
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of reactive oxygen species (ROS)-self-immolative polyoxalate prodrug nanoparticles for hormone dependent cancer therapy with minimized side effects†
Anita
Höcherl
 *a,
Eliézer
Jäger
*a,
Alessandro
Jäger
a,
Martin
Hrubý
a,
Rafał
Konefał
a,
Olga
Janoušková
a,
Jiří
Spěváček
a,
Yaming
Jiang
b,
Peter W.
Schmidt
b,
Timothy P.
Lodge
b and
Petr
Štěpánek
a
*a,
Eliézer
Jäger
*a,
Alessandro
Jäger
a,
Martin
Hrubý
a,
Rafał
Konefał
a,
Olga
Janoušková
a,
Jiří
Spěváček
a,
Yaming
Jiang
b,
Peter W.
Schmidt
b,
Timothy P.
Lodge
b and
Petr
Štěpánek
a
aInstitute of Macromolecular Chemistry v.v.i., Academy of Sciences of the Czech Republic, Heyrovsky Sq. 2, 162 06 Prague 6, Czech Republic
bDepartment of Chemistry, University of Minnesota, 207 Pleasant Street SE, Minneapolis, MN, USA. E-mail: hocherl@imc.cas.cz; jager@imc.cas.cz
First published on 3rd March 2017
Abstract
A new reactive oxygen species (ROS)-sensitive, self-immolative biodegradable polyoxalate prodrug based on the anticancer chemotherapeutic hormone analog diethylstilbestrol was synthesized via one-pot step-growth polymerization. The nanoparticles prepared from this prodrug undergo self-immolative degradation releasing the chemotherapeutic drug in ROS-rich environments, e.g., in cancer cells. This new ROS self-immolative polyprodrug backbone eliminates the need for a linker between polymer chain and drug, resulting in a more specific drug release and minimized toxic side effects to non-ROS-producing cells as proven by in vitro experiments. The strategy enables re-utilization of a successful chemotherapeutic agent that has been clinically under-utilized due to dose-related side effects.
Introduction
Cancer is a leading cause of mortality worldwide, accounting for 8.2 million deaths in 2012, and it is expected that annual cancer cases will rise from 14 million in 2012 to 22 million within the next two decades.1 In men, one of the five most common causes of death is due to cancers of the prostate.1 Diethylstilbestrol (DEB, synthetic non-steroidal estrogen) has been a standard approach to the treatment of advanced prostate cancer for 50 years.2 DEB is highly effective in prostate cancer treatment, with studies reporting that DEB has a similar efficacy to orchiectomy, at a daily dosage from 1 mg (low dose regime) to 3 mg.2a–d DEB was also successfully employed in estrogen additive therapy for women with estrogen receptor (ER)-positive metastatic breast cancer,3 with studies showing that the survival of patients treated with daily 5 mg DEB was significantly higher than with Tamoxifen.2d However, DEB was eventually withdrawn from the clinical repertoire due to persistent severe dose-related toxicities, mainly cardiotoxicity.2,3In recent years, the re-utilization of neglected and/or toxic drugs becomes possible by virtue of the encapsulation of drugs into lipid- or polymer-based nanoparticles (NPs) as e.g., Doxorubicin encapsulated in liposomes (Doxil®) or through the synthesis of polymer–drug(protein) conjugates, e.g., PEGaspargase (Oncaspar®).4 Both approaches have proven clinically efficient in the treatment of several diseases. Despite this good efficiency, encapsulation can lead to drug leakage from the NPs during storage and blood circulation,4 