
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycan-decorated HPMA copolymers as high-affinity lectin ligands†
P.
Bojarová
*a,
P.
Chytil
*b,
B.
Mikulová
a,
L.
Bumba
a,
R.
Konefał
b,
H.
Pelantová
a,
J.
Krejzová
a,
K.
Slámová
a,
L.
Petrásková
a,
L.
Kotrchová
b,
J.
Cvačka
c,
T.
Etrych
b and
V.
Křen
a
aInstitute of Microbiology, Czech Academy of Sciences, Vídeňská 1083, CZ-14220 Prague 4, Czech Republic. E-mail: bojarova@biomed.cas.cz
bInstitute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovský Sq. 2, CZ-162 06 Prague 6, Czech Republic. E-mail: chytil@imc.cas.cz
cThe Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, CZ-166 10 Prague 6, Czech Republic
First published on 21st March 2017
Abstract
Novel conjugates of N-(2-hydroxypropyl) methacrylamide (HPMA) copolymers tethered with chitooligosaccharidic epitopes of varying lengths were shown to be potent ligands of a model lectin, wheat germ agglutinin (WGA). The azide-functionalized oligosaccharidic epitopes were prepared by the action of Tyr470Asn mutant β-N-acetylhexosaminidase from Talaromyces flavus in a single reaction step and were conjugated to HPMA copolymer precursors in a defined pattern and density through Cu+-catalyzed azide–alkyne cycloaddition. The soluble, biocompatible, and structurally flexible synthetic glycopolymers were studied for their binding to WGA in a competitive enzyme-linked lectin assay (ELLA), and the kinetics of interaction were analyzed by surface plasmon resonance (SPR). To the best of our knowledge, this study presents the first HPMA copolymers derivatized with long oligosaccharides that demonstrate high affinity to a lectin target. The binding affinities in the low nanomolar and subnanomolar ranges place the prepared glycopolymers among the best WGA ligands reported to date. This study demonstrates the targeting potential of these glycopolymers for therapeutically relevant lectins.
Introduction
Nature-derived proteins are usually decorated with specific patterns of covalently linked carbohydrate moieties that encode a variety of physiological and pathological processes such as cell signaling and communication, cell adhesion, pathogen recognition, inflammation, and immune response.1 This sugar code is prominently deciphered by virtually ubiquitous glycan-binding proteins named lectins. Because monovalent lectin–glycan interactions are relatively weak, the biological response in vivo needs to be amplified through multivalent displays of sugar ligands. The resulting multivalent complexes show a higher collective affinity (i.e., avidity) than the simple sum of their individual binding events, exhibiting the so-called multivalency or cluster glycoside effect.2Current glycomics can mimic nature-like multivalent sugar displays in the form of synthetic glycoconjugates based on a polymer, dendrimer or solid scaffolds with miscellaneous compositions, architectures, and flexibilities.3,4 The orientation and density of the carbohydrate epitopes as well as the type and length of the connecting spacers are crucial parameters to be considered for the appropriate multiplication of the weak monovalent glycan–lectin interaction. Selective and efficient lectin targeting results in useful glyco-therapeutics and diagnostic tools, agents for targeted drug delivery, cell imaging or for other biological and biomedical applications.4
Wheat germ agglutinin (WGA) is a conventional model lectin used for studying multivalent carbohydrate–lectin interactions and structure–affinity relationships in tailored multivalent glycoconjugates.5 It is abundantly present in the endosperm of wheat (Triticum vulgaris) and in other natural sources including some mammalian cell membranes. WGA binds N-acetyl-D-glucosamine (GlcNAc) residues and their β1,4-linked oligomers as well as sialic acid. In a solution at physiological pH, WGA exists mostly as a homodimer with the molecular weight of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Each monomer comprises four hevein-like domains (A–D), each containing a unique binding site for GlcNAc, with the shortest mutual distance of 13–14 Å.6 Recent studies reported by Renaudet and coworkers presented several complex dendrimeric inhibitors of WGA based on cyclopeptides7 and polyester scaffolds containing up to 96 GlcNAc units;8 these inhibitors demonstrated binding affinities towards WGA in the nanomolar range.
000 g mol−1. Each monomer comprises four hevein-like domains (A–D), each containing a unique binding site for GlcNAc, with the shortest mutual distance of 13–14 Å.6 Recent studies reported by Renaudet and coworkers presented several complex dendrimeric inhibitors of WGA based on cyclopeptides7 and polyester scaffolds containing up to 96 GlcNAc units;8 these inhibitors demonstrated binding affinities towards WGA in the nanomolar range.
For the synthesis of glycopolymer scaffolds, various strategies including living radical, cationic/anionic, and ring opening metathesis polymerizations of carbohydrate-bearing monomeric blocks have been adopted in the last ten years.9 An alternative approach, also applicable for more complex glycan epitopes, is to conjugate functionalized glycan chains with appropriate functional groups distributed ad arbitrium along the polymer backbone. Advantageously, this strategy can exploit the elegant click chemistry10 approach – Cu+-catalyzed azide–alkyne cycloaddition (CuAAC),11 yielding triazole from an azide and a terminal alkyne.
The synthetic polymer carriers based on N-(2-hydroxypropyl) methacrylamide (HPMA) copolymers represent the most attractive agents for specific drug delivery and targeting12 due to their non-immunogenicity, non-toxicity, biocompatibility, and water solubility.
If drugs or therapeutics for in vivo applications are bound to these polymers (affording the so-called polymer prodrugs), the occurrence of possible adverse effects is diminished and/or retarded. A typical application is in the treatment of cancer, especially that manifested with solid tumors, in which the polymer-prodrugs accumulate through the enhanced permeability and retention (EPR) effect.13 However, the typical uptake efficiency barely exceeds 10% of the used dose. Conjugation of polymer prodrugs to ligands that are specific to selected tumor receptors is the main idea of the active targeting treatment approach. Lectin receptors constitute very promising targets;4 the conjugation of polymers to specific glycan epitopes results in enhanced selectivity of binding to these receptors, affording high-affinity multivalent ligands with potential biomedical uses.
In the past, several studies have been devoted to exploring the ability of HPMA copolymers decorated with simple sugars to serve as ligands of endogenous lectins14 such as the asialoglycoprotein receptor or galectin-3. The main problem in the construction of these glycopolymers has always been the synthesis of highly specific tailored carbohydrate epitopes, especially those of an oligosaccharidic nature, to be efficiently conjugated to the polymer carrier. To the best of our knowledge, the only known oligosaccharide-bearing HPMA-based copolymer, recently reported by Roy et al.,15 carried a trimannoside prepared through a complex multistep synthetic procedure.
