
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free macrocyclisation by nucleophile-mediated oxa-Michael addition polymerisation of divinyl sulfone and alcohols†
Simone
Strasser
,
Christina
Wappl
and
Christian
Slugovc
 *
*
Institute for Chemistry and Technology of Materials, Graz University of Technology, NAWI Graz, Stremayrgasse 9, A 8010 Graz, Austria. E-mail: slugovc@tugraz.at
First published on 16th February 2017
Abstract
The oxa-Michael addition reaction has been widely studied and employed in organic synthesis, but has not been developed as a tool for polymer synthesis. Herein, the 4-dimethylaminopyridine initiated polyaddition reaction of divinyl sulfone and several di- or multifunctional alcohols has been studied. This atom-economical polymerisation reaction proceeds quickly and quantitatively under solvent-free conditions producing predominantly macrocyclic products following a zwitter-ionic mechanism. Multifunctional alcohols yield duroplastic polymers. Considering the ready availability of alcohols, the oxa-Michael addition polymerisation is an interesting alternative to the well-established thiol-Michael addition polymerisation.
Introduction
Organocatalysed polymerisations have gained ever increasing attention over the last few years mainly because they hold promise to enable environmentally more friendly processes using less toxic and cheaper chemicals.1 An important polymerisation reaction based on organocatalysis is the thiol-Michael addition polymerisation2 which is nowadays a widely applied tool in polymer and materials chemistry.3 The thiol-Michael addition exists in two mechanistic variants, i.e. the base-catalysed and the nucleophile-initiated variant.4 In the latter case a nucleophile is added to the electron deficient olefin generating a strongly basic zwitter-ionic species which subsequently deprotonates the thiol. The resulting thiolate then attacks another electron deficient olefin forming the desired thioether bond.5 Thiols are good reactants in this transformation because they are relatively acidic and thiolates are fairly good nucleophiles. Nevertheless the use of thiols entails some drawbacks such as bad odour and toxicity, side-product formation via oxidative disulfide formation and oxidative instability of the formed thioether groups. The use of alcohols instead of thiols therefore constitutes an interesting alternative, in particular also because the number of readily available alcohols is much higher compared to thiols. However, alcohols are less acidic than thiols6 and alkoxides are generally poorer nucleophiles than thiolates rendering an oxa-Michael addition polymerisation more challenging. Indeed, although the oxa-Michael reactions are widely used in small molecule synthesis,7 