and most of the polymer–drug(protein) conjugates require the use of specific linkers and long synthetic pathways, e.g., through acetal,5 ester6 or hydrazone7 bonds. Consequently, polymer systems gain much more relevance in biomedical applications if they are tailored to be degradable in response to a stimulus, e.g., light, pH change, or more recently, the elevated presence of ROS in cancer tissues.8 The increased H2O2 production of cancer cells causes locally high peroxide concentrations, which can even exceed the oxidative burst of activated macrophages.8d This is due to imbalances in the ROS production (especially H2O2), and it leads to oxidative stress and activation of inflammation events, which can damage tissues and organs.9
Oxidation-responsive polymers and their relevant NPs including poly(propylene sulfide)s,10 ferrocene-based,11 boronic ester-based,12 thioketal-based,13 or polyoxalate-based14 polymers have been prepared and studied for their potential in biomedical applications. Among these, the preparation of polymer systems sensitive to H2O2 through the cleavage of oxalate bonds is a particularly straightforward approach. Polymers containing oxalate bonds have been recently tested in murine models demonstrating interesting properties as antioxidants,14a,b as antioxidant-theranostic agents,14c ischemia/reperfusion-targeted nanotherapeutics,14d and as nanoreactor systems for in vivo imaging of H2O2.14c,d Further attractive properties of the oxalate based polymers NPs include their degradation-induced luminescence (discussed hereafter) with tunable emission by the encapsulation of selected dyes, excellent specificity to H2O2 over other ROS species, deep-tissue imaging capabilities, and continuous detection of in situ produced H2O2 for long times.14
However no application for the treatment of cancer cells with this promising system has been reported yet. Herein, we describe a one-pot synthesis route to a novel self-immolative H2O2-sensitive polyprodrug material that provides biocompatibile and biodegradable nanoparticles with triggered degradation and chemotherapeutic drug release in ROS-rich environments (Scheme 1). The concept of polyprodrugs avoids the use of chemically specific linkers for drug release, by integrating the drug molecules into the polymer carrier backbone with stimuli-responsive bond and hyper-fast chain-breakage capabilities.15
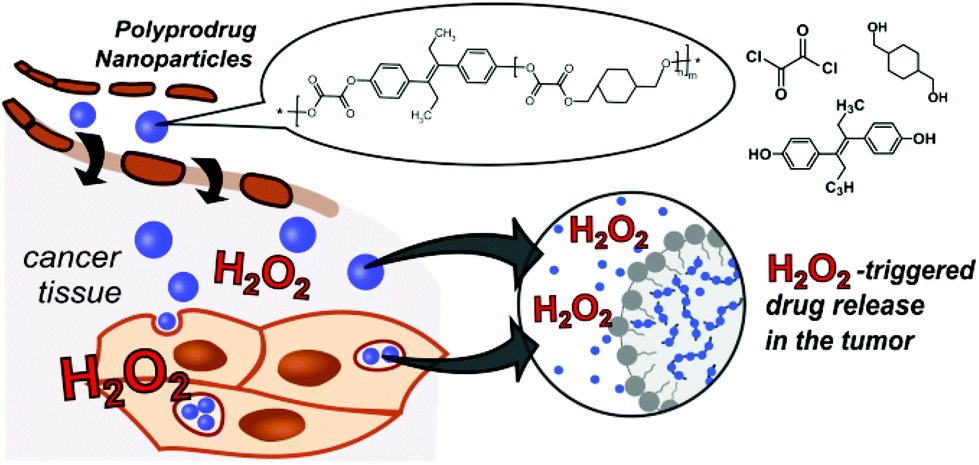 | ||
| Scheme 1 Polyoxalate prodrug NPs (PDEB NPs) with self-immolative polymer degradation and DEB release in H2O2-rich tumor microenvironments. | ||
Results and discussion
Polymers PDEB1, PDEB2, PDEB3
The three ROS self-immolative polyprodrug backbones with varied DEB amounts were synthesized by a one-pot step-growth polymerization. The reaction conditions were optimized and fine-tuned according to the molar ratio of the diols and the oxalyl chloride, in accordance with the reaction pathway detailed in the ESI† (see ESI† for methods). Briefly, diethylstilbestrol and 1,4-cyclohexanedimethanol (CHD) were dissolved in tetrahydrofuran (THF), and triethylamine was added dropwise at 4 °C. Afterwards oxalyl chloride dissolved in THF was added dropwise, and the polymerization reaction was continued for 6 hours at room temperature under nitrogen atmosphere. The