This study describes the synthesis of thirteen HPMA copolymers decorated with chitooligosaccharide epitopes of varying lengths and their capacity to interact with WGA as a model lectin. Azide-functionalized chitooligomers of one to five GlcNAc units were prepared in a single transglycosylation reaction catalyzed by the Tyr470Asn mutant of β-N-acetylhexosaminidase from Talaromyces flavus;16 they were conjugated in a defined density to propargyl-functionalized HPMA copolymers by the CuAAC reaction. The glycopolymers exhibited extremely strong binding to WGA, as shown in the competitive ELLA assays and in SPR studies; the affinities of the best conjugates were in the subnanomolar range, which ranks them among the strongest ligands reported to date.8 The present structures show a promising path to achieve high-affinity biocompatible glycopolymer carriers targeted at lectin-displaying structures.
Experimental
Materials and methods
:1) was used at the flow rate of 30 μL min−1, and the samples were injected using a 2 μL loop. The mass spectra of negatively charged ions were internally calibrated using deprotonated palmitic acid as a lock mass. Data were acquired and processed using Xcalibur software (Thermo Fisher Scientific).
The 1H and 13C NMR spectra of chitooligomer standards 26–29 were compared with the previously reported data;17 very good agreement was found for the carbon chemical shifts. Due to the strong overlap of proton signals, the assignment of individual GlcNAc units in 5–9 was mainly achieved using HSQC-TOCSY experiments, supported by the information extracted from the HSQC and COSY spectra. The β-anomeric configuration of the sugar units was determined from the magnitudes of the JH-1,H-2 coupling constants. The up-field shifted resonances of all the C-2 carbons were in accordance with N-acetylation at this position. The glycosidic linkage position was deduced from the downfield glycosylation shifts of the involved C-4 carbons and unambiguously confirmed using the heteronuclear correlations of these carbons with the anomeric protons of subsequent GlcNAc units. The attachment of the azidoethyl group was also confirmed by HMBC correlations.
The glycopolymers and their polymer precursors were investigated via a Bruker Avance III 600 spectrometer operating at 600.2 MHz with DMSO-d6 or D2O as the solvent. The width of the 90° pulse was 10 μs, with a relaxation delay of 10 s. The acquisition time was 3.63 s with 200 (16 for kinetics) scans. The structure and purity of the monomers and the contents of the polymer-bound propargyl groups and carbohydrates were determined in DMSO-d6. For the calculations, integral intensities of δ (ppm) = 3.67 (1 H, br s, CH–OH) or δ (ppm) = 4.71 (1 H, br s, CH–OH) of the HPMA monomer unit were used. To calculate the contents of propargyl groups in the polymer precursors 12a and 12b, these signals were compared with the integral intensities of δ (ppm) = 3.85 (2 H, br, CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C); to calculate the contents of carbohydrate 3 in the glycopolymers 16–18, the signals were compared with the integral intensity δ (ppm) ≈ 7.84 (1 H, s, CH of triazole). Moreover, to calculate the contents of carbohydrate 2 in the glycopolymers 13–15, carbohydrate 5 in glycopolymers 19–20, carbohydrate 6 in glycopolymers 21–22, carbohydrate 7 in glycopolymers 23–24, and carbohydrate 8 in glycopolymer 25, the signals were compared with the integral intensity of δ (ppm) ≈ 7.80 (1 H, br, CH of triazole and NH–acetyl).
C); to calculate the contents of carbohydrate 3 in the glycopolymers 16–18, the signals were compared with the integral intensity δ (ppm) ≈ 7.84 (1 H, s, CH of triazole). Moreover, to calculate the contents of carbohydrate 2 in the glycopolymers 13–15, carbohydrate 5 in glycopolymers 19–20, carbohydrate 6 in glycopolymers 21–22, carbohydrate 7 in glycopolymers 23–24, and carbohydrate 8 in glycopolymer 25, the signals were compared with the integral intensity of δ (ppm) ≈ 7.80 (1 H, br, CH of triazole and NH–acetyl).
The course of azide–alkyne cycloaddition was monitored in situ by 1H NMR. Polymer 12a (20 mg, 13.8 μmol of propargyl groups), carbohydrate 3 (4.00 mg, 13.8 μmol), and sodium ascorbate (1.32 mg, 6.9 μmol) were dissolved in D2O (0.6 mL); the solution was then placed in a NMR cuvette and bubbled with argon. After this, a solution of the catalyst CuSO4·5H2O (1.72 mg, 6.9 μmol) in D2O (50 μL), bubbled with argon, was added to the reaction mixture. The 1H NMR spectra were obtained before the reaction and 10, 30, 45, and 60 min after the addition of Cu+ catalyst.
:1, v/v) and analyzed via a TSKgel Amide-80 5 μm HILIC column (250 × 4.6 mm, Tosoh Bioscience, DE). Binary gradient elution was used: mobile phase A = 100% acetonitrile; mobile phase B = water; gradient: 22% B for 0–7 min, 22–35% B for 7–20 min; 35% B for 20–25 min; and 35–22% B for 25–26 min. The flow rate was 1 mL min−1 at 27 °C and the injection volume was 1 μL; the samples were detected at 200 nm. Retention times were as follows: 2-acetamido-2-deoxy-β-D-glucopyranosyl azide 2, 5.54 min; 2-azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranoside 3, 5.20 min; p-nitrophenyl 2-acetamido-2-deoxy-β-D-glucopyranoside 4, 4.09 min; dimer 5, 8.01 min; trimer 6, 12.89 min; tetramer 7, 17.07 min; pentamer 8, 21.48 min; and hexamer 9, 24.29 min.
The purity of the monomers for polymer synthesis was determined using a C18 reverse-phase Chromolith Performance RP-18e column (4.6 × 100 mm, Merck Millipore) with diode array detection. The mobile phase was water/acetonitrile/0.1% TFA with a gradient of 5–95% v/v acetonitrile and a flow rate of 5 mL min−1.
The molecular weights and dispersities of the polymers were determined using a Shimadzu HPLC system equipped with a gel permeation chromatography (GPC) column (TSKgel G3000SWxl, 300 × 7.8 mm; 5 μm), connected to refractive index (RI) Optilab®-rEX and multiangle light scattering (MALS) detectors (DAWN HELEOS II, Wyatt Technology Co., USA). A mixture of methanol/0.3 M sodium acetate buffer, pH 6.5 (4:1, v/v) at the flow rate of 0.5 mL min−1 was used as the mobile phase.
Synthetic procedures
:MeOH, 7:3, v/v). Deprotection according to Zemplén afforded the title compound 3 as a white solid (0.87 g, 3 mmol; 27% yield). MS (ESI−) m/z 289.1 [M − H]−, 325.1 [M + Cl]−, 335.1 [M + HCOO]−, 349.1 [M + CH3COO]−. [α]20589 −46.7 (c 0.242 in H2O). For 1H and 13C NMR data, see the ESI, Table S1 and Fig. S1a and b.†
:H2O:NH4OH aq., 7:2:1, v/v/v). After ca. 90% conversion was reached in the reaction mixture (after 2.5–3.5 h), 50–100% of the original amount of donor 4 was added, and the reaction was monitored further.
