
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A broad scope of aliphatic polyesters prepared by elimination of small molecules from sustainable 1,3-dioxolan-4-ones†
Stefan A.
Cairns
,
Amelie
Schultheiss
and
Michael P.
Shaver
 *
*
School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: michael.shaver@ed.ac.uk
First published on 17th April 2017
Abstract
Biodegradable aliphatic polyesters built from sustainable feedstocks are one of the most promising solutions to address the pollution and oil-dependence challenges of modern plastics, but remain limited in monomer scope and thus the accessible polymer properties. We report a family of monomers that are built from renewable resources and use the elimination of small molecules to access aliphatic polyesters, circumventing challenging monomer syntheses to make these functionalised polymers. The driving force for ring opening polymerisation is the elimination of formaldehyde or acetone from easy-to-synthesise 1,3-dioxolan-4-ones to produce an array of structurally divergent polyesters. The polymers are prepared with high retention of stereochemistry, meaning isotactic polymers are easily prepared from natural enantiopure feedstocks. Reaction kinetics, structure/property relationships, copolymers of traditional cyclic esters, and direct recycling of waste paraformaldehyde showcase the scope of this new reaction in polymer chemistry.
Introduction
Biodegradable polyesters remain at the forefront of research into sustainable polymers, with the readily synthesised poly(lactic acid) now realising the commercial potential of green plastics.1–4 Accessing a broader scope of these aliphatic polyesters remains a challenge, especially when retaining the simple syntheses required for commercial applications. We thus target a family of monomers that are easily prepared from inexpensive, sustainable feedstocks and polymerise to form a range of structurally-diverse polyesters.Aliphatic polyesters are prepared by polycondensation of α-hydroxy acids (Fig. 1, A) or, more commonly, from the ring-opening polymerisation of cyclic esters (ROP, Fig. 1, B).1,2 The latter is preferred due to control over molecular weights and dispersity (Đ) and thus polymer properties,3,4 with ROP driven by release of ring strain.5,6 While the ROP of lactide (R = CH3) and glycolide (R = H) are commonplace, especially in biomedicine7 and food packaging,8 broadening this strategy to other cyclic esters is difficult. Baker, Möller and coworkers were pioneers in expanding the scope of cyclic diesters,9,10 showing the importance of this functional group choice by tuning polymer glass transition temperatures (Tgs) from −37 °C to 104 °C.11,12 Unfortunately, the diester synthesis is challenging for these alternative substituents, affording monomers in low yields after complex separations.
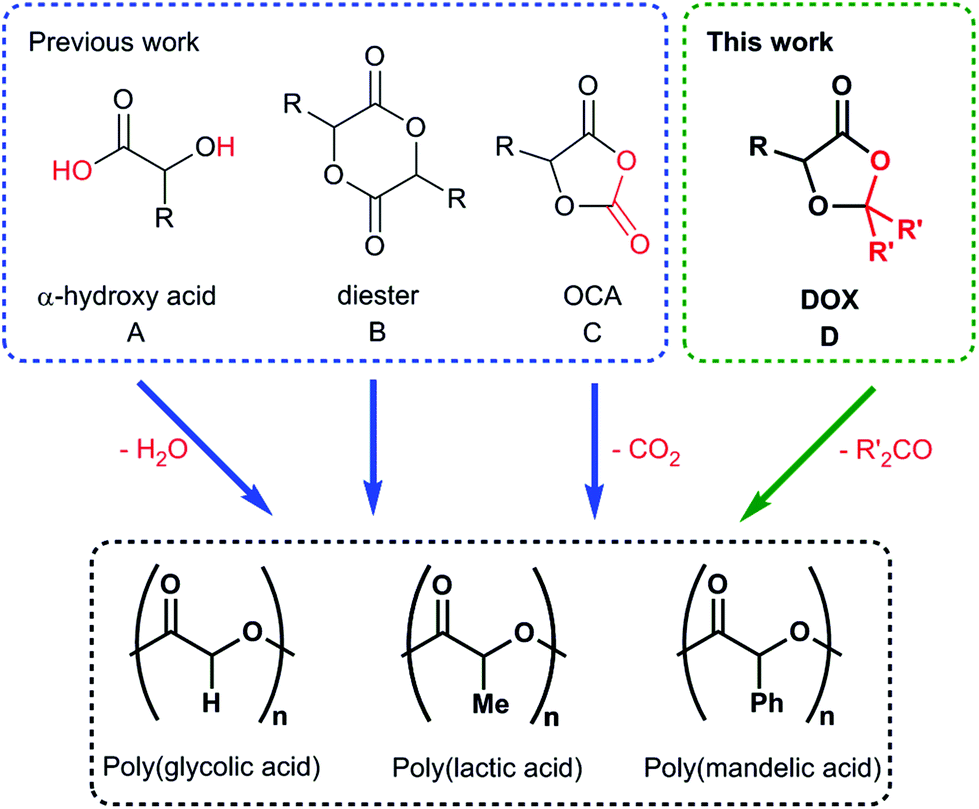 | ||
| Fig. 1 Synthetic routes to poly(α-hydroxy acid)s. A polycondensation, B ring opening polymerisation of diesters, C ring opening polymerisation of O-carboxyanhydrides eliminating carbon dioxide and D ring opening polymerisation of 1,3-dioxolan-4-ones eliminating acetone or formaldehyde. | ||
To circumvent the issue of monomer access, Bourissou and coworkers devised an alternate route to these important poly-(α-hydroxy acid)s.13 The ring-opening polymerisation of O-carboxy-anhydrides (Fig. 1, C) is driven by the release of CO2. Catalyst choice can control stereochemistry, and Buchard and coworkers exploited the strategy to prepare isotactic poly(mandelic acid) as a polystyrene mimic – a highly important target in sustainable polymer synthesis.14 While accessing the desired broad scope of polyesters, the monomer synthesis requires phosgene gas or diphosgene, creating concerns over monomer cost and toxicity at larger scales.15,16