resulting polyprodrugs were obtained by extraction using DCM and precipitation in cold hexane. Polyprodrugs with varied amounts of DEB (PDEB1 ∼2.3 wt%; PDEB2 ∼7.5 wt% and PDEB3 ∼13.0 wt% of DEB) were obtained by varying the DEB, oxalyl chloride and CHD ratio (see Fig. S3 and ESI† for methods). The PDEBs were obtained as yellowish solids and the chemical structure was identified with 1H NMR spectroscopy (Fig. 1A top and Fig. S1, ESI†). Successful polymer synthesis was confirmed by 1H NMR (Fig. 1A top) and by size exclusion chromatography (SEC) analysis (Fig. S2, ESI†). The weight-average molecular weight (Mw) of polymer PDEB1 was 11.8 kDa with reasonable dispersity Mw/Mn ≈ 1.4 as determined by SEC (Fig. S2, ESI†). No important differences in Mw were observed for PDEB2 and PDEB3 (Fig. S2, ESI†). The 1H NMR spectrum of PDEB1 (Fig. 1A top, see ESI† for methods) shows the characteristic signals for protons belonging to the repeat units (see ESI† for peak assignments). Significantly lower mobility was revealed for aromatic protons of DEB units (signals a, b in Fig. 1A) in DMF-d7 solutions of PDEB polyprodrugs in comparison with solution of the neat DEB from 1H spin–spin relaxation times T2 (see ESI† for measurements details), which in polyprodrugs (T2 = 0.76 s) were twice shorter in comparison with the neat DEB (T2 = 1.42 s). This fact together with the disappearance of the signal of DEB OH protons at 9.5 ppm in the spectra of the polyprodrugs (see ESI, Fig. S1B†) indicate that DEB units are covalently incorporated in the polymer chain. The degradation of PDEB1 as evaluated by 1H NMR spectroscopy was complete after 3 days of incubation (Fig. 1A middle after 24 h and bottom after 72 h) as the broad peaks in 1H NMR related to the polyprodrug are replaced by sharp peaks of the low-molecular-weight degradation products (monomers), confirming the self-immolative depolymerization triggered by H2O2.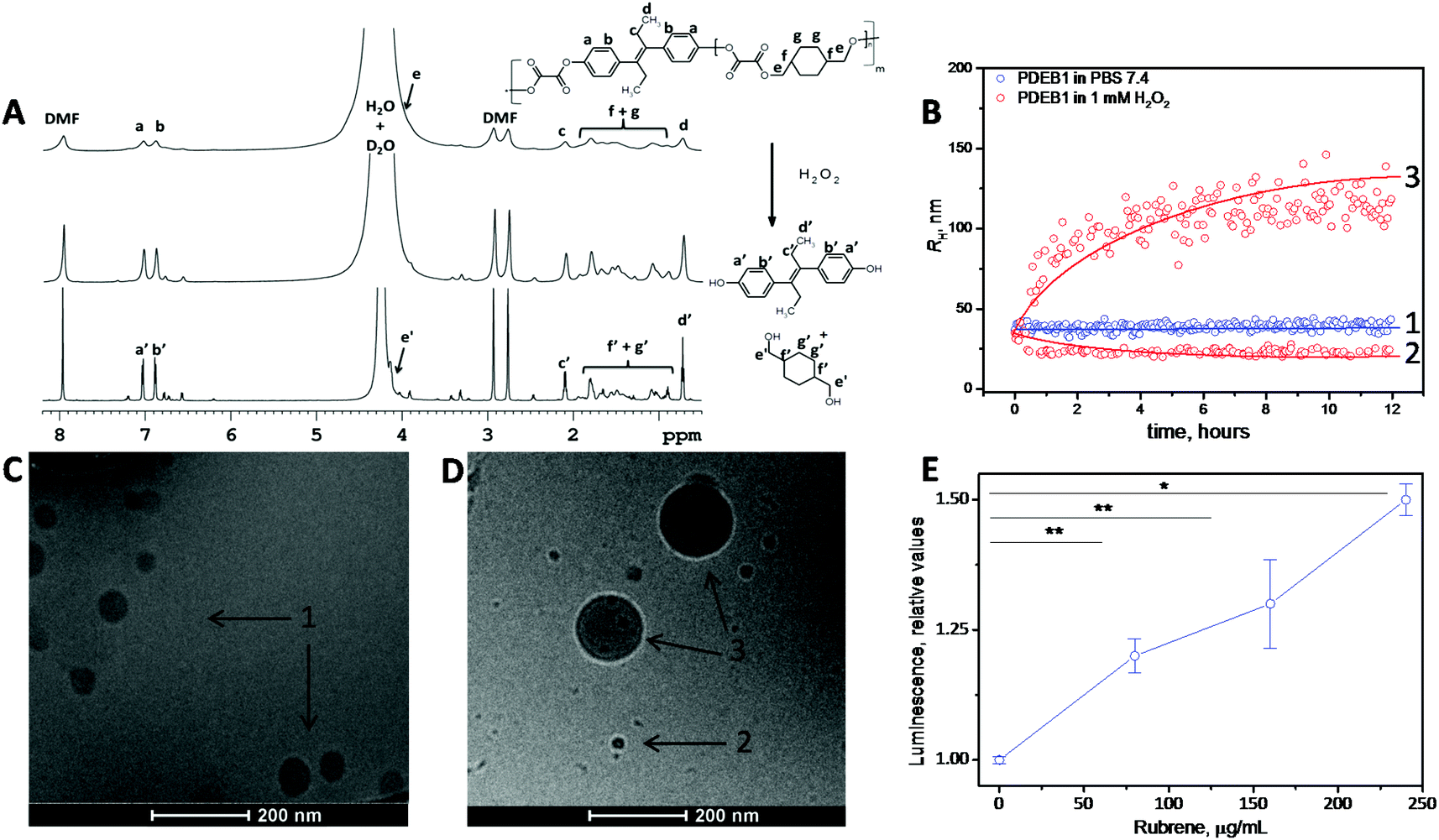 | ||
| Fig. 1 (A) 1H NMR spectra of the synthesized ROS self-immolative PDEB1 polyprodrug (before H2O2 addition in PBS/DMF-d7) (top) and the degradation upon incubation with 1 mM of H2O2 after 24 h (middle) and 72 h (bottom). (B) Average RH for PDEB1 polyprodrug NPs (blue – 1) with no H2O2 addition and after 12 h of incubation (red – 2 and 3) in 1 mM of H2O2 at 37 °C in PBS. (C) Cryo-TEM of the PDEB1 polyprodrug NPs before incubation with H2O2 and (D) after incubation with 1 mM H2O2 for 24 h. (E) Luminescence in MCF7 cancer cells after 2 h incubation with rubrene-loaded PDEB1 NPs, indicating the ROS-triggered cleavage of the oxalate groups in the polymer backbone. Sample ‘0 μg·mL−1 rubrene’ corresponds to the NP-free control sample (*p < 0.01; **p < 0.03). The arrows in (C) and (D) correspond to the average RH from the DLS in (B). | ||
Nanoparticles