For the preparative reaction (Scheme 1), donor 4 (51 mg, 0.15 mmol), acceptor 3 (87 mg, 0.30 mmol), and the Tyr470 Asn mutant of the β-N-acetylhexosaminidase from T. flavus (4.73 U, 1.4 mg, 167 μL) were incubated in 50 mM citrate-phosphate buffer (pH 5.0, 3.0 mL) at 35 °C and 1000 rpm under monitoring by HPLC and TLC as described above. After 2.5 h (ca. 90% conversion), another portion of 4 (51 mg, 0.15 mmol) was added. After 8 h, the reaction was stopped by boiling for 2 min, and the mixture was centrifuged (13500 rpm; 10 min) after cooling down to room temperature. The supernatant was concentrated in vacuo to ca. 2 mL volume and loaded onto a Biogel P-2 column (2.6 × 100 cm, Bio-Rad, USA). Water was used as the mobile phase at the elution rate of 10 mL h−1. The fractions containing the products were obtained, traces of remaining p-nitrophenol were extracted with ethyl acetate (3 × 10 mL), and the samples were lyophilized; products 5–9 were obtained as white solids. Pure acceptor 3 was partially recovered (35 mg). For the respective NMR data, see the ESI, Tables S2–S6 and Fig. S2–S6.† 2-Azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside (5), 32 mg, 0.065 mmol; HRMS (ESI−): found m/z 492.19479 (expected 492.19473 for [M − H], C18H30O11N5); [α]20589 −34.7 (c 0.190 in H2O). 2-Azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside (6) 19 mg, 0.028 mmol; HRMS (ESI−): found m/z 695.27420 (expected 695.27410 for [M − H]−, C26H43O16N6); [α]20589 −21.9 (c 0.187 in H2O). 2-Azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside (7), 10 mg, 0.011 mmol; HRMS (ESI−): found m/z 898.35309 (expected 898.35347 for [M − H], C34H56O21N7); [α]20589 −22.3 (c 0.175 in H2O). 2-Azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside (8), 2.0 mg, 1.81 μmol; HRMS (ESI−): found m/z 1101.43077 (expected 1101.43285 for [M − H]−, C42H69O26N8); [α]20589 −12.3 (c 0.089 in H2O). 2-Azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranosyl-(1→4)-2-acetamido-2-deoxy-β-D-glucopyranoside (9), 2.7 mg, 2.07 μmol; HRMS (ESI−): found m/z 1304.51088 (expected 1304.51222 for [M − H]−, C50H82O31N9). The total isolated yield of the functionalized oligosaccharides 5–9 was 60% (based on the consumed acceptor 3).
 | ||
| Scheme 1 Chemoenzymatic synthesis of the functionalized oligosaccharide precursors 5–9. | ||
:1, v/v, at 70 °C for 16 h. The molar ratio of monomers/chain transfer agent/initiator was 350:2:1; the molar ratios of monomers 10/11 were 9:1 for 12a and 4:1 for 12b. The terminal dithiobenzoate group was removed by a method described elsewhere.25 Aminolysis of thiazolidine-2-thione groups by propargylamine was carried out in dimethylformamide (DMF) in the presence of N,N-diisopropylethylamine (DIPEA) as a base. Detailed synthetic procedures have been described in the ESI, section 3.1.†
 | ||
| Scheme 2 Synthesis of glycopolymers 13–25. AIBN, 2,2′-azobisisobutyronitrile; DIPEA, N,N-diisopropylethylamine. | ||
Glycopolymers 13–25 were prepared by the reaction of propargyl groups of polymer precursor 12a or 12b with the respective azido-functionalized GlcNAc (2 and 3) or functionalized chitooligosaccharides 5–8 by Cu+-catalyzed azide–alkyne cycloaddition in water (Scheme 2). Glycopolymers 15 and 18 were synthesized from the polymer carrier 12b, containing a higher amount of propargyl groups (21.0 mol%). The other glycopolymers were synthesized from 12a. The sample preparation of glycopolymer 17 is as follows. A solution of CuSO4·5H2O (2.15 mg, 8.6 μmol) in water (25 μL) was added to a mixture of copolymer 12a (25 mg, 17.2 μmol of propargyl groups), sodium ascorbate (1.71 mg, 8.6 μmol), and carbohydrate 3 (5.00 mg, 17.2 μmol) dissolved in water (225 μL). The reaction mixture was bubbled with argon before and after the addition of Cu+ to the reaction mixture and was vortexed. After 30 min, PBS buffer with 5% EDTA disodium salt (750 μL) was added, and the polymer was purified by gel filtration via a PD10 desalting column containing Sephadex G-25 resin (GE Healthcare Life Sciences) using water as the mobile phase. The polymer fraction was lyophilized to obtain glycopolymer 17 (28.8 mg; 96% yield). The yields of glycopolymers 13–25 ranged approximately from 90 to 95%. For a detailed description of glycoconjugates, see Table 1.