applications in polyaddition reactions are scarce.8 Recently we have communicated on the triphenylphosphine mediated oxa-Michael addition of divinyl sulfone (DVS) and alcohols and found that the reaction is particularly fast under solvent-free conditions allowing for polyaddition reactions with di- and trifunctional alcohols.9We herein wish to present our results on the oxa-Michael addition polymerisation of the substrates shown in Scheme 1 and introduce 4-dimethylaminopyridine (DMAP) as a preferred nucleophile.10
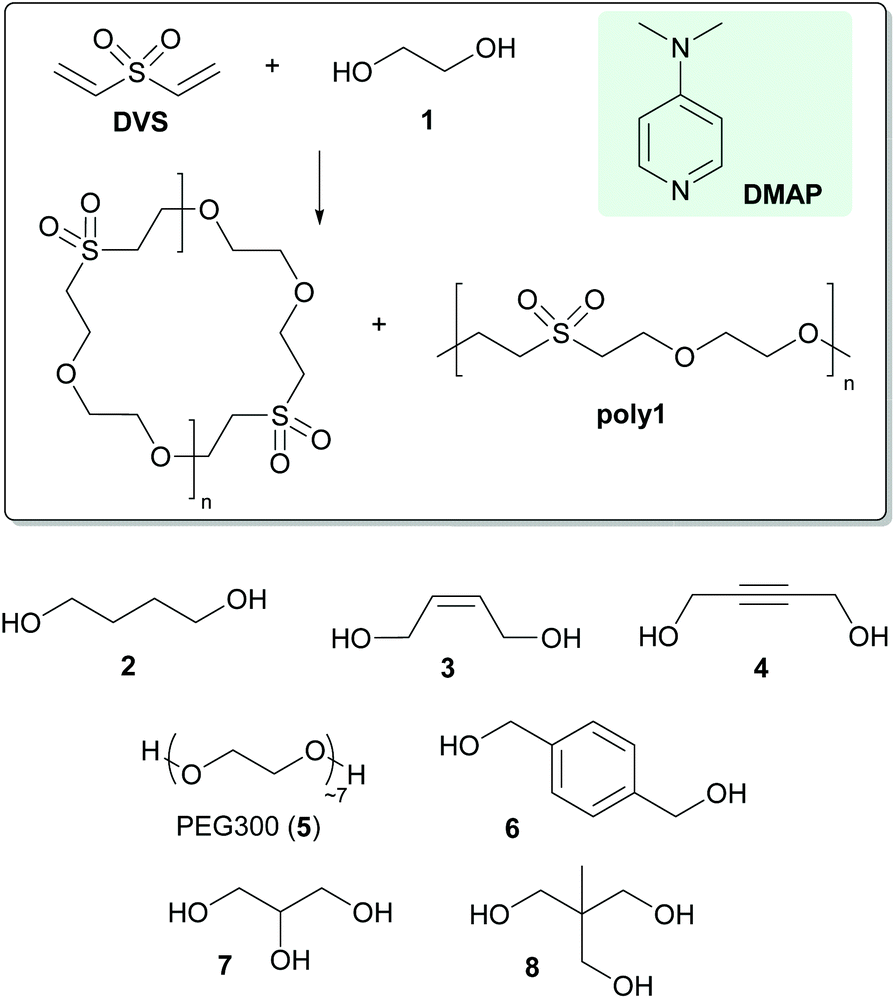 | ||
| Scheme 1 Substrates and the nucleophile used for oxa-Michael addition polymerisations. | ||
Experimental
Materials and methods
Divinyl sulfone (DVS), 4-dimethylaminopyridine (DMAP) and all alcohols used were purchased from Aldrich and were used as received. Only ethylene glycol was distilled prior to use and PEG300 was dried under oil-pump vacuum for 24 h. Column chromatography was done using silica gel (60, 0.03–0.2 mm, product no. P090.5) purchased from Lactan, Austria. Aluminum sheets with silica gel (60 F254) for thin layer chromatography (TLC) were purchased from Merck KGaA, Germany.Nuclear magnetic resonance (NMR) experiments were done on a Bruker Avance 300 MHz spectrometer (1H: 300.36 MHz; 13C: 75.53 MHz) at 25 °C. Chemical shifts are given in ppm relative to a tetramethylsilane standard. Deuterated solvents were obtained from Cambridge Isotope Laboratories Inc. and spectra were referenced against the residual proton signals according to the literature.11
Thermogravimetric analyses (TGA) were performed with a Netzsch Simultaneous Thermal Analyzer STA 449C (crucibles: aluminium from Netzsch). A helium flow of 35 mL min−1 was used in combination with a protective flow of helium of 8 mL min−1. The heating rate until a final temperature of 550 °C was 10 °C min−1.
Gel permeation chromatography (GPC) was carried out on a system provided by WGE Dr Bures operated with THF (separating columns from MZ-Gel SD plus, linear 5μ; UV and RI detector SEC 3010). Poly(styrene) standards purchased from Polymer Standard Service were used for calibration.
Dynamic mechanical analyses (DMA) were performed on a DMA Q 800 from TA Instruments Waters GmbH in the 3-point bending mode (10 mm clamping lengths) of specimens with sample dimensions of 3 × 5 × 20 mm with a frequency of oscillation of 1 Hz and an amplitude of 25 μm in a temperature range from −4 to 50 °C (heating rate: 3 °C min−1).
Differential scanning calorimetry (DSC) analyses were performed on a DSC 8500 instrument from Perkin Elmer in a temperature range from −20 to 100 °C with a heating rate of 20 °C min−1 for the first run and with 20 °C min−1 in the second run (Tg values were retrieved from the second heating run).