We sought a new synthetic methodology that would access the same broad functional group tolerance as O-carboxy-anhydrides, but derive from sustainable, scalable and inexpensive resources. We report herein the ring-opening polymerisation of renewable 1,3-dioxolan-4-ones (DOX), eliminating formaldehyde or ketones to afford structurally divergent polyesters (Fig. 1, D).
Hillmyer and coworkers provided some key inspiration for this work in their preparation of polyesteracetals, observing that polymerisation of six membered 2-methyl-1,3-dioxane-4-one would eliminate acetaldehyde under high catalyst loadings.17 A recent report by Miller and coworkers suggested that copolymerisation of DOX (R,R′ = H) with lactide (LA) formed polyesteracetals.18 In both of these papers, small molecule elimination was a reaction to be avoided. We hypothesised that we could instead exploit acetal elimination, using these DOX monomers to purposefully target polyesters.
Results and discussion
Before developing a broad scope synthesis, we began by showing that the published SnOct2 catalysed system can be used to prepare poly(lactic acid)-co-(glycolic acid), PLGA, upon expulsion of the acetal functionality (scheme, Table 1), especially at higher reaction temperatures. The elimination of formaldehyde under these reaction conditions was confirmed by FTIR of the white solid produced during the reaction (see Fig. S01†). These SnOct2 catalysed reactions gave polymers with broad Đs and very low DOX incorporation.|
|
|||||||
|---|---|---|---|---|---|---|---|
LA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOX :DOX |
[Cat] | Time (h) | PLAa (%) | PGAa (%) | M n,th (Da) |
M
nb (Da) |
Đ |
| I = BnOH. M:Cat:I = 100:1:1. Temperature = 40 °C. Monomer concentration = 1 M in toluene.a Monomer conversion determined by 1H NMR spectroscopy.b Đ and Mn determined by gel permeation chromatography. |
|||||||
| 50:50 |
1 | 7 | 96 | 90 | 10000 |
11100 |
2.2 |
| 50:50 |
2 | 1 | 91 | 10 | 7000 | 6700 | 1.1 |
| 50:50 |
2 | 24 | 99 | 71 | 9800 | 9100 | 1.6 |
| 50:50 |
3 | 7 | 12 | 4 | 1300 | — | — |
| 50:50 |
4 | 7 | 99 | 83 | 9700 | 4000 | 1.8 |
| 60:40 |
1 | 7 | 98 | 96 | 10800 |
14100 |
1.6 |
| 70:30 |
1 | 7 | 67 | 100 | 8600 | 6100 | 1.4 |
| 80:20 |
1 | 7 | 82 | 100 | 10700 |
8300 | 1.2 |
| 90:10 |
1 | 7 | 84 | 77 | 11700 |
7300 | 1.1 |

Expanding the catalyst scope facilitated a much improved copolymerisation of the unsubstituted DOX monomer. As homopolymerisation of DOX affords an insoluble white solid, presumably intractable poly(glycolic acid), optimisation focused on the copolymerisation of DOX and L-LA. Common catalysts MeAl(salen)tBu,tBu,Pr (1),19,20 diazabicycloundecene (DBU, 2) and triazabicyclodecene (TBD, 3)21,22 were explored, along with ZnEt2,17 (4), used by Hillmyer and coworkers in polyesteracetal synthesis (Table 1). While all the catalysts tested were capable of polymerising the L-LA monomer, 1 and 4 facilitated incorporation of high quantities of DOX to generate PLGA, while 1 gave the most comparable values between the number average molecular weight determined by SEC and the theoretically calculated weight. The organocatalysts were surprisingly poor, with 2 favouring L-LA homopolymerisation and 3 showing near complete inhibition. The ability of 1 and 4 to best control this polymerisation suggests a coordination–insertion mechanism, as both organometallic Zn complexes and salen supported Al catalysts operate as such in cyclic ester ROP. Using 1, the ratio of monomer incorporation could be tuned (Table 1) from 10–50% glycolic acid linkages. Control varied with monomer ratio, evidenced by increasing dispersity, suggesting transesterification was prevalent at high DOX loadings.