| Compound |
M
na (g mol−1) |
Đ | Sugar motif | n | IC50 (nM) |
r
pc |
r p/nd |
|---|---|---|---|---|---|---|---|
| a The molecular weights (Mn) and dispersities (Đ) of the polymers were determined using GPC with MALS and RI detection. b Average number of glycans per polymer chain (glycan content, mol%); n = 1, monovalent standard. c Relative potency, i.e. IC50 (monovalent standard)/IC50 (multivalent glycopolymer). d Relative potency per glycan. | |||||||
| 1 | GlcNAc | 1 | (17400 ± 2800) × 103 |
1 | 1 | ||
| 26 | (GlcNAc)2 | 1 | (800 ± 170) × 103 | 1 | 1 | ||
| 27 | (GlcNAc)3 | 1 | (107 ± 39) × 103 | 1 | 1 | ||
| 28 | (GlcNAc)4 | 1 | 70000 ± 3000 |
1 | 1 | ||
| 29 | (GlcNAc)5 | 1 | 41000 ± 9000 |
1 | 1 | ||
| 13 | 21000 |
1.13 | GlcNAc-N | 8.1 (6.3%) | 49600 ± 9900 |
351 | 43 |
| 14 | 22900 |
1.12 | GlcNAc-N | 12.3 (9.1%) | 10100 ± 1400 |
1723 | 140 |
| 15 | 27300 |
1.11 | GlcNAc-N | 21.9 (15.1%) | 360 ± 100 | 48333 |
2207 |
| 16 | 24600 |
1.05 | GlcNAc-O | 9.3 (6.2%) | 5100 ± 2300 | 3412 | 367 |
| 17 | 25200 |
1.06 | GlcNAc-O | 13.6 (9.4%) | 350 ± 120 | 49714 |
3655 |
| 18 | 29100 |
1.07 | GlcNAc-O | 24.0 (16.4%) | 7.6 ± 2.3 | 2289474 |
95395 |
| 19 | 22800 |
1.12 | (GlcNAc)2-O | 7.1 (5.4%) | 3.4 ± 1.1 | 235294 |
33140 |
| 20 | 24600 |
1.13 | (GlcNAc)2-O | 11.8 (9.4%) | 1.88 ± 0.47 | 425532 |
36062 |
| 21 | 23800 |
1.12 | (GlcNAc)3-O | 6.9 (5.3%) | 3.68 ± 0.98 | 29076 |
4214 |
| 22 | 26600 |
1.14 | (GlcNAc)3-O | 10.6 (8.2%) | 1.34 ± 0.64 | 79851 |
7533 |
| 23 | 25900 |
1.33 | (GlcNAc)4-O | 6.6 (4.8%) | 2.58 ± 0.46 | 27131 |
4111 |
| 24 | 29200 |
1.12 | (GlcNAc)4-O | 9.9 (7.2%) | 2.78 ± 0.19 | 25180 |
2543 |
| 25 | 22800 |
1.09 | (GlcNAc)5-O | 2.9 (2.2%) | 11.6 ± 1.6 | 3534 | 1219 |
Enzyme-linked lectin assay (ELLA)
The competitive ELLA assay was principally performed, as has been previously described,7 after optimizing the reactant volumes, concentrations, and incubation times as follows. Typically, 96-well Nunc-immuno™ MaxiSorp™ microtiter flat-bottom plates were coated with PAA-GlcNAc (GlcNAc polyacrylamide, 0.3 μg mL−1 in 50 mM Na2CO3/NaHCO3 buffer (pH 9.6); 100 μL per well) for 1 h at 37 °C. The coated wells were then washed with T-PBS buffer (i.e., phosphate buffered saline (pH 7.4) containing 0.05% v/v Tween 20; 4 × 300 μL per well). The wells were blocked with 1.5% BSA in T-PBS (200 μL per well) for 4 h at 37 °C and washed with T-PBS (4 × 300 μL). Each inhibitor (chitooligomer standards 1, 26–29 or glycopolymers 13–25, in serial dilutions) was pre-incubated with WGA-HRP (wheat germ agglutinin labeled with horseradish peroxidase, 0.2 μg mL−1) at the ratio of 1:1 in PBS buffer (pH 7.4) at 37 °C and 400 rpm for 1 h. The inhibitor serial dilutions were determined such that both plateaus of the sigmoidal inhibition curve were well defined. The preincubated mixtures were transferred to the blocked wells and incubated for 1 h at 37 °C. After washing with T-PBS (4 × 300 μL), the activity of WGA-HRP bound to the GlcNAc-coated wells was determined via a SIGMAFAST™ OPD tablet set (0.4 mg mL−1o-phenylene diamine dihydrochloride and 0.4 mg mL−1 urea hydrogen peroxide in 0.05 M phosphate-citrate buffer, pH 5.0; 100 μL per well). The OPD reaction was stopped by adding H2SO4 (30% v/v, 50 μL per well) and the absorbance was measured at 490 nm via a Tecan Sunrise microplate absorbance reader (Tecan Group Ltd, Switzerland). WGA-HRP preincubated with PBS buffer instead of inhibitor was used as a blank, corresponding to 100% binding (no inhibition). The inhibition (%) was calculated as follows:where A is the absorbance. It was plotted against the concentration of the respective inhibitor and fitted to a four-parameter logistic equation using Prism 7.0 (GraphPad Software Inc, La Jolla, USA) to afford a sigmoidal dose–response inhibition curve. The half maximal inhibitor concentration IC50 was calculated as the inhibitor concentration (based on the molarity of the glycopolymer) at which 50% inhibition of the binding of WGA-HRP to the GlcNAc-coated wells was reached. The resulting IC50 values (Table 1) were calculated from three independent experiments.

Surface plasmon resonance (SPR)
SPR measurements were performed at 25 °C using a GLC sensor chip mounted on a ProteOn XPR36 Protein Interaction Array System (Bio-rad, Hercules, CA, USA). The wheat germ agglutinin (WGA) protein was diluted to a final concentration of 10 μg mL−1 in phosphate buffered saline (PBS) and captured to the chip using a ProteOn amine coupling kit (Bio-Rad, Hercules, CA, USA) at the flow rate of 30 μL min−1. The analyte glycopolymers were serially diluted in the running buffer (PBS supplemented with 0.005% Tween 20) and injected in parallel over the immobilized WGA lanes at the flow rate of 30 μL min−1. The WGA surface was typically regenerated with an injection of 50 mM HCl for 1 min. The sensograms were corrected for sensor background by interspot referencing (the sites within the 6 × 6 array that are not exposed to ligand immobilization but are exposed to analyte flow) and double referenced by subtraction of the analyte using a blank injection. The data were globally analyzed by simultaneously fitting both the association and the dissociation phases for five different analyte concentrations using a 1:1 Langmuir-type binding and the bivalent analyte model to determine the association and dissociation rate constants. The Langmuir-type model assumes the interaction between the ligand (L) and the analyte (A), resulting in the direct formation of the final complex (LA):where ka and kd are the association and the dissociation rate constants, respectively. The bivalent analyte model assumes a two-step association process:

where the first binding event is described by ka1 and kd1, whereas ka2 and kd2 describe the association and dissociation of the second binding event, respectively. An apparent equilibrium dissociation constant, KD, for the 1
 :1 Langmuir-type binding was determined as follows:
:1 Langmuir-type binding was determined as follows:and that for the bivalent analyte binding model was determined as follows:


Results and discussion
Chemoenzymatic synthesis of functionalized chitooligomers 5–9
