
An indacenodithiophene-based semiconducting polymer with high ductility for stretchable organic electronics†

Abstract
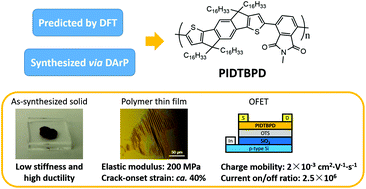
An alkyl-substituted indacenodithiophene-based donor–acceptor π-conjugated polymer (PIDTBPD) with low stiffness and high ductility is reported. The polymer was synthesized after DFT calculations predicted that it would have a kinked backbone conformation while showing strong intramolecular charge transfer (ICT), suggestive of the fact that it would be beneficial to the polymer's elasticity and charge mobility. Atom-efficient direct arylation polymerization (DArP) was exploited to synthesize the polymer. Mechanical studies indicate that PIDTBPD has relatively rapid stress-relaxation properties, which lead to a low elastic modulus of 200 MPa and high crack-onset strain of ca. 40% (lower limit). A moderate charge carrier mobility of 2 × 10−3 cm2 V−1 s−1 with a current on/off ratio of 2.5 × 106 was obtained from the fabricated OFETs. Further experiments were performed to elucidate the structural aspects of this polymer: UV-Vis and PL spectra suggest that minimal conformational change occurs in the polymer between its diluted solution and thin film states; DSC measurements indicate that the polymer's Tg is below −20 °C, allowing it to be in a rubbery state at room temperature; and XRD studies support this observation suggesting that the polymer is mostly amorphous at room temperature.
- This article is part of the themed collection: Pioneering Investigators
 Please wait while we load your content...
Please wait while we load your content...