These copolymerisations are easily extended to other monomer combinations. Copolymerisation of DOX with ε-caprolactone (εCL) and β-butyrolactone (βBL) catalysed by 1 is efficient at ambient temperatures, with Đs ranging from 1.2 to 1.8 (Tables S1 and S2†). At high DOX loadings, longer reaction times were required and led to broader molecular weight distributions, especially in sluggish βBL polymerisations. In all cases, DOSY NMR experiments confirmed the formation copolymers by the presence of a single diffusion coefficient (Fig. S2–S4†). The polymer is likely a random copolymer, evidenced by the presence of heterodiads in the 13C spectra. Importantly, these methods permit the introduction of isolated glycolic acid units, a unique advantage over copolymerisations of LA and glycolide.
With optimised conditions in hand we developed the monomer scope for this potential route to aliphatic polyesters. Using a modified literature procedure,23,24 a family of 1,3-dioxolan-4-ones are easily prepared from the often renewable α-hydroxy acids and less toxic paraformaldehyde or acetone in good isolated yields (Fig. 2). Interestingly, the synthetic accessibility of these new monomers complements the established cyclic diester synthesis, with the highest yields obtained for the challenging mandelic acid derivatives.
 | ||
| Fig. 2 Synthesis of 1,3-dioxolan-4-one monomers. | ||
For these substituted monomers, homopolymerisation is facile (Fig. 3, Tables 2 and S03–S05†), leading to the formation of the desired aliphatic polyester. For example, poly(lactic acid) is synthesised by the ROP of MeDOX with the concomitant elimination of paraformaldehyde. Catalyst 1 performed best, even over ZnEt2, generating well controlled PLA (Đ = 1.2). The bulky salen ligand may protect the coordination sphere of the Lewis acidic complex, assisting to control the polymerisation and afford the desired polymer; this is evidenced, in part, by 1's broad monomer scope in cyclic ester ROP.25
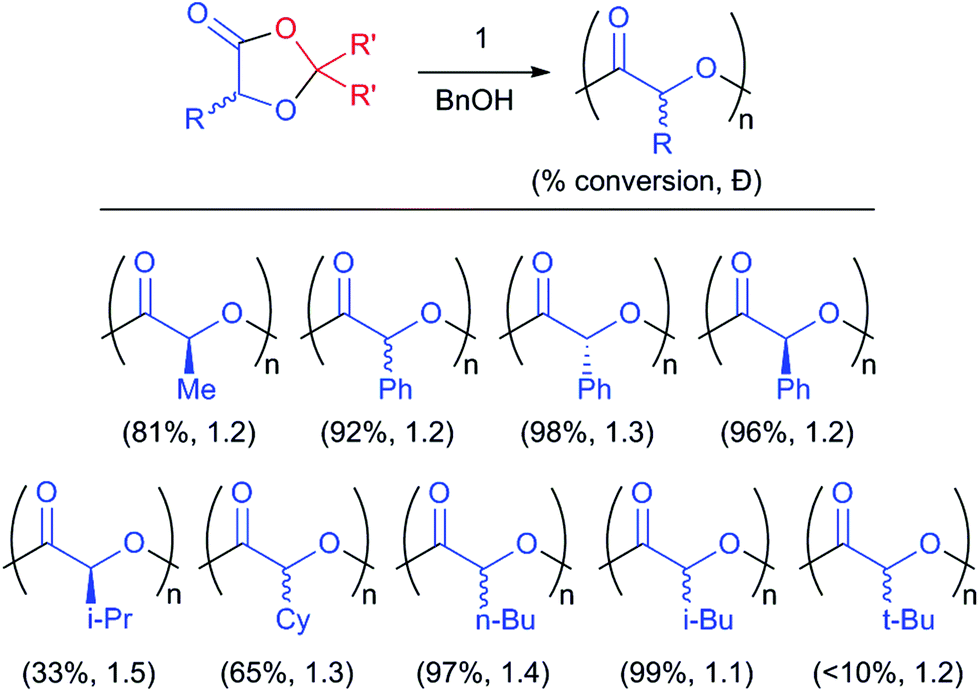 | ||
| Fig. 3 ROP of 1,3-dioxolan-4-ones to poly(α-hydroxy acid)s. | ||
| M | M:Cat:I |
Cat | t (h) | Conv.a (%) |
M
n,thb (Da) |
M
nc (Da) |
Đ |
|---|---|---|---|---|---|---|---|
| I = BnOH. Monomer conc. = 1 M in toluene.a Monomer conversion % determined by crude sample 1H NMR spectroscopy.b Mn,th = (M:BnOH) × MW(monomer) × (conv (%)) + MW(end group).c Đ and Mn determined by gel permeation chromatography.d In the absence of solvent at 180 °C. |
|||||||
| MeDOX | 100:1:1 |
4 | 48 | 88 | 6400 | 4300 | 1.5 |
| MeDOX | 100:1:1 |
5 | 24 | 28 | 2100 | — | — |
| MeDOX | 100:1:1 |
1 | 24 | 81 | 5900 | 8300 | 1.2 |
| S-PhDOX | 50:1:1 |
4 | 4 | 27 | 3700 | 2000 | 1.2 |
| S-PhDOX | 50:1:1 |
5 | 4 | 65 | 4470 | 3400 | 2.2 |
