, all MIC values of the P(ManEMA)10–P(Lys) and P(ManEMA)20–P(Lys) conjugates were lower than that of the α-polylysine. In addition, on the basis of the mass fraction of lysine units (Table 3), the P(ManEMA)10–P(Lys) conjugate exhibited MIC values lower than the linear P(Lys), namely at 7 μg mL−1 toward E. coli, 25 μg mL−1 toward P. aeruginosa, and 13 μg mL−1 toward both Staphylococci. Bacteria expressing type I fimbriae, such as many of the E. coli strains, exhibit a high affinity toward mannose-containing materials. The pendant mannose moieties on the glycopolymer arms have presumably increased the binding affinity of the P(ManEMA)–P(Lys) conjugate for bacterial cells, resulting in a higher interaction frequency.
| Sample | Mass fraction of lysine units | HC50c, μg mL−1 |
IC50, μg mL−1 | Selectivityd | ||||
|---|---|---|---|---|---|---|---|---|
| Theoreticala | Experimentalb | E. coli | P. aeruginosa | S. aureus | S. epidermidis | |||
| a Theoretical values were derived from the molar feed ratio of glycopolymers and α-polylysine by assuming 100% conversion of ‘click’ reaction. b Experimentally determined values were derived from peak area integrations of the 1H NMR spectra. c All HC50 values were larger than the highest concentration of compound tested at 8192 μg mL−1. d Selectivity values were calculated as HC50/MIC, whereas selectivity values in bracket were calculated as IC50/MIC. | ||||||||
| P(Lys) | 100% | 100% | >8192 | >20 | >819 (>2.0) | >256 (>0.6) | >512 (>1.3) | >512 (>1.3) |
| P(ManEMA)10–P(Lys) | 46% | 39% | >8192 | >33 | >455 (>1.8) | >128 (>0.5) | >256 (>1.0) | >256 (>1.0) |
| P(ManEMA)20–P(Lys) | 29% | 21% | >8192 | >40 | >128 (>0.6) | >64 (>0.3) | >128 (>0.6) | >128 (>0.6) |
| P(ManEMA)30–P(Lys) | 23% | 13% | >8192 | >280 | >32 (>1.1) | >16 (>0.5) | >64 (>2.1) | >64 (>2.1) |
| P(GluEMA)10–P(Lys) | 46% | 39% | >8192 | >58 | >128 (>0.9) | >128 (>0.9) | >256 (>1.8) | >256 (>1.8) |
| P(GluEMA)20–P(Lys) | 29% | 22% | >8192 | >170 | >32 (>0.7) | >32 (>0.7) | >64 (>1.3) | >64 (>1.3) |
| P(GluEMA)30–P(Lys) | 23% | 15% | >8192 | >360 | >32 (>1.4) | >32 (>1.4) | >64 (>2.8) | >64 (>2.8) |
| P(GalEMA)10–P(Lys) | 46% | 40% | >8192 | >49 | >128 (>0.8) | >64 (>0.4) | >256 (>1.5) | >128 (>0.8) |
| P(GalEMA)20–P(Lys) | 29% | 24% | >8192 | >84 | >64 (>0.7) | >64 (>0.7) | >64 (>0.7) | >64 (>0.7) |
| P(GalEMA)30–P(Lys) | 23% | 15% | >8192 | >348 | >32 (>1.4) | >32 (>1.4) | >64 (>2.7) | >32 (>1.4) |
In order to investigate the enhanced binding affinity contributed by the pendant mannose moieties, the MIC of linear P(Lys) and the P(ManEMA)10–P(Lys) conjugate was tested in the presence of methyl α-D-mannopyranoside (MeMan), a nanomolar FimH antagonist.40,41 There is no difference in the MIC value of the linear P(Lys), regardless of the presence of MeMan (Fig. 3). Interestingly, the MIC value of the P(ManEMA)10–P(Lys) conjugate in the presence of MeMan is 25 μg mL−1, higher than that in the absence of MeMan. To further elaborate the binding interference, E. coli suspension was incubated with fluorescence isothiocyanate (FITC)-labelled P(ManEMA)10–P(Lys) and MeMan at varying concentrations. In the flow cytometric histogram (Fig. 4), the highest fluorescence intensity was detected in the bacterial suspension without MeMan. With the increase of the MeMan concentration, the overall fluorescence intensity shifted to lower values. The mean fluorescence intensity also decreased gradually with the increase in MeMan concentration, implying that MeMan molecules interfered with the binding capacity of P(ManEMA)10–P(Lys) on bacteria. From the fluorescence micrographs of the bacterial suspension (Fig. 5), the increase in MeMan concentration reduced the number of bacteria bound by FITC-labelled conjugates. It has been reported that gold nanoparticles modified with glycopolymer and cationic polymer exhibited specific adhesion toward E. coli, and could be recycled via addition of mannose to detach the nanocomposites from the bacterial cells.42 This result implies that the pendant mannose moieties increased the affinity of the P(ManEMA)10–P(Lys) conjugate for bacterial surfaces, and binding competition occurred between P(ManEMA)10–P(Lys) and MeMan.