For elegant, gentle, well-defined, and high-yielding coupling of oligosaccharide epitopes to HPMA copolymers via Cu+-catalyzed azide–alkyne cycloaddition, we used two functionalities: an azido-moiety directly attached to the C-1 of GlcNAc, as in 2-acetamido-2-deoxy-β-D-glucopyranosyl azide (2), and a more flexible 2-azidoethyl group, as in 2-azidoethyl 2-acetamido-2-deoxy-β-D-glucopyranoside (3) and its oligomers (5–9). Functionalized carbohydrate precursors 2 and 3 were essentially prepared as has been previously described,19 with minor modifications. For the preparation of 2-azidoethyl-derivatized chitooligosaccharides containing 2–6 GlcNAc moieties (5–9), we employed a one-step transglycosylation reaction catalyzed by the Tyr470Asn mutant of the β-N-acetylhexosaminidase from Talaromyces flavus, recently developed in our laboratory.16 This mutant enzyme is the first reported transglycosidase from family 20 of glycoside hydrolases and is one of very few mutant glycosidases employing a substrate-assisted catalytic mechanism.26 By substituting the active-site Tyr470 with asparagine, the natural hydrolytic activity was practically abolished; the enzyme almost exclusively functioned in the transglycosylation mode, producing β-1,4-linked oligosaccharides of up to six GlcNAc units with 100% selectivity using the readily available p-nitrophenyl 2-acetamido-2-deoxy β-D-glucopyranoside donor (4) (Scheme 1). This enzymatic synthesis of oligosaccharides 5–9 was especially challenging because O-ethylazido glycosides are favorable substrates for hydrolysis by glycosidases. Only minor traces of hydrolytic products were observed during transglycosylation with our mutant enzyme. The mutant enzyme was recombinantly expressed in Pichia pastoris in a yield of 10 mg of the pure enzyme from 100 mL of culture media. After tuning the reaction conditions (HPLC) and scale-up, a total of 66 mg of functionalized chitooligomeric products 5–9 were isolated by gel permeation chromatography after one synthetic step. In addition to the desired functionalized products 5–9, minor amounts of p-nitrophenyl chitooligomers were formed in the reaction. Hexasaccharide 9 could be isolated only in low amounts; therefore, only disaccharide 5, trisaccharide 6, tetrasaccharide 7, and pentasaccharide 8 were used for coupling to the HPMA copolymers.Synthesis and structural characterization of glycopolymers 13–25
Polymer precursors containing propargyl groups, suitable for covalent attachment of functionalized saccharides, were synthesized by copolymerization of HPMA (10) with monomer 11 containing amino-reactive thiazolidine-2-thione groups according to a previously described procedure.22,23 Then, aminolysis of the thiazolidine-2-thione groups by propargylamine was performed to yield the polymer precursors 12a and 12b. Precursors 12a and 12b contained 10.2 mol% and 21.0 mol% propargyl groups, respectively, which corresponded to the desired amounts of carbohydrates for conjugation. The RAFT polymerization technique enabled us to prepare these polymer carriers with a narrow distribution of molecular weights (dispersity Đ = 1.08 for 12a and 1.09 for 12b). The molecular weights of the polymer carriers (Mn = 20200 g mol−1 for 12a and 21000 g mol−1 for 12b) corresponded to the typical sizes of the HPMA-based copolymer conjugates used for in vivo applications.
In total, thirteen conjugates of functionalized oligosaccharides with HPMA copolymers were prepared by means of the efficient CuAAC click reaction. Fig. 1 shows the reaction progress during the synthesis of glycopolymer 17, as monitored by 1H NMR. Within 10 min after adding the Cu+ catalyst, compound 3 was completely bound to polymer precursor 12a, as demonstrated by the disappearance of peaks 8 and 9, belonging to the propargyl moiety, and by the formation of peaks 10 and 11, related to the triazole moiety (Fig. 1).
 | ||
| Fig. 1 Progress of the CuAAC click reaction during the synthesis of glycopolymer 17. The reaction was monitored in situ by 1H NMR spectroscopy in D2O. (a) Reaction mixture before catalyst addition; (b) 10 min after catalyst addition; and (c) 1 h after catalyst addition. The small peaks marked as (l) belong to the carbohydrate protons. | ||
GlcNAc azide 1 yielded glycoconjugates 13–15 (containing 6.3, 9.1, and 15.1 mol% of 1, respectively). 2-Azidoethyl GlcNAc 3 afforded glycoconjugates 16–18 (containing 6.2, 9.4, and 16.4 mol% of 3, respectively). Glycopolymers 19 and 20 originated from dimer 5 (with 5.4 and 9.4 mol% of 5, respectively), glycopolymers 21 and 22 from trimer 6 (with 5.3 and 8.2 mol% of 6, respectively), and glycopolymers 23 and 24 from tetramer 7 (4.8 and 7.2 mol% of 7, respectively). Pentamer 8 yielded glycopolymer 25 (2.2 mol%). In general, lower amounts and simpler structures of saccharides were nearly quantitatively bound to the polymer precursors, in contrast to the longer chitooligomer chains. This was probably caused by steric hindrance decreasing the content of bound saccharide, especially in the case of pentamer 8. The molecular weights of the glycopolymers slightly increased as compared to those of their respective polymer precursors. This increase may be ascribed to the presence of the saccharide moieties in the polymer structure. As expected, the attachment of saccharides did not influence the polymer dispersity.
Inhibitory potency of glycopolymers 13–25 in the ELLA assay
Glycopolymers 13–25 were assayed for their abilities to inhibit the binding of wheat germ agglutinin (WGA) to immobilized GlcNAc in a competitive enzyme-linked lectin assay (ELLA). The inhibitory potencies were compared to those of the respective monovalent carbohydrate standards, namely GlcNAc (1), and its chitooligosaccharides: N,N′-diacetylchitobiose (26) to N,N′,N′′,N′′′,N′′′′-pentaacetylchitopentaose (29). Wheat germ agglutinin is a well-known homodimeric lectin that can bind GlcNAc as well as its β1,4-linked oligomers; therefore, it was a perfectly suitable model for our study. For detection purposes, WGA was used in the form of a conjugate with horseradish peroxidase (WGA-HRP). The inhibition of the binding of WGA-HRP to immobilized GlcNAc by glycopolymers 13–25 or by standard carbohydrates 1, 26–29 was plotted against their concentrations; the obtained sigmoidal curves enabled calculation of the half maximal inhibitor concentration IC50 (i.e., the inhibitor concentration at which 50% inhibition of the binding of WGA-HRP to immobilized GlcNAc was reached), as shown in Fig. 2 and Table 1. The sugar-free HPMA copolymer precursors 12a and 12b showed no inhibition.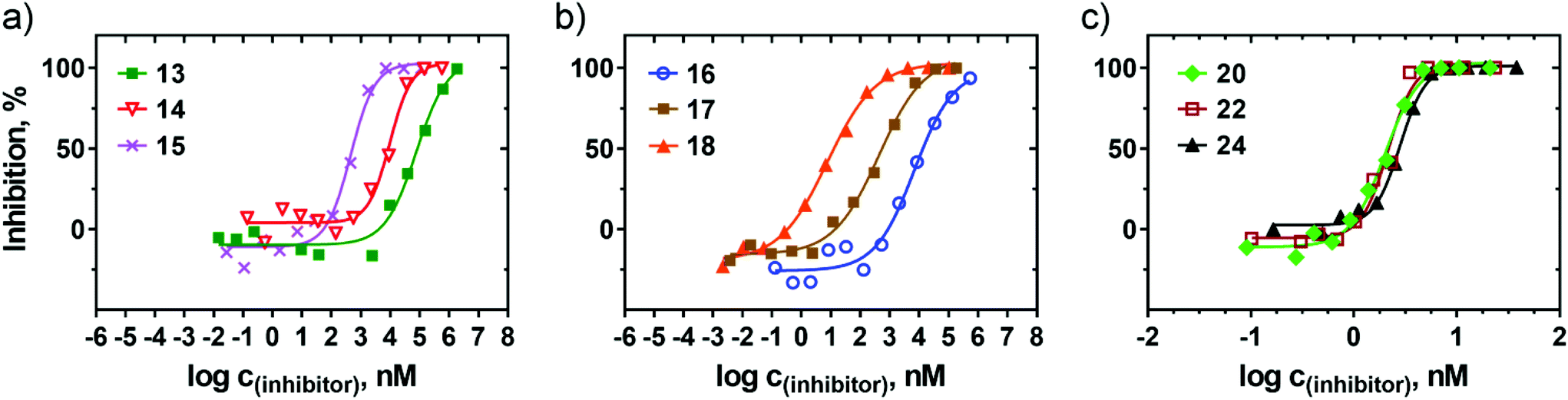 | ||