| S-PhDOX | 50:1:1 |
1 | 4 | 89 | 6080 | 3700 | 1.3 |
| S-PhDOX | 200:1:1 |
1 | 120 | 83 | 22370 |
8400 | 1.1 |
| R-PhDOX | 200:1:1 |
1 | 120 | 96 | 25860 |
7700 | 1.5 |
| R-PhDOX | 500:1:1 |
1 | 72d | 55 | 37000 |
18700 |
1.2 |
Acetal elimination is confirmed by 1H and 13C{1H} NMR spectroscopy and MALDI-ToF experiments showing an expected repeat unit m/z = 72 (Fig. S5 and S6†). Importantly, the ROP occurs with no racemisation, affording isotactic PLA with a Tm of 153 °C and Tg of 59 °C (Fig. S7†). Loadings as high as 500 eq. of MeDOX per catalyst facilitated the formation of higher molecular weight polyesters, with a small increase in Đ to 1.4.
Importantly, this allows us to target other isotactic polymers. In particular, the ROP of R- or S-PhDOX affords isotactic poly(mandelic acid), to date only prepared via an expensive phosgene-derived O-carboxyanhydride.12,26 This key bio-degradable polystyrene mimic is formed,27 at M:Cat ratios of 200:1, with conversions up to 96% and Đ as low as 1.1. No epimerisation is observed, affording highly isotactic polymers with excellent thermal properties (Fig. 4). While transesterification reactions persist, the method is much improved from condensation polymerisations28–30 of α-bromophenyl acetic acid or mandelic acid which require harsh conditions to yield atactic oligomers. Again, acetal elimination was confirmed by NMR studies, while MALDI-ToF experiments confirm a repeat unit of m/z = 134 (Fig. S07 and S08†). While formation of poly(mandelic acid) was slow, polymerisations were robust, with 500 equivalents of PhDOX polymerised in the absence of solvent at 180 °C, maintaining a narrow Đ of 1.2. These harsher temperatures do lead to atactic PMA.
 | ||
| Fig. 4 Contrasting 1H NMR spectra of the methine regions of isotactic poly(mandelic acid) (left) and atactic poly(mandelic acid) (right). | ||
Alkyl substituted polyesters are also easily targeted through ROP of the corresponding i-propyl, n-butyl, i-butyl, t-butyl and cyclohexyl DOX monomers (Fig. 3 and Table S5†). The proximity of the steric bulk to the ester functional group controlled the rate of polymerisation, with ring-opening of t-BuDOX so sluggish as to not form tractable polymers. Polymerization of DOX monomers have led to number average weights remaining lower than theoretically calculated molecular weights suggesting chain exchange reactions known to be promoted by ROP catalysts 1, 4 and 5 prevail in this new ROP.31,32 The kinetics of these polymerisations confirm that polymerisations are well controlled, with the pseudo first order rate constants trending for MeDOX > n-BuDOX > i-BuDOX (kobs = 3.0 × 10−5 s−1, 1.6 × 10−5 s−1, 8.3 × 10−6 s−1), correlating with proximal steric bulk (Fig. 5).