 | ||
| Fig. 3 Optical density of overnight E. coli cultures in MHB containing P(Lys) and P(ManEMA)10–P(Lys), with and without the presence of 0.2 M MeMan. | ||
 | ||
| Fig. 4 Flow cytometric histogram of E. coli in PBS (control) and PBS containing FITC-labelled P(ManEMA)10–P(Lys), in the absence and presence of MeMan. Error bar denotes standard deviation obtained from three sample replicates. Asterisk (*) denotes significant difference with p-value < 0.05 (Tukey's test). | ||
 | ||
| Fig. 5 Fluorescence micrographs of E. coli in PBS containing FITC-labelled P(ManEMA)10–P(Lys), in the absence (a) and presence of (b) 0.1 M, (c) 0.2 M, and (d) 0.3 M MeMan. | ||
3.3. Hemolytic activity and cytotoxicity of the four-arm star glycopolymer–polypeptide conjugates
A hemolysis assay was carried out to study the hemotoxicity of the glycopolymer–polypeptide conjugates. Each compound was exposed to rabbit whole blood to identify the HC50 value, the compound concentration which induces lysis of 50% of the red blood cells (Table 3). The linear P(Lys) and all glycopolymer–polypeptide conjugates did not exhibit the HC50 characteristic at any concentration below 8192 μg mL−1. This result was in agreement with that reported previously. Up to 10000 μg mL−1 of linear polylysine prepared by the NCA ROP technique was non-hemolytic toward human erythrocytes.22 It appears that the absence of a hydrophobic domain might contribute to the remarkable hemocompatibility of the linear P(Lys) and glycopolymer–polypeptide conjugates, as hydrophobic groups are known to be responsible for inducing hemolytic activity.
In the cytocompatibility study, metabolic activity of mouse 3T3 fibroblast cells in the presence of antimicrobial compounds was determined using an MTT assay. From the cell viability curves (Fig. 6), all of the glycopolymer–polypeptide conjugates exhibit higher cytocompatibility than the linear α-polylysine. Conjugates with longer glycopolymer arms also provided higher cell viability, indicating that α-polylysine arms are mainly responsible for the cytotoxic effect of the conjugates. The half-maximum inhibitory concentration (IC50) of P(ManEMA)10–P(Lys) increased significantly from that of P(Lys), and continued to increase logarithmically with longer glycopolymer arms (Table 3). This cytotoxicity effect is associated with the charge density of the conjugates, and the result is in agreement with the trend in ζ increase. The compounds with higher ζ values were more cytotoxic, because the pendant positive charges of α-polylysine most likely intensified the interaction with the negatively charged cell surfaces and interfered with the cell functions and stability.