| Fig. 2 Competitive inhibition of WGA-HRP binding to GlcNAc by multivalent glycopolymers 13–25, as determined by ELLA. (a) Inhibition by glycopolymers 13–15 (6.3–15.1 mol% of N-bound GlcNAc, respectively). (b) Inhibition by glycopolymers 16–18 (6.2–16.4 mol% of O-bound GlcNAc, respectively). (c) Inhibition by glycopolymers 20, 22, and 24 (containing comparable amounts of the di-, tri-, and tetrasaccharide glycans, respectively). | ||
The results clearly show a vast difference between the two linkers tested – whereas the GlcNAc-HPMA conjugates containing the shorter and more rigid azido linker (13–15) demonstrated IC50 values around the μM range (from 49 μM to 360 nM, depending on the carbohydrate content), the more flexible 2-azidoethyl linker brought a significant improvement in the inhibition potency, with an IC50 as low as 7.6 nM for 18 (16 mol% GlcNAc). In the case of both linkers, a significant decrease in IC50 of roughly one order of magnitude was observed with the increasing GlcNAc content from 6 to 9 mol% and again from 9 to 15 mol%. This reflects an easier structural orientation of the GlcNAc units attached to the polymer chain towards the lectin binding sites. The relative inhibitory potency per glycan (rp/n, Table 1) is a measure of potency enhancement induced by the cluster glycoside effect. In this respect, glycopolymer 18 proved to be the best compound in the series, demonstrating a multivalency factor of over 95000 compared to that of the monovalent GlcNAc standard.
All chitooligosaccharide-HPMA conjugates (19–25, containing glycans with two to five GlcNAc moieties) exhibited excellent inhibition potencies in the low nanomolar range: glycopolymer 22, derivatized with 8.2 mol% of trimer 6, had an IC50 of 1.34 nM. Note that no significant improvement of inhibition potency was observed with the increasing molar content of glycans in the oligosaccharide-presenting series, in contrast to the GlcNAc conjugates. This may be because the longer oligosaccharide chains, combined with the high flexibility of the polymer backbone, may structurally adapt to lectin binding requirements even at low carbohydrate contents (7–10 glycans per polymer chain). This is especially outstanding in the case of N,N′,N′′,N′′′,N′′′′-pentaacetylchitopentaose-HPMA conjugate 25, where only three glycans per polymer molecule resulted in an IC50 of 11.6 nM – a similar result to that of conjugate 18, carrying twenty-four GlcNAc units. This is also an excellent result compared to that of the previously reported dendrimers, which generally required several dozens of GlcNAc units per molecule to reach comparable inhibition potencies.8 It may be concluded that the derivatization of HPMA copolymer with longer chitooligomer chains results in highly efficient WGA inhibitors, even with a strikingly low carbohydrate content in the molecule. This parameter is especially important due to the fact that a high molar content of the carbohydrate portion may result in altered physicochemical properties and pharmacokinetics of the glycopolymer compared to that of the sugar-void analogue.
Kinetics of interaction of glycopolymers 13–25 with WGA
The binding of glycopolymers 13, 15, 17, 18, and 19–25 to WGA was further investigated by surface plasmon resonance (SPR), and the results are summarized in Table 2 and Fig. 3. WGA was immobilized to a sensor chip by amine coupling chemistry, and serially diluted glycopolymers were injected in parallel over the sensor surface. To minimize non-specific binding and the mass transfer effect, three coupling concentrations of WGA leading to the refractive index changes of 500, 1000, and 1500 relative units (RU) were tested. Initial estimates of the koff values showed a concentration dependence at the coupling levels of 1000 and 1500 RU, indicating a mass transfer effect. The coupling level of 500 RU resulted in clear responses at the flow rate of 30 μL min−1, and these conditions were chosen for the subsequent experiments. The real time kinetics of the interactions between each glycopolymer and WGA are shown in Fig. 3. The concentration-dependent binding curves revealed a clear SPR response, maintaining the typical association and dissociation phases of the sensograms. The binding of all the tested glycopolymers was exclusively conferred by the sugar moieties because no binding of sugar-free propargyl-functionalized HPMA copolymer 12a to the WGA surface was detected (data not shown). The kinetic parameters of the interactions were calculated from global fitting of the concentration-dependent binding curves. As shown in Fig. 3, the kinetics of the interactions of N-linked glycopolymers 13 and 15 did not fit any available binding models, indicating a rather complex interaction of the N-linked glycopolymers with WGA. The estimated dissociation constants (KD), derived from the steady state (Req) values of the association phase of the sensograms, were about 10 μM and 100 nM for 13 and 15, respectively. The significant increase in the binding affinity of glycopolymer 15 could be attributed to the higher glycan content (15.1% for 15vs. 6.3% for 13), suggesting that the binding affinity of the N-linked glycopolymers primarily originated from the degree of derivatization of the HPMA copolymer with GlcNAc units. It appears that the shortness and rigidity of the azido linker in the N-linked glycopolymers prevents rotational freedom of the sugar moieties; hence, the N-linked glycopolymers represent low-affinity WGA ligands (Table 2). | ||
| Fig. 3 Surface plasmon resonance (SPR) kinetic binding analysis of the interactions between the chitooligosaccharide-modified HPMA copolymers and WGA. WGA was immobilized on the sensor chip, and the indicated concentrations of the N- or O-linked glycopolymers were injected in parallel over the WGA-coated chip at the flow rate of 30 μL min−1 for the association and dissociation phases of the sensograms. The fitted curves (bivalent analyte model) are superimposed as black lines on top of the sensograms. | ||
| Glycopolymera | k a1 × 105 (M−1 s−1) | k d1 × 10−5 (s−1) | k a2 (RU s−1) | k d2 (s−1) |
K
Db (nM) |
|
|---|---|---|---|---|---|---|
a Compound number, type of glycan attached, and its content in the glycopolymer (mol%).