 | ||
| Fig. 5 Kinetic plots of MeDOX, n-BuDOX, i-BuDOX and t-BuDOX polymerisations. | ||
In support of our mechanistic hypothesis (Fig. 6), these studies suggest that sterics, not electronics, dictate ROP rates, impacting monomer coordination to the bulky Al-salen catalyst. Short polymerisations examined by NMR without workup show that the Al catalyst remains attached to the growing polymer chains, evidenced by matching diffusion coefficients in DOSY NMR studies for catalyst/polymer signals (Fig. S12†). This coordination–insertion mechanism would necessitate BnO-chain ends arising from initiation by the added BnOH, as confirmed by MALDI-ToF experiments (Fig. S8†). Interestingly, while too infrequent to be observed in NMR, the MALDI suggests that the retention of select acetal linkages may be possible, an important factor in improving polymer degradation. Their presence may be limited to the chains detected in MALDI experiments, and not in the bulk of the sample, as no evidence of their presence is noted by increasing NMR scans or sensitivity. As other systems do give increased acetal retention,18 the defined coordination sphere imposed by the Al salen framework may be key in promoting formaldehyde elimination. A full mechanistic study is a future focus for our group beyond this initial communication.
 | ||
| Fig. 6 Proposed mechanism for the coordination–insertion ROP of DOX monomers by MeAl(salen) catalysts. | ||
To further expand this elimination-driven ROP, monomers paired with non-toxic acetone were explored. Specifically, we synthesised 2,2,5-trimethyl-1,3-dioxolan-4-one (Me3DOX) and 2,2-dimethyl-5-phenyl-1,3-dioxolan-4-one (Me2PhDOX) by refluxing the appropriate α-hydroxy acid and p-toluenesulfonic acid (10%) in acetone (Fig. 3). Elimination of acetone to yield the desired polyester was slower, as the polymerisation of Me3DOX at 120 °C reached 95% conversion after 7 days (albeit in a closed vessel with no purposeful elimination of acetone) to afford a poly(lactic acid) with accurate molecular weights and low Đ. Polymerisation of Me2PhDOX required more forcing conditions, polymerising neat at 180 °C, giving a polymer with accurate molecular weights and low dispersity. Again, the resulting poly(mandelic acid) racemises under these harsh conditions to give an atactic polymer (Fig. S09†).
The scope of this simple reaction is significant, introducing functionalities ranging from poorly packed alkyl chains to rigid aryl rings. While future work will focus on the inclusion of heteroatoms or reactive functionalities, this first family of polyesters aims to alter the thermal properties of these aliphatic polyesters. In initial studies, glass transition temperatures ranged from 22 °C (R = i-Bu) to 106 °C (R = Ph) (Table S6†).
Finally, it is important to note that these monomers provide an easy way to close the loop on this green polymer synthesis, with the produced paraformaldehyde recycled into the monomer synthesis with no purification and no reduction in isolated monomer yields (see ESI†).
Conclusions
In summary, we report the first examples of ROP driven by the elimination of formaldehyde or acetone. The DOX monomer family is surprisingly versatile, broad in scope and can be produced from renewable resources. Lewis-acidic aluminum catalysts supported by the salen ligand proved best in early catalyst screens, with Đ as low as 1.08. The flexibility of this new route is exemplified in both homopolymerisations of a wide array of substituted monomers and copolymerisation with traditional cyclic esters. Polymerisations proceed without racemisation, affording isotactic poly(lactic acid) and poly(mandelic acid), the former having exceptional thermal properties for a polyester. We are now working to expand both monomer and catalyst scope and investigate the mechanism of this important new route to aliphatic polyesters.Experimental
General considerations