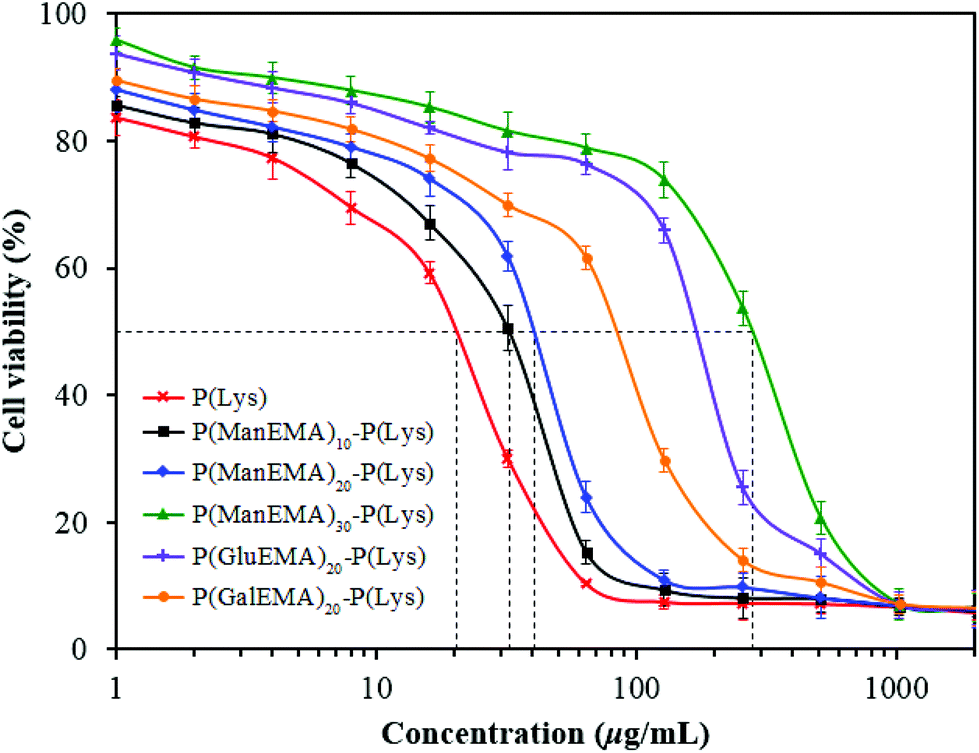 | ||
| Fig. 6 Viability of the 3T3 fibroblasts after incubation with the P(Lys), P(ManEMA)10–P(Lys), P(ManEMA)20–P(Lys), P(ManEMA)30–P(Lys), P(GluEMA)20–P(Lys), and P(GalEMA)20–P(Lys) conjugates. Error bar denotes standard deviation obtained from three sample replicates. | ||
The hemo- and cytoselectivity values, HC50/MIC and IC50/MIC, were calculated to determine the therapeutic index of the antimicrobial compounds (Table 3). The HC50/MIC values of linear α-polylysine and all the glycopolymer–polypeptide conjugates are generally higher than those of many other antimicrobial peptides, such as indolicidin,43 melittin,44 magainin and protegrin,45 which have HC50/MIC values of approximately 8, 8, 5 and 3, respectively, when calculated using MIC toward S. aureus. The hemoselectivity of P(Lys) and P(GlyEMA)10–P(Lys) is even higher than that of polymyxin B, a topical antibiotic with an HC50/MIC value of approximately 141 against S. aureus.46 The IC50/MIC values of the conjugates do not appear to change appreciably in comparison to that of the linear α-polylysine, except for the P(GlyEMA)30–P(Lys) conjugates which show increased cytoselectivity when calculated using MIC towards the Gram-positive bacteria.