b Apparent equilibrium dissociation constant (KD) for the bivalent analyte binding model was calculated as follows:  c Estimated dissociation constant (KD) derived from the steady state (Req) values of the association phase of the sensograms.
c Estimated dissociation constant (KD) derived from the steady state (Req) values of the association phase of the sensograms.
|
||||||
| 13 | GlcNAc-N (6.3%) | N.D. | N.D. | N.D. | N.D. | >10000c |
| 15 | GlcNAc-N (15.1%) | N.D. | N.D. | N.D. | N.D. | >100c |
| 17 | GlcNAc-O (9.4%) | 1.6 ± 1.5 | 2528 ± 850 | 140 ± 95 | 272 ± 94 | 104 ± 35 |
| 18 | GlcNAc-O (16.4%) | 3.5 ± 1.2 | 610 ± 290 | 0.9 ± 0.7 | 2.5 ± 1.1 | 13 ± 5 |
| 19 | (GlcNAc)2-O (5.4%) | 6.1 ± 2.4 | 71 ± 35 | 0.005 ± 0.004 | 0.1 ± 0.1 | 1.1 ± 0.6 |
| 20 | (GlcNAc)2-O (9.4%) | 5.7 ± 1.8 | 34 ± 21 | 26 ± 12 | 603 ± 213 | 0.6 ± 0.3 |
| 21 | (GlcNAc)3-O (5.3%) | 3.6 ± 1.5 | 45 ± 26 | 0.003 ± 0.02 | 0.1 ± 0.1 | 1.2 ± 0.5 |
| 22 | (GlcNAc)3-O (8.2%) | 4.1 ± 1.8 | 21 ± 13 | 6.8 ± 2.8 | 43 ± 24 | 0.4 ± 0.2 |
| 23 | (GlcNAc)4-O (4.8%) | 3.5 ± 1.3 | 72 ± 36 | 0.5 ± 0.3 | 2.4 ± 1.2 | 1.7 ± 0.8 |
| 24 | (GlcNAc)4-O (7.2%) | 3.1 ± 2.1 | 9.5 ± 0.4 | 23 ± 14 | 51 ± 21 | 0.2 ± 0.1 |
| 25 | (GlcNAc)5-O (2.2%) | 1.3 ± 0.9 | 102 ± 35 | 1.9 ± 1.1 | 52 ± 18 | 7.6 ± 3.1 |
The kinetics of interaction of the O-linked glycopolymers with WGA did not fit a simple 1:1 Langmuir binding model; however, the data were described well using a bivalent analyte model (Fig. 3). This model describes the interaction of an immobilized ligand (WGA) with an analyte (glycopolymer) that carries two identical and independent binding sites. The bivalent analyte model results in two sets of rate constants, one for each binding step, where the second binding step is directed by the first binding step. This cooperative binding is called avidity and usually reflects the polyvalent nature of a glycopolymer. The calculated association and dissociation rate constants (ka1, ka2, kd1, and kd2) for the binding of the O-linked glycopolymers to WGA are listed in Table 2. The data show that the O-linked glycopolymers bind WGA with KD in the nanomolar and subnanomolar ranges, which ranks them among the best ligands of WGA ever reported.8 The overall comparison of the KD values reveals that the binding properties of the O-linked glycopolymers carrying glycans consisting of two (19, 20), three (21, 22), and four (23, 24) carbohydrate units are relatively similar, with binding affinities approximately 2 to 8 times higher for the glycopolymers with higher glycan molar contents (20, 22, 24) than for those with lower glycan contents (19, 21, 23). In particular, the KD for the high-content (7.2 mol%) glycopolymer 24 derivatized with chitotetraose reached the value of 0.2 nM, which is the highest binding affinity reported in our study. This high binding affinity can be attributed to the very slow dissociation rate of the 24-WGA complex because the association rates of the O-linked glycopolymers remained comparable. The O-linked glycopolymer 25 carrying chitopentaose was characterized by a faster dissociation rate of the encounter complex, yielding a decrease of the KD value to 7.4 nM, which is very likely due to the relatively low glycan content (2.2 mol%) in 25. The comparatively lowest binding affinities of the O-linked glycopolymers were established for those decorated with single GlcNAc units (17 and 18). The predominant mechanism contributing to their lower binding capability was the very fast dissociation rate of the encounter complex; this was especially pronounced in glycopolymer 17, which has a lower GlcNAc content (9.4 mol%; cf. 18 with 16.4 mol% GlcNAc).
Overall, these results clearly show that high-affinity binding of the chitooligosaccharide-modified HPMA copolymers to WGA is caused by the avidity effect of multiple sugar moieties in the glycopolymers. The first association rate constants ka1, representing the initial stage of the binding process, appear to be similar for all the glycopolymer–WGA interactions, indicating that the single GlcNAc unit is sufficient to elicit the interaction of the HPMA copolymer with the lectin surface. On the other hand, single GlcNAc units are not sufficient to maintain a stable complex due to a much faster dissociation (kd1) of the glycopolymers derivatized with single GlcNAc units as compared to those with di-, tri or tetrasaccharide units, suggesting significant beneficial effects of additional sugar units for the stability of the glycopolymer–WGA interaction. The density of glycans on the polymer carrier (given by their molar content) much more strongly influences the glycopolymer binding affinity in the case of derivatization with single GlcNAc units than with longer chitooligomer chains, probably due to the high flexibility and spatial adaptability of longer glycans.
Structure–affinity relationship: influence of the spacer and glycan lengths
The data acquired from both the ELLA and SPR assays clearly demonstrate that the degree of derivatization of the polymer chain with glycans as well as the type of spacer (its chemical character, length, and flexibility) decidedly influence the binding properties of the glycopolymers to WGA. The pronounced impact of the glycan content on the binding affinity is visible in glycopolymers tethered with single GlcNAc units (especially in the series 16–18), probably due to the limited flexibility and adaptability of the displayed monosaccharides to the lectin structural requirements. In the oligosaccharide-decorated glycopolymers, the contribution of the increased density of the presented glycans to the binding improvement reaches scarcely one order of magnitude. The type of spacer (N-bound glycan in 13–15, or O-bound via an ethyl linker in 16–18) results in a difference of 1 to 2 orders of magnitude in binding.The most outstanding improvement in the binding of the glycopolymers to WGA was brought about by the increased length of the glycan chain, especially when comparing the monomeric and dimeric GlcNAc: glycopolymer 19 (with dimeric GlcNAc) binds more than three orders of magnitude (IC50) more strongly to WGA than its counterpart 16 (with mono-GlcNAc). As suggested by the analysis of the binding kinetics, longer glycan chains stabilize the glycopolymer–WGA complex better and slow down its dissociation. The binding parameters KD acquired for oligosaccharide-decorated glycopolymers 20, 22, and 24 are in the subnanomolar range (0.6, 0.4, and 0.2 nM, respectively) and rank these glycopolymers among the best WGA ligands ever reported. Note that just ten glycans per polymer molecule are sufficient to achieve this impressive binding efficiency.