All experiments involving moisture- and air-sensitive compounds were performed under a nitrogen atmosphere using an MBraun LABmaster sp glovebox system equipped with a −35 °C freezer and [H2O] and [O2] analysers or using standard Schlenk techniques. Gel permeation chromatography (GPC) was used to determine polymer molecular weights and dispersities and was carried out in THF at a flow rate of 1 mL min−1 at 35 °C on a Malvern Instruments Viscotek 270 GPC Max triple detection system with 2 × mixed bed styrene/DVB columns (300 × 7.5 mm). GPC analysis was performed using OmniSEC 5.0 software. PLA, PHB and PCL dn/dc values used were 0.050,33 0.065,34 and 0.072 ml g−1.35 The dn/dc value of PMA was experimentally determined to be 0.11 ml g−1. All other homopolymer molecular weights were determined versus polystyrene standards. The molecular weights of copolymers containing DOX were calculated by 1H NMR spectroscopy by relative integration of benzylic end-group resonances to polymer resonances. 1H and 13C NMR spectra were recorded at 300 K with Bruker Avance spectrometers (400, 500 or 600 MHz) in CDCl3 or C6D6. Mass spectrometry was performed on a Bruker UltraflexExtreme MALDI-ToF spectrometer. MALDI-ToF samples were prepared using the following matrices; 2,5-dihydroxybenzoic acid for poly(lactic acid) and poly(hexahydromandelic acid), dithranol for poly(mandelic acid) and sodium or potassium trifluroacetate was used as the ionisation source. Differential scanning calorimetry (DSC) was carried out using a DSC 404 F3 Pegasus® DSC instrument using a heat (25–200 °C)/cool (200–25 °C)/heat (25–500 °C) cycle at a rate of 10 °C min−1. Values of Tg and Tm were obtained from the second heating scan.Materials
L-Lactide was provided by Corbion and was purified by three vacuum sublimations prior to polymerisations. Benzyl alcohol, β-butyrolactone, ε-caprolactone, and d-chloroform were purified by stirring over CaH2 followed by distillation under an inert atmosphere. Deuterated solvents were dried over molecular sieves or Na/benzophenone and subsequently distilled under an inert atmosphere. Toluene and hexanes were dried using an Innovative Technologies purification system consisting of alumina and copper catalyst. Following drying solvents were degassed by three freeze–pump–thaw cycles prior to use. MeAl(salen)tBu,tBu,Pr was synthesised by following established protocols.Monomer synthesis
Two general monomer synthesis procedures were followed:1,3-Dioxolan-4-one (DOX). Colourless oil, 26% yield. 1H NMR (500 MHz, CDCl3) δ 5.50 (s, 2H, OCH2O), 4.20 (s, 2H, CCH2O) ppm. 13C NMR (126 MHz, CDCl3) δ 171.20, 96.10, 62.45 ppm.
5-Methyl-1,3-dioxolan-4-one (MeDOX). Colourless oil, 75% yield. 1H NMR (500 MHz, CDCl3) δ 5.50 (s, 1H, OCH2O), 5.38 (s, 1H, OCH2O), 4.27 (q, J = 6.8 Hz, 1H, OCCHO), 1.47 (d, J = 6.8 Hz, 3H, CCH3). 13C NMR (151 MHz, CDCl3) δ 121.97, 42.36, 42.34, 18.32, −35.26.
5-Phenyl-1,3-dioxolan-4-one (PhDOX). Colourless oil, 96% yield. 1H NMR (500 MHz, CDCl3) δ 7.52–7.37 (m, 5H, Ar), 5.67 (s, 1H, OCH2O), 5.58 (s, 1H, OCH2O), 5.23 (s, 1H, CCCHO) ppm. 13C NMR (126 MHz, CDCl3) δ 171.38, 133.30, 129.24, 128.90, 126.52, 94.57, 74.49 ppm.
5-Isopropyl-1,3-dioxolan-4-one (i-PrDOX). Colourless oil, 90% yield. 1H NMR (500 MHz, CDCl3) δ 5.51 (s, 1H, OCH2O), 5.42 (d, J = 0.9 Hz, 1H, OCH2O), 4.04 (dt, J = 4.4, 0.7 Hz, 1H, OCCHO), 2.18 (heptd, J = 6.8, 4.2 Hz, 1H), 1.09 (d, J = 6.9 Hz, 3H), 1.01 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 172.35, 94.48, 29.88, 18.26, 16.65.
5-Cyclohexyl-1,3-dioxolan-4-one (CyDOX). Colourless oil, 86% yield. 1H NMR (500 MHz, CDCl3) δ 5.50 (s, 1H, OCH2O), 5.42 (s, 1H, OCH2O), 4.03 (d, J = 4.0 Hz, 1H, OCCHO), 1.91–1.73 (m, 3H), 1.72–1.58 (m, 2H), 1.34–1.09 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 172.59, 94.74, 39.43, 28.93, 26.92, 26.08, 26.02, 25.87.
5-nButyl-1,3-dioxolan-4-one (n-BuDOX). Colourless oil, 60% yield. 1H NMR (500 MHz, CDCl3) δ 5.48 (s, 1H, OCH2O), 5.38 (s, 1H, OCH2O), 4.16 (m, 1H, OCCHO), 1.85 (m, 1H, CH2CH2CH2CH3), 1.77–1.65 (m, 1H, CH2CH2CH2CH3), 1.52–1.27 (m, 4H, CH2CH2CH3), 0.89 (t, J = 7.2 Hz, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 172.98, 94.03, 73.02, 29.92, 26.88, 22.12, 13.66.