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were recorded on two different machines: (a) on a Micromass TofSpec 2E Time-of-Flight mass spectrometer. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as a matrix substance and tetrahydrofuran (THF) as a solvent. Sample solutions have been prepared by mixing solutions of DCTB (10 mg mL−1), sodium trifluoroacetate (1 mg mL−1) and the samples (5 mg mL−1) in a ratio of 7/2/2 (v/v/v). Calibration was done externally with polyethylene glycol standards (5 mg mL−1). (b) On a Waters Micromass MALDI micro MX Time-of-Flight mass spectrometer. 1,8,9-Anthracenetriol (dithranol) was used as the matrix substance and THF as the solvent. Sample solutions have been prepared by mixing solutions of dithranol (10 mg mL−1), sodium trifluoroacetate (1 mg mL−1) and the samples (5 mg mL−1 in chloroform) in a ratio of 7/2/2 (v/v/v). Calibration was done externally with polyethylene glycol standards (5 mg mL−1) GC MS: mass spectra were recorded on a Waters Micromass GCT Premier mass spectrometer internally calibrated with heptacosa. Of a sample solution (1 mg mL−1) 1 μL was injected into a gas chromatograph (Agilent, 7890A, operated with He, injection temperature: 300 °C; split: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100; column: DB 5MS from J&W Scientific; temperature program: starts at 40 °C, heating rate 50 °C min−1, end temperature 300 °C for 5 min).
:100; column: DB 5MS from J&W Scientific; temperature program: starts at 40 °C, heating rate 50 °C min−1, end temperature 300 °C for 5 min).
Synthetic procedures
1H-NMR (300 MHz, CDCl3, 25 °C): δ 6.78 (dd, 1H, 3JHH(E) = 16.6 Hz, 3JHH(Z) = 9.9 Hz, CH), 6.39 (d, 1H, 3JHH = 16.6 Hz, CH2(E)), 6.12 (d, 1H, 3JHH = 9.9 Hz, CH2(Z)), 3.91 (t, 124H, –SO2–CH2CH2–O–), 3.64 (s, 127H, –O–CH2CH2–O–), 3.35 (t, 123H, –SO2–CH2CH2–O). 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 138.0 (–SO2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 129.2 (–SO2–CHCH2), 72.7 (HO–CH2CH2–O–), 70.4 (–O–CH2CH2–O–), 70.1 (HO–CH2CH2–O–), 64.7 (–SO2–CH2CH2–O–), 55.0 (–SO2–CH2CH2–O–) ppm.
CH2), 129.2 (–SO2–CHCH2), 72.7 (HO–CH2CH2–O–), 70.4 (–O–CH2CH2–O–), 70.1 (HO–CH2CH2–O–), 64.7 (–SO2–CH2CH2–O–), 55.0 (–SO2–CH2CH2–O–) ppm.
1H-NMR (300 MHz, CDCl3, 25 °C): δ 6.71 (dd, 1H, 3JHH = 16.7 Hz, 3JHH = 9.9 Hz, CH), 6.39 (d, 1H, 3JHH = 16.7 Hz, CH2(E)), 6.09 (d, 1H, 3JHH = 9.9 Hz, CH2(Z)), 3.84 (t, 31H, –SO2–CH2CH2–O–), 3.63 (t, 1.4H, CH2CH2CH2CH2–OH), 3.48 (s, 31H, –O–CH2CH2CH2CH2–O–), 3.30 (t, 31H, –SO2–CH2CH2–O), 1.63 (s, 31H, –O–CH2CH2CH2CH2–O–). 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 137.9 (–SO2–CHCH2), 129.0 (–SO2–CHCH2), 71.4 (HO–CH2CH2CH2CH2–O–), 71.1 (–O–CH2CH2CH2CH2–O–), 64.3 (–SO2–CH2CH2–O–), 62.5 (HO–CH2CH2CH2CH2–O–), 55.1 (–SO2–CH2CH2–O–), 29.7 (–O–CH2CH2CH2CH2–O–), 26.2 (–O–CH2CH2CH2CH2–O–) ppm.