4. Conclusion
Preparation of four-arm star glycopolymers and polypeptide (α-polylysine), involving the use of atom transfer radical polymerization, N-carboxyanhydride ring-opening polymerization, and copper-catalyzed azide–alkyne ‘click’ cycloaddition techniques, was demonstrated. In comparison to the linear polypeptide, the glycopolymer–polypeptide conjugates exhibited a lower cytotoxic effect toward 3T3 fibroblast cells. All the conjugates, as well as the linear polypeptide, showed ultralow hemolytic activity, by being non-hemolytic up to a concentration of 8192 μg mL−1. Moreover, the glycopolymer–polypeptide conjugates showed a broad spectrum of antimicrobial activity toward Gram-positive and Gram-negative bacteria. On a peptide-content basis, the conjugation of the short-arm mannose-based glycopolymer to the polypeptide increased bactericidal efficacy, with only a half reduction in the selectivity value. The improved antibacterial effect was attributable to the increased binding affinity with bacterial surfaces, due to the pendant mannose moieties on the glycopolymer domains. The conjugates also withstood competition when binding the bacteria in the presence of free mannopyranoside. Conjugation with glycopolymer is therefore a promising strategy to increase the bactericidal activity and biocompatibility of antimicrobial polypeptides.Acknowledgements
This work was funded and supported by a Singapore Ministry of Education Tier 3 Grant (MOE2013-T3-1-002). We would like to thank Mr Liu Peng and Mr Xu Gang for their help with the flow cytometry assay.References
- Y. Liu and J. A. Imlay, Science, 2013, 339, 1210–1213 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- N. El-Gendy, J. Qian, K. Eshelman, M. Rivera and C. Berkland, Biomacromolecules, 2015, 16, 1480–1488 CrossRef CAS PubMed.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- A. T. Y. Yeung, S. L. Gellatly and R. E. W. Hancock, Cell. Mol. Life Sci., 2011, 68, 2161 CrossRef CAS PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, Trends Biotechnol., 2011, 29, 464–472 CrossRef CAS PubMed.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS PubMed.
- E. Cosimi, O. D. Engl, J. Saadi, M.-O. Ebert and H. Wennemers, Angew. Chem., Int. Ed., 2016, 55, 13127–13131 CrossRef CAS PubMed.
- S. Martens, J. Van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182–14185 CrossRef CAS PubMed.
- L. A. Carpino, S. Ghassemi, D. Ionescu, M. Ismail, D. Sadat-Aalaee, G. A. Truran, E. M. E. Mansour, G. A. Siwruk, J. S. Eynon and B. Morgan, Org. Process Res. Dev., 2003, 7, 28–37 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS PubMed.
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC.
- C. Zhou, X. Qi, P. Li, W. N. Chen, L. Mouad, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomacromolecules, 2010, 11, 60–67 CrossRef CAS PubMed.
- Y. Xi, T. Song, S. Tang, N. Wang and J. Du, Biomacromolecules, 2016, 17, 3922–3930 CrossRef CAS PubMed.
- R. F. Epand, W. L. Maloy, A. Ramamoorthy and R. M. Epand, Biochemistry, 2010, 49, 4076–4084 CrossRef CAS PubMed.
- A. Strassburg, F. Kracke, J. Wenners, A. Jemeljanova, J. Kuepper, H. Petersen and J. C. Tiller, Macromol. Biosci., 2015, 15, 1710–1723 CrossRef CAS PubMed.
- C. Krumm, S. Harmuth, M. Hijazi, B. Neugebauer, A.-L. Kampmann, H. Geltenpoth, A. Sickmann and J. C. Tiller, Angew. Chem., Int. Ed., 2014, 53, 3830–3834 CrossRef CAS PubMed.
- S. P. Le-Masurier, H. T. T. Duong, C. Boyer and A. M. Granville, Polym. Chem., 2015, 6, 2504–2511 RSC.
- M. Liu, K. Wang, X. Zhang, X. Zhang, Z. Li, Q. Zhang, Z. Huang and Y. Wei, Tetrahedron, 2015, 71, 5452–5457 CrossRef CAS.
- E. H. H. Wong, M. M. Khin, V. Ravikumar, Z. Si, S. A. Rice and M. B. Chan-Park, Biomacromolecules, 2016, 17, 1170–1178 CrossRef CAS PubMed.
- H. Park, S. Walta, R. R. Rosencrantz, A. Korner, C. Schulte, L. Elling, W. Richtering and A. Boker, Polym. Chem., 2016, 7, 878–886 RSC.