5-iButyl-1,3-dioxolan-4-one (i-BuDOX). Colourless oil, 60%. 1H NMR (500 MHz, CDCl3) δ 5.50 (s, 1H), 5.39 (s, 1H), 4.20 (ddd, J = 9.3, 3.9, 0.9 Hz, 1H, OCCHO), 1.87 (dh, J = 8.1, 6.6 Hz, 1H, CH2CH(CH3)), 1.75–1.59 (m, 2H, CH2CH(CH3)), 0.96 (dd, J = 6.8, 5.5 Hz, 6H, CH3). 13C NMR (126 MHz, CDCl3) δ 173.43, 93.93, 77.25, 77.00, 76.75, 71.83, 38.95, 25.03, 22.79, 21.63.
:1, 50 mL) and refluxed in a Dean–Stark apparatus, periodically removing water formed over 6 h. The reaction mixture was cooled and washed with sodium bicarbonate aq. (10%), water and sodium chloride aq. (sat.). The organic layer was dried with magnesium sulphate and the solvent was removed from the organic layer in vacuo. The crude product was then stirred over calcium hydride for 16 h before being purified by vacuum distillation or toluene recrystallisation to obtain pure products.
2,2,5-Trimethyl-1,3-dioxolan-4-one (Me3DOX). Colourless oil, 60% yield. 1H NMR (500 MHz, CDCl3) δ 4.43 (q, J = 6.8 Hz, 1H, CHCH3), 1.56 (s, 3H, OOCCH3), 1.49 (s, 3H, OOCCH3), 1.42 (d, J = 6.8 Hz, 3H, CHCH3). 13C NMR (126 MHz, CDCl3) δ 173.78, 110.36, 110.29, 110.06, 70.40, 27.41, 25.56, 17.36.
2,2-Dimethyl-5-phenyl-1,3-dioxolan-4-one (Me2PhDOX). White crystalline solid, 95% yield. 1H NMR (500 MHz, CDCl3) δ 7.52–7.46 (m, 2H, Ar), 7.48–7.36 (m, 4H, Ar), 5.42 (s, 1H, OOCCH), 1.75 (s, 3H, OOCCH3), 1.70 (s, 3H, OOCCH3). 13C NMR (126 MHz, CDCl3) δ 171.42, 134.45, 128.95, 128.73, 126.42, 110.94, 75.89, 27.26, 26.19.
Representative polymerisation
MeAl[salen]tButBuPr (5.5 mg, 0.01 mmol), BnOH (1 μL, 0.01 mmol) and MeDOX (88 mg, 1.0 mmol) in toluene (1 mL) was added to an oven dried ampoule. The ampoule was then sealed and heated to 120 °C for 24 h. The reaction was then quenched by addition of two drops of MeOH and samples were taken for crude 1H NMR. The remainder was added dropwise to cold methanol and upon cooling to −35 °C for two days a white solid was precipitated. The white solid was filtered, dried to constant weight and used for GPC analysis.Copolymerisation data
Substituted RDOX homopolymerisations data
R = Methyl, poly(lactic acid): 1H NMR (500 MHz, CDCl3) δ 5.16 (q, J = 7.1 Hz, 1H), 1.58 (d, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.59, 69.01, 16.65.R = Phenyl, poly(mandelic acid): 1H NMR (601 MHz, CDCl3) δ 7.48–7.01 (m, 5H), 6.15–5.92 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 166.76, 132.33, 129.34, 128.64, 127.80, 74.75.
R = Cyclohexyl, poly(hexahydromandelic acid): 1H NMR (500 MHz, CDCl3) δ 5.05–4.87 (m, 1H), 2.32–0.68 (m, 11H). 13C NMR (126 MHz, CDCl3) δ 168.70, 39.51, 28.69, 27.18, 25.89.
R = iPropyl, poly(vandelic acid): 1H NMR (500 MHz, CDCl3) δ: 4.9 (m, 55.7H), 4.1 (d, 2H), 2.3 (b, 59.2H) 1.39 (s, 9H), 1.1 (m, 350.6H). (CDCl3, 125 MHz) δ: 168.6, 128, 125.
R = n-Butyl, poly(2-nButylhexanoic acid): 1H NMR (601 MHz, CDCl3) δ 5.21–5.03 (m, 1H), 2.03–1.85 (m, 2H), 1.45–1.30 (m, 5H), 0.98–0.82 (m, 7H). 13C NMR (126 MHz, CDCl3) δ 169.17, 72.49, 33.66, 27.03, 22.36, 13.78.