With the aim to reduce the required dose, to minimize toxic side effects and to increase the specificity of DEB for cancer tissue, Tween-stabilized NPs based on the ROS self-immolative polyprodrug backbone PDEB1 were prepared (see ESI† for methods). Due to the incorporated oxalate linkers in the polyprodrug backbone, in an ROS-rich environment the polyprodrug will be self-immolatively degraded and the chemotherapeutic DEB released. We recently developed a NP drug-delivery system with H2O2-sensitive groups, which was able to deliver its drug cargo selectively to cancer cells (exploiting the higher level of peroxide in cancer cells compared to non-cancer cells).12a This approach is here refined with the insertion of the peroxide-sensitive oxalates as cleavable breakpoints integrated into the backbone as self-immolative polyprodrugs for selective drug release.PDEB1 was chosen for in vitro evaluation (PDEB1 ∼2.3 wt% of DEB, Fig. S3 and eqn (S3), ESI†) due to the superior stability of the PDEB1 NPs (see ESI† for methods). Dynamic light scattering (DLS) demonstrated NP stability in simulated physiological conditions and proved their peroxide-triggered degradation (Fig. 1B), and the degradation-triggered DEB release (Fig. S4, ESI†) specifically in simulated ROS-rich environments (H2O2). Note that the NPs were prepared to an average size of ca. 72 nm in diameter (hydrodynamic radius, 2RH = DH ≈ 72 nm), e.g., within a range known to be ideal for efficient tumor accumulation due to the EPR effect.4,7
H2O2-Triggered polymer degradation
Under exposure to physiologically relevant levels of H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8a,12a (250 μM, Fig. S4, ESI†) the oxalate groups of PDEB1 are oxidized and subsequently hydrolyzed triggering a self-immolative polymer backbone degradation releasing the chemotherapeutic DEB (see Scheme 1, Fig. 1B, S3 and S4, ESI†). The degradation of PDEB1 in the presence of H2O2 was also probed by cryo-TEM (Fig. 1C and D). Applying several different techniques, 1H NMR spectra, DLS and the cryo-TEM images we showed that PDEB1 polyprodrug NPs degraded into small molecules and oligomers in a time- and H2O2-dependent manner. The larger objects observed in DLS and cryo-TEM during degradation (DLS in red Fig. 1B and cryo-TEM in Fig. 1D) most probably are related to the hydrophobicity of the polymer fragments that re-aggregate in solution due to the presence of the stabilizing agent and/or to NPs swollen due to the H2O2 attack.16 Polymer degradation proceeds more extensively with increasing incubation time and H2O2 concentration (Fig. 1B and C and S5, ESI†). It is well established that the degradation of polyoxalate-based polymers induces chemiluminescence in peroxide-rich environments.14,17 In this way the polymer degradation in cancer cells was demonstrated using PDEB1 polyprodrug NPs loaded with the dye rubrene (Fig. 1E), where the ROS-triggered cleavage of oxalate bounds induced rubrene luminescence (see Scheme S1, ESI† for proposed mechanism). The chemiluminescence was observed in MCF7 cancer cells (Fig. 1E) as well as in macrophages (see Fig. S6, ESI†), and the intensity of rubrene chemiluminescence correlated with the particle concentration, making the PDEB1 polyprodrug NPs useful for theranostics as well.14c,17