Conclusions
Our study presents a series of biocompatible water-soluble HPMA-copolymers decorated with chitooligosaccharide chains composed of 1–5 GlcNAc moieties that exhibit outstanding affinities to a model lectin, wheat germ agglutinin. The best glycopolymers in the set show a subnanomolar dissociation constant KD and thus can be classified among the strongest WGA-binding conjugates ever reported. The real-time interaction kinetics determined by SPR revealed that longer oligosaccharide chains (≥2) efficiently prevent the dissociation of glycopolymers from WGA, stabilizing their mutual interaction and resulting in high-affinity binding. The flexibility of the HPMA copolymer carrier enables cooperative binding to multiple binding sites, which enormously increases the relative binding potency per one glycan (up to 95000 times in glycopolymer 18) compared to that of the free monovalent ligand. These results demonstrate the immense potential of the HPMA copolymers derivatized with complex carbohydrate chains for targeting purposes. The advanced chemoenzymatic techniques thus pave the way for the production of tailored high-affinity lectin ligands.
Acknowledgements
Support by the Czech Science Foundation (GA15-02578J), the Ministry of Health of the Czech Republic (16-28594A), the Ministry of Education, Youth and Sports of the Czech Republic (EATRIS-CZ, LM2015064, and LD15085), and the National Sustainability Program I (POLYMAT LO1507) is gratefully acknowledged.Notes and references
- G. A. Rabinovich, Y. van Kooyk and B. A. Cobb, Ann. N. Y. Acad. Sci., 2012, 1253, 1–15 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- P. Bojarová, R. R. Rosencrantz, L. Elling and V. Křen, Chem. Soc. Rev., 2013, 42, 4774–4797 RSC.
- P. Bojarová and V. Křen, Biomater. Sci., 2016, 4, 1142–1160 RSC; B. Lepenies, J. Yin and P. H. Seeberger, Curr. Opin. Chem. Biol., 2010, 14, 404–411 CrossRef CAS PubMed.
- V. Wittmann and R. J. Pieters, Chem. Soc. Rev., 2013, 42, 4492–44503 RSC.
- C. S. Wright, J. Biol. Chem., 1992, 287, 14345–14352 Search PubMed.
- M. Fiore, N. Berthet, A. Marra, E. Gillon, P. Dumy, A. Dondoni, A. Imberty and O. Renaudet, Org. Biomol. Chem., 2013, 11, 7113–7122 CAS.
- M. Ghirardello, K. Öberg, S. Staderini, O. Renaudet, N. Berthet, P. Dumy, Y. Hed, A. Marra, M. Malkoch and A. Dondoni, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2422–2433 CrossRef CAS.
- A. M. Eissa and N. R. Cameron, Adv. Polym. Sci., 2013, 253, 71–114 CrossRef CAS; M. Hetzer, G. Chen, C. Barner-Kowollik and M. H. Stenzel, Macromol. Biosci., 2010, 10, 119–126 CrossRef PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS; R. A. Evans, Aust. J. Chem., 2007, 60, 384–395 CrossRef.
- K. Ulbrich and V. Šubr, Adv. Drug Delivery Rev., 2010, 62, 150–166 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- P. J. Julyan, L. W. Seymour, D. R. Ferry, S. Daryani, C. M. Boivin, J. Doran, M. David, D. Anderson, C. Christodoulou, A. M. Young, S. Hesslewood and D. J. Kerr, J. Controlled Release, 1999, 57, 281–290 CrossRef CAS PubMed; A. David, P. Kopečková, T. Minko, A. Rubinstein and J. Kopeček, Eur. J. Cancer, 2004, 40, 148–157 CrossRef PubMed.
- D. Roy, B. Ghosn, E.-H. Song, D. M. Ratner and P. S. Stayton, Polym. Chem., 2013, 4, 1153–1160 RSC.
- K. Slámová, J. Krejzová, P. Marhol, L. Kalachova, N. Kulik, H. Pelantová, J. Cvačka and V. Křen, Adv. Synth. Catal., 2015, 357, 1941–1950 CrossRef.
- S. Singh, J. Packwood, C. J. Samuel, P. Critchley and D. H. G. Crout, Carbohydr. Res., 1995, 279, 293–305 CrossRef CAS PubMed; H. Ihara, S. Hanashima, T. Okada, R. Ito, Y. Yamaguchi, N. Taniguchi and Y. Ikeda, Glycobiology, 2010, 20, 1021–1033 CrossRef PubMed.
- P. Fialová, A. T. Carmona, I. Robina, R. Ettrich and P. Sedmera, Tetrahedron Lett., 2005, 46, 8715–8718 CrossRef.
- K. Adamiak, T. Anders, M. Henze, H. Keul, M. Möller and L. Elling, J. Mol. Catal. B: Enzym., 2012, 84, 108–114 CrossRef CAS.
- D. Horton, Org. Synth., 1966, 46, 1 CrossRef CAS.
- J. A. Rupley, Biochim. Biophys. Acta, 1964, 83, 245–255 CAS.
- P. Chytil, T. Etrych, J. Kříž, V. Šubr and K. Ulbrich, Eur. J. Pharm. Sci., 2010, 41, 473–482 CrossRef CAS PubMed.
- V. Šubr and K. Ulbrich, React. Funct. Polym., 2006, 66, 1525–1538 CrossRef.
- P. Šácha, T. Knedlík, J. Schimer, J. Tykvart, J. Parolek, V. Navrátil, P. Dvořáková, F. Sedlák, K. Ulbrich, J. Strohalm, P. Majer, V. Šubr and J. Konvalinka, Angew. Chem., Int. Ed., 2016, 55, 2356–2360 CrossRef PubMed.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- K. Slámová, P. Bojarová, L. Petrásková and V. Křen, Biotechnol. Adv., 2010, 28, 682–693 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural characterization of functionalized chitooligomers 3, 5–9 (NMR data and spectra, MS spectra, HPLC chromatograms); structural identification of chitooligomer standards 26–29; synthesis of polymer precursors 12a and 12b; structural characterization of glycopolymers 13–25 (NMR spectra). See DOI: 10.1039/c7py00271h |
| This journal is © The Royal Society of Chemistry 2017 |