R = i-Butyl, poly(2-iButylhexanoic acid): 1H NMR (601 MHz, CDCl3) δ 5.09 (dd, J = 9.4, 4.2 Hz, 1H), 1.85–1.73 (m, 4H), 0.94 (dd, J = 20.2, 6.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.70, 71.34, 39.31, 24.50, 22.96, 21.41.
Acknowledgements
We gratefully acknowledge the support of EU funding bodies, the Marie-Curie Actions Programme (FP7-PEOPLE-2013-CIG-618372), the University of Edinburgh and EaStCHEM. We thank Corbion for providing us with lactide monomer. We thank Fergus McWhinnie and Annabel Serpico for useful discussions on enzymatic biodegradation.Notes and references
- J. P. Swanson, M. R. Martinez, M. A. Cruz, S. G. Mankoci, P. J. Costanzo and A. Joy, Polym. Chem., 2016, 7, 4693–4702 RSC.
- J. Natarajan, G. Madras and K. Chatterjee, RSC Adv., 2016, 6, 40539–40551 RSC.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- A. Salerno and C. D. Pascual, RSC Adv., 2013, 3, 17355 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- A. Duda and S. Penczek, Macromolecules, 1990, 23, 1636 CrossRef CAS.
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056 CrossRef PubMed.
- S.-Y. Lu and I. Hamerton, Prog. Polym. Sci., 2002, 27, 1661 CrossRef CAS.
- G. L. Baker, E. B. Vogel and M. R. Smith III, Polym. Rev., 2008, 48, 64–84 CrossRef CAS.
- T. Trimaille, M. Möller and R. Gurny, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4379 CrossRef CAS.
- M. Yin and G. L. Baker, Macromolecules, 1999, 32, 7711 CrossRef CAS.
- F. Jing, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 9304 CrossRef CAS.
- O. Thillaye du Boullay, E. Marchal, B. Martin-Vaca, F. P. Cossío and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 16442 CrossRef CAS PubMed.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858 CrossRef CAS PubMed.
- LCt50 = 1.7 ppm per 4 h (rat). J. Pauluhn, Inhalation Toxicol., 2011, 23, 65 CrossRef CAS PubMed.
- L. F. Haber, The Poisonous Cloud: Chemical Warfare in the First World War, Oxford University Press, 1986 Search PubMed.
- A. E. Neitzel, M. A. Petersen, E. Kokkoli and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 1156 CrossRef CAS.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768 RSC.
- J. P. Macdonald, M. P. Parker, B. W. Greenland, D. Hermida-Merino, I. W. Hamley and M. P. Shaver, Polym. Chem., 2014, 6, 1445 RSC.
- F. Sinclair, L. Chen, B. W. Greenland and M. P. Shaver, Macromolecules, 2016, 49, 6826–6834 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade and R. M. Waymouth, et al. , Macromolecules, 2006, 39, 8574 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS PubMed.
- P. Salomaa, S. Laiho, S. J. Cyvin, P. C. Kvande and E. Meisingseth, Acta Chem. Scand., 1963, 17, 103 CrossRef CAS.
- Y. Asabe, S. Takitani and Y. Tsuzuki, Bull. Chem. Soc. Jpn., 1973, 46, 661 CrossRef CAS.
- J. P. Macdonald and M. P. Shaver, ACS Symposium series: Green Polymer Chemistry: Biobased Materials and Biocatalysis, 2015, vol. 10, p. 147 Search PubMed.
- To purchase and prepare 1 kg Ph(OCA) in an academic lab would cost $2982.61 USD vs. $156 USD with further price reductions at higher scale. See ESI† for further detail.
- Biodegradation of poly(mandelic acid) with proteinase K led to a 12% weight loss in 24 h. See ESI† for further detail.
- A. G. Pinkus, R. Subramanyam, S. L. Clough and T. C. Lairmore, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 4291 CrossRef CAS.
- A. J. Domb, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1973 CrossRef CAS.
- J. K. Whitesell and J. A. Pojman, Chem. Mater., 1990, 2, 248 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, R. I. Pugh and A. J. P. White, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15343 CrossRef CAS PubMed.
- G. Schwach, J. Coudane, R. Engel and M. Vert, Polym. Bull., 1994, 32, 617–623 CrossRef CAS.
- G. L. Fiore, J. L. Klinkenberg and C. L. Fraser, Macromolecules, 2008, 41, 9397 CrossRef CAS.
- Y. Hori and T. Hagiwara, Int. J. Biol. Macromol., 1999, 25, 237 CrossRef CAS PubMed.
- X. Zhou and L. Hong, Colloid Polym. Sci., 2013, 291, 2155 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py00254h |
| This journal is © The Royal Society of Chemistry 2017 |