8a,12a (250 μM, Fig. S4, ESI†) the oxalate groups of PDEB1 are oxidized and subsequently hydrolyzed triggering a self-immolative polymer backbone degradation releasing the chemotherapeutic DEB (see Scheme 1, Fig. 1B, S3 and S4, ESI†). The degradation of PDEB1 in the presence of H2O2 was also probed by cryo-TEM (Fig. 1C and D). Applying several different techniques, 1H NMR spectra, DLS and the cryo-TEM images we showed that PDEB1 polyprodrug NPs degraded into small molecules and oligomers in a time- and H2O2-dependent manner. The larger objects observed in DLS and cryo-TEM during degradation (DLS in red Fig. 1B and cryo-TEM in Fig. 1D) most probably are related to the hydrophobicity of the polymer fragments that re-aggregate in solution due to the presence of the stabilizing agent and/or to NPs swollen due to the H2O2 attack.16 Polymer degradation proceeds more extensively with increasing incubation time and H2O2 concentration (Fig. 1B and C and S5, ESI†). It is well established that the degradation of polyoxalate-based polymers induces chemiluminescence in peroxide-rich environments.14,17 In this way the polymer degradation in cancer cells was demonstrated using PDEB1 polyprodrug NPs loaded with the dye rubrene (Fig. 1E), where the ROS-triggered cleavage of oxalate bounds induced rubrene luminescence (see Scheme S1, ESI† for proposed mechanism). The chemiluminescence was observed in MCF7 cancer cells (Fig. 1E) as well as in macrophages (see Fig. S6, ESI†), and the intensity of rubrene chemiluminescence correlated with the particle concentration, making the PDEB1 polyprodrug NPs useful for theranostics as well.14c,17
Cell viability testing
Taking into account previous works reporting the significantly higher cell viability for particles prepared from polyoxalates lacking DEB,18 herein the cell viability experiments were performed with PDEB NPs. In vitro evaluation of the self-immolative PDEB1 polyprodrug NPs was performed in selected cell lines (see Fig. 2). Cell model systems for hormone therapy-treatable (LNCaP) and therapy-resistant (PC3) prostate cancer were used to evaluate the potential of PDEB1 in prostate cancer treatment. Like all cancer cells, these cells have higher ROS levels, and PC3 and LNCaP produce up to 3-times more H2O2 than non-cancerous cells,8d thus being well suited to demonstrate the ROS-sensitive drug release of PDEB1. MCF7 cells were selected as a breast cancer model system, based on recent studies showing satisfactory results of DEB in the treatment of post-menopausal breast cancer.2d,3 Human fibroblasts (HF) were used as a non-cancerous reference system. Cell viability after 5 days of incubation with PDEB1 polyprodrug NPs was evaluated using the AlamarBlue Assay (Fig. 2, see ESI† for methods and for DEB toxicity mechanism). The lower toxicity of the free, highly hydrophobic, drug compared to the drug-carrier system is a known effect and it is attributed both to the extensive adsorption of the hydrophobic drug molecules onto serum components such as serum albumin, whereas NPs drug carriers transport the drug directly into the cells19a,b and to the enhanced endocytotic uptake of polymer NPs when compared with the free drugs.19c–h HF cells produce a significantly lower level of ROS compared to the cancer cells,8 hence the PDEB1 polyprodrug is degraded. However, as clearly visible in Fig. 2A, the ROS-sensitive polyprodrug NPs exerted a much higher toxicity on the cancer cells, MCF7, than on HF, while the free drug was equally toxic to cancer and non-cancer cells. Note that PDEB1 is specifically efficient versus the cancer cells in the low micromolar range, i.e., at a concentration approximating the DEB blood plasma level in breast cancer therapy.2d Also for prostate cancer cells (Fig. 2B) LNCaP and even for the hormone-resistant PC3, PDEB1 polyprodrug NPs significantly lowered the required dose compared to the free drug. Similarly, the improved toxicity of PDEB1 was observed with cancer cell lines.2c PDEB1 also showed superior toxicity over the free drug.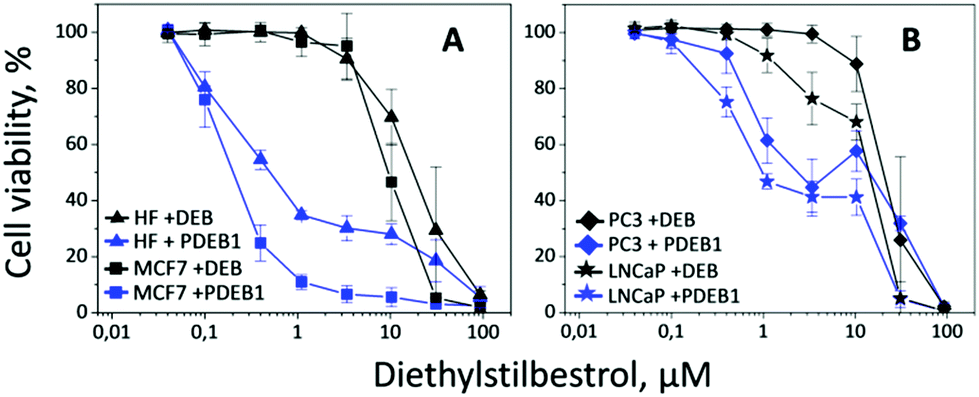 | ||