- D. Shi, M. Ran, H. Huang, L. Zhang, X. Li, M. Chen and M. Akashi, Polym. Chem., 2016, 7, 6779–6788 RSC.
- Z. Bar-Shavit, R. Goldman, I. Ofek, N. Sharon and D. Mirelman, Infect. Immun., 1980, 29, 417–424 CAS.
- P. M. Chaudhary, S. Sangabathuni, R. V. Murthy, A. Paul, H. V. Thulasiram and R. Kikkeri, Chem. Commun., 2015, 51, 15669–15672 RSC.
- M. N. Liang, S. P. Smith, S. J. Metallo, I. S. Choi, M. Prentiss and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13092–13096 CrossRef CAS PubMed.
- N. Sharon, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 527–537 CrossRef CAS PubMed.
- J. Lu, C. Fu, S. Wang, L. Tao, L. Yan, D. M. Haddleton, G. Chen and Y. Wei, Macromolecules, 2014, 47, 4676–4683 CrossRef CAS.
- J.-L. Reymond, M. Bergmann and T. Darbre, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- Y. Shi, R. W. Graff, X. Cao, X. Wang and H. Gao, Angew. Chem., Int. Ed., 2015, 54, 7631–7635 CrossRef CAS PubMed.
- S. G. Spain, L. Albertin and N. R. Cameron, Chem. Commun., 2006, 4198–4200 RSC.
- S. Zhang and Y. Zhao, Macromolecules, 2010, 43, 4020–4022 CrossRef CAS.
- M. A. Wikler, Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: Approved standard, Clinical Laboratory Standards Institute, Pennsylvania, 2009 Search PubMed.
- H. Shi, L. Liu, X. Wang and J. Li, Polym. Chem., 2012, 3, 1182–1188 RSC.
- D. Roy, B. Ghosn, E.-H. Song, D. M. Ratner and P. S. Stayton, Polym. Chem., 2013, 4, 1153–1160 RSC.
- M. Kar, B. Malvi, A. Das, S. Panneri and S. S. Gupta, J. Mater. Chem., 2011, 21, 6690–6697 RSC.
- D. Pranantyo, L. Q. Xu, E.-T. Kang, M. K. Mya and M. B. Chan-Park, Biomacromolecules, 2016, 17, 4037–4044 CrossRef CAS PubMed.
- D. Huesmann, A. Birke, K. Klinker, S. Türk, H. J. Räder and M. Barz, Macromolecules, 2014, 47, 928–936 CrossRef CAS.
- M. Hartmann, H. Papavlassopoulos, V. Chandrasekaran, C. Grabosch, F. Beiroth, T. K. Lindhorst and C. Röhl, FEBS Lett., 2012, 586, 1459–1465 CrossRef CAS PubMed.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C.-S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS PubMed.
- Y. Yuan, F. Liu, L. Xue, H. Wang, J. Pan, Y. Cui, H. Chen and L. Yuan, ACS Appl. Mater. Interfaces, 2016, 8, 11309–11317 CAS.
- H. M. Jindal, C. F. Le, M. Y. Mohd Yusof, R. D. Velayuthan, V. S. Lee, S. M. Zain, D. M. Isa and S. D. Sekaran, PLoS One, 2015, 10, e0128532 Search PubMed.
- W. L. Zhu, Y. H. Nan, K. S. Hahm and S. Y. Shin, J. Biochem. Mol. Biol., 2007, 40, 1090–1094 CAS.
- S. Rotem, I. Radzishevsky and A. Mor, Antimicrob. Agents Chemother., 2006, 50, 2666–2672 CrossRef CAS PubMed.
- S. Ruden, K. Hilpert, M. Berditsch, P. Wadhwani and A. S. Ulrich, Antimicrob. Agents Chemother., 2009, 53, 3538–3540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary synthesis procedures and characterization figures. See DOI: 10.1039/c7py00441a |
| This journal is © The Royal Society of Chemistry 2017 |