| Fig. 2 Cell viability after 5 days incubation with the free drug DEB (black) and with the polyprodrug PDEB1 NPs (blue). The cell lines of HF (triangle) and MCF7 (square) are shown in (A), and of LNCaP (star) and PC3 (diamond) in (B). | ||
Microscopy studies
Light microscopy revealed initial morphological changes and M-phase cell cycle arrest after 24 h incubation (and significant nuclear aneuploidy at >72 h) with PDEB1 polyprodrug NPs, while the free drug had no visible effect (data not shown). For the evaluation of morphological changes the cells were further studied after 24 h incubation with free DEB and PDEB1 NPs using confocal laser scanning microscopy (CLSM) (Fig. 3 and S7, ESI†). CLSM images in Fig. 3 show that the non-cancer cells (HF) were the least affected by PDEB1 polyprodrug NPs. HF displayed only an altered, less fibrous cell morphology and occasional nuclear fragmentation. Compared to that, cancer cell lines showed significant morphological changes such as nuclear fragmentation (indicating mitotic arrest) and aneuploidy (indicating G2/M arrest) in a high number of cells after incubation with PDEB1. The free drug did not have any visible toxic effects. Specifically LNCaP cells were strongly affected by PDEB1 in that most cells were detached, forming clusters (see Fig. S8†). This indicates a specific sensitivity of LNCaP to PDEB1 polyprodrug that results in disrupted cell adhesion, thereby promising additional cytotoxic effects in long-term, which adds to the attractiveness of PDEB1 polyprodrug for application in prostate cancer therapy. For further details on the microscopy studies, see ESI and Fig. S9.†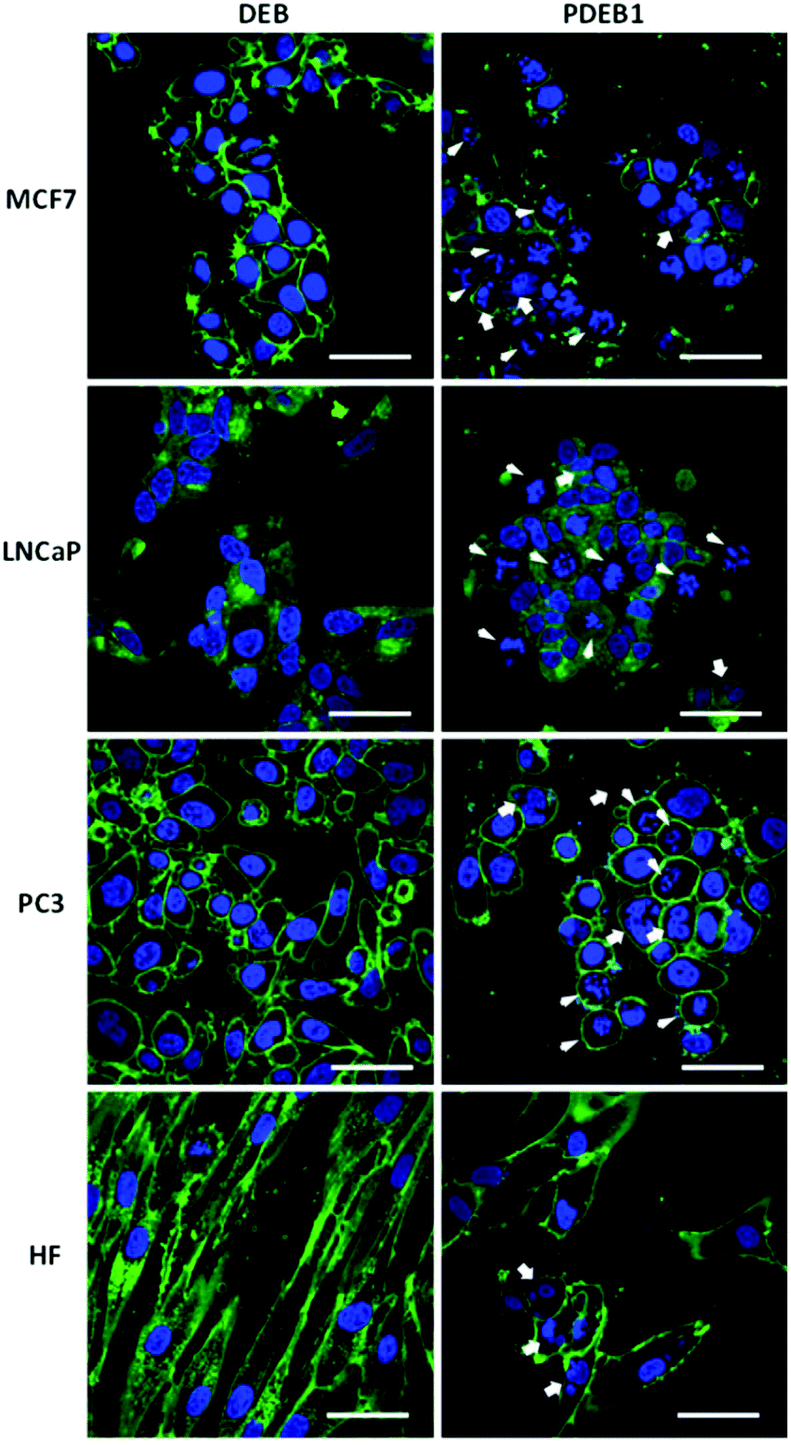 | ||
| Fig. 3 Cells were incubated for 24 h with free DEB or PDEB1 NPs, at a total concentration of 3.4 μM DEB, to visualize nuclear fragmentation (arrow) and cells in mitotic arrest (arrowhead); nuclei were stained using Hoechst 33342 (blue) and the cytoplasmic membrane was labelled with CellMask™ (green). Scale bar = 50 μm. | ||
Conclusions
The CLSM data correlates with the viability analysis, demonstrating again the enhanced toxicity of the PDEB1 prodrug against cancer cells. Note that the cytotoxicity of the PDEB1 polyprodrug NPs to HF cells was comparable to the toxicity of NPs prepared with the FDA-approved polymer polylactic acid PLA (Fig. S10, see ESI† for methods). The presented system shows great promise to achieve drug release specifically into tumors and lower the required drug dose in estrogen-dependent hormone therapy. In summary, for the first time a one-pot synthetic pathway to synthesize a new ROS self-immolative polyprodrug backbone based on the chemotherapeutic hormone analogue DEB is presented. The prepared NPs demonstrate higher activity against prostate and breast cancer cells in vitro, with lower activity in non-cancer cells due to the selectivity to ROS-rich environments. Particle degradation in ROS-rich environments can be followed by chemiluminescence, opening the possibility for usage as theranostics. Finally the strategy presented here eliminates the need for a biodegradable linker. The NPs make it possible to reduce the side-effects of a highly effective and successful therapeutic molecule that was utilized for the treatment of hormone-dependent cancers, through a simple and efficient strategy.Acknowledgements
The financial support of the Ministry of Education, Youth and Sports (grants # LH14079, POLYMAT #LO1507), the Czech Science Foundation (grant # 16-02870S, 15-13853S and 17-09998S), and the National Science Foundation through the University of Minnesota MRSEC, Award DMR-1420013 is gratefully appreciated.Notes and references
- World Health Organization (WHO), http://www.who.int/mediacentre/factsheets/fs297/en/ (July, 2016).
- (a) S. B. Malkowicz, Urology, 2001, 58, 108 CrossRef CAS PubMed; (b) E. D. Crawford, Rev. Urol., 2004, 6, S3 Search PubMed; (c) R. Turo, M. Smolski, R. Esler, M. L. Kujawa, S. J. Bromage, N. Oakley, A. Adeyoju, S. C. W. Brown, R. Brough, A. Sinclair and G. N. Collins, Scand. J. Urol., 2014, 48, 4 CrossRef CAS PubMed; (d) P. P. Peethambaram, J. N. Ingle, V. J. Suman, L. C. Hartmann and C. L. Loprinzi, Breast Cancer Res. Treat., 1999, 54, 117 CrossRef CAS PubMed; (e) H. A. Kemp, G. F. Read, D. Riad-Fahmy, A. W. Pike, S. J. Gaskell, K. Queen, M. E. Harper and K. Griffiths, Cancer Res., 1981, 41, 4693 CAS.
- (a) R. Twombly, J. Natl. Cancer Inst., 2011, 103, 920 CrossRef PubMed; (b) J. N. Ingle, D. L. Ahmann, S. J. Green, J. H. Edmonson, H. F. Bisel, L. K. Kvols, W. C. Nichols, E. T. Creaan, R. G. Hahn, J. Rubin and S. Fryak, N. Engl. J. Med., 1981, 304, 16 CrossRef CAS PubMed.
- (a) T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320 CAS; (b) E. Jäger and F. C. Giacomelli, Curr. Top. Med. Chem., 2015, 15, 328 CrossRef.
- J. Zou, G. Jafr, E. Themistou, Y. Yap, Z. A. P. Wintrob, P. Alexandridis, A. C. Ceacareanu and C. Cheng, Chem. Commun., 2011, 47, 4493 RSC.
- A. G. Cheetham, Y.-C. Ou, P. Zhang and H. Cui, Chem. Commun., 2014, 50, 6039 RSC.
- (a) K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338 CrossRef CAS PubMed; (b) E. Jäger, A. Jäger, P. Chytil, T. Etrych, B. Říhová, F. C. Giacomelli, P. Štěpánek and K. Ulbrich, J. Controlled Release, 2013, 165, 153 CrossRef PubMed.
- (a) M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722 CrossRef CAS PubMed; (b) S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1 CrossRef CAS PubMed; (c) T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS; (d) B. Kumar, S. koul, L. Khandrika, R. B. Meacham and H. K. Koul, Cancer Res., 2008, 68, 177 CrossRef PubMed.
- (a) A. Mantovani, P. Allavena, A. Sica and F. Balkwill, Nature, 2008, 454, 436 CrossRef CAS PubMed; (b) S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radicals Biol. Med., 2010, 49, 1603 CrossRef CAS PubMed; (c) N. Houstis, E. D. Rosen and E. S. Lander, Nature, 2006, 440, 944 CrossRef CAS PubMed.
- B. L. Allen, J. D. Johnson and J. P. Walker, ACS Nano, 2011, 5, 5263 CrossRef CAS PubMed.
- (a) M. Hmyene, A. Yassar, M. Escorne, A. Percheron-Guegan and F. Garnier, Adv. Mater., 1994, 6, 564 CrossRef CAS; (b) J. Tian, J. Chen, C. Ge, X. Liu, J. He, P. Ni and Y. Pan, Bioconjugate Chem., 2016, 27, 1518 CrossRef CAS PubMed.
- (a) E. Jäger, A. Höcherl, O. Janoušková, A. Jäger, M. Hrubý, R. Konefal, M. Netopilík, J. Pánek, M. Šlouf, K. Ulbrich and P. Štěpánek, Nanoscale, 2016, 8, 6958 RSC; (b) C. de G. Lux, S. Joshi-Barr, T. Nguyen, E. Mahmoud, E. Schopf, N. Fomina and A. Almutairi, J. Am. Chem. Soc., 2012, 134, 15758 CrossRef PubMed.
- (a) G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2016, 1600124 Search PubMed; (b) D. S. Wilson, G. Dalmasso, L. Wang, S. V. Sitaraman, D. Merlin and N. Murthy, Nat. Mater., 2010, 9, 923 CrossRef CAS PubMed.
- (a) D. Jeong, C. Kang, E. Jung, D. Yoo, D. Wu and D. Lee, J. Controlled Release, 2016, 233, 72 CrossRef CAS PubMed; (b) S. Kim, H. Park, Y. Song, D. Hong, O. Kim, E. Jo, G. Khang and D. Lee, Biomaterials, 2011, 32, 3021 CrossRef CAS PubMed; (c) C. Kang, W. Cho, M. Park, J. Kim, S. Park, D. Shin, C. Song and D. Lee, Biomaterials, 2016, 85, 195–203 CrossRef CAS PubMed; (d) D. Lee, S. Park, S. Bae, D. Jeong, M. Park, C. Kang, W. Yoo, M. A. Samad, Q. Ke, G. Khang and P. M. Kang, Sci. Rep., 2015, 5, 16592 CrossRef PubMed.
- (a) X. Hu, G. Liu, Y. Li, X. Wang and S. Liu, J. Am. Chem. Soc., 2015, 137, 362 CrossRef CAS PubMed; (b) X. Hu, J. Hu, J. Tian, Z. Ge, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617 CrossRef CAS PubMed.
- (a) A. Jäger, E. Jäger, F. Surman, A. Höcherl, B. Angelov, K. Ulbrich, M. Drechsler, V. M. Garamus, C. Rodriguez-Emmenegger, F. Nallet and P. Štěpánek, Polym. Chem., 2015, 6, 4946 RSC; (b) S. Petrova, E. Jäger, R. Konefał, A. Jäger, C. G. Venturini, J. Spěváček, E. Pavlova and P. Štěpánek, Polym. Chem., 2014, 5, 3884 RSC; (c) F. C. Giacomelli, P. Štěpánek, V. Schmidt, E. Jäger, A. Jäger and C. Giacomelli, Nanoscale, 2012, 4, 4504 RSC.
- (a) D. Lee, S. Khaja, J. C. Velasquez-Castano, M. Dasari, C. Sun, J. Petros, W. R. Taylor and N. Murthy, Nat. Mater., 2007, 6, 765 CrossRef CAS PubMed; (b) D. Hong, B. Song, H. Kim, J. Know, G. Khang and D. Lee, Ther. Delivery, 2011, 2, 1407 CrossRef CAS.
- (a) S. Cho, O. Hwang, I. Lee, G. Lee, D. Yoo, G. Khang, P. M. Kang and D. Lee, Adv. Funct. Mater., 2012, 22, 4038 CrossRef CAS; (b) S. Kim, K. Seong, O. Kim, H. Seo, M. Lee, G. Khang and D. Lee, Biomacromolecules, 2010, 11, 555 CrossRef CAS PubMed; (c) H. Park, S. Kim, S. Kim, Y. Song, K. Seung, D. Hong, G. Khang and D. Lee, Biomacromolecules, 2010, 11, 2103 CrossRef CAS PubMed.
- (a) D. A. Smith, L. Di and E. H. Kerns, Nat. Rev. Drug Discovery, 2010, 9, 929 CrossRef CAS PubMed; (b) A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583 CrossRef CAS PubMed; (c) G. Gaucher, R. H. Marchessault and J.-C. Leroux, J. Controlled Release, 2010, 143, 2 CrossRef CAS PubMed; (d) D. Li, Y. Zhang, S. Jin, J. Guo, H. Gao and C. Wang, J. Mater. Chem. B, 2014, 2, 5187 RSC; (e) F. Danhier, N. Lecouturier, B. Vroman, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, J. Controlled Release, 2009, 133, 11 CrossRef CAS PubMed; (f) H. S. Yoo, E. A. Lee and T. G. Park, J. Controlled Release, 2002, 82, 17 CrossRef CAS; (g) M. J. Vicent, R. Tomlinson, S. Brocchini and R. Duncan, J. Drug Targeting, 2004, 12, 491 CrossRef CAS PubMed; (h) V. Giménez, C. James, A. Armiñán, R. Schweins, A. Paul and M. J. Vicent, J. Controlled Release, 2012, 159, 290 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py00270j |
| This journal is © The Royal Society of Chemistry 2017 |