1H-NMR (300 MHz, CDCl3, 25 °C): δ 8.21 (d, 1.3H, residual DMAP), 6.71 (dd, 1H, 3JHH = 16.6 Hz, 3JHH = 9.9 Hz, CH), 6.48 (d, 1.3H, residual DMAP), 6.40 (d, 1H, 3JHH = 16.6 Hz, CH2(E)), 6.10 (d, 1H, 3JHH = 9.9 Hz, CH2(Z)), 5.72 (s, 36.9H, –CH2CHCHCH2–), 4.19 (d, 1.8H, HO–CH2CHCHCH2–), 4.09 (d, 72.7H, –O–CH2CHCHCH2–O–), 3.85 (t, 72.3H, –SO2–CH2CH2–O–), 3.32 (s, 72.6H, –SO2–CH2CH2–O), 2.99 (d, 4.6H, residual DMAP). 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 149.8 (DMAP), 137.9 (–SO2–CHCH2), 133.3 (HO–CH2CHCHCH2–O–), 129.3 (–O–CH2CHCHCH2–O–), 129.1 (–SO2–CHCH2), 127.2 (HO–CH2CHCHCH2–O–), 106.7 (DMAP), 66.9 (–O–CH2CHCHCH2–O–), 63.9 (–SO2–CH2CH2–O–), 58.6 (–CH2–SO2–CHCH2), 55.1 (–SO2–CH2CH2–O–), 39.1 (DMAP) ppm.
1H-NMR (300 MHz, CDCl3, 25 °C): δ 8.20 (d, 2.2H, residual DMAP), 6.73 (dd, 1H, 3JHH = 16.6 Hz, 3JHH = 9.9 Hz, CH), 6.48 (d, 2.2H, residual DMAP), 6.41 (d, 1H, 3JHH = 16.6 Hz, CH2(E)), 6.11 (d, 1H, 3JHH = 9.9 Hz, CH2(Z)), 4.25 (s, 160.3H, –O–CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2–O–), included in the integral before 4.29 (HO–CH2CCCH2–O–), 3.96 (t, 155H, –SO2–CH2CH2–O–), 3.35 (s, 156H, –SO2–CH2CH2–O), 2.99 (d, 4.6H, residual DMAP). 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 149.7 (DMAP), 137.8 (–SO2–CHCH2), 129.5 (–SO2–CHCH2), 127.2 (HO–CH2CHCHCH2–O–), 106.7 (DMAP), 86.2 (HO–CH2CCCH2–O–), 82.5 (–O–CH2CCCH2–O–), 80.5 (HO–CH2CCCH2–O–), 63.4 (–O–CH2CCCH2–O–), 58.7 (–SO2–CH2CH2–O–), 54.9 (–SO2–CH2CH2–O–), 50.9 (HO–CH2CCCH2–O–), 39.1 (DMAP) ppm. 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 149.7 (DMAP), 137.8 (–SO2–CHCH2), 129.5 (–SO2–CHCH2), 127.2 (HO–CH2CHCHCH2–O–), 106.7 (DMAP), 86.2 (HO–CH2CCCH2–O–), 82.5 (–O–CH2CCCH2–O–), 80.5 (HO–CH2CCCH2–O–), 63.4 (–O–CH2CCCH2–O–), 58.7 (–SO2–CH2CH2–O–), 54.9 (–SO2–CH2CH2–O–), 50.9 (HO–CH2CCCH2–O–), 39.1 (DMAP) ppm.
CCH2–O–), included in the integral before 4.29 (HO–CH2CCCH2–O–), 3.96 (t, 155H, –SO2–CH2CH2–O–), 3.35 (s, 156H, –SO2–CH2CH2–O), 2.99 (d, 4.6H, residual DMAP). 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 149.7 (DMAP), 137.8 (–SO2–CHCH2), 129.5 (–SO2–CHCH2), 127.2 (HO–CH2CHCHCH2–O–), 106.7 (DMAP), 86.2 (HO–CH2CCCH2–O–), 82.5 (–O–CH2CCCH2–O–), 80.5 (HO–CH2CCCH2–O–), 63.4 (–O–CH2CCCH2–O–), 58.7 (–SO2–CH2CH2–O–), 54.9 (–SO2–CH2CH2–O–), 50.9 (HO–CH2CCCH2–O–), 39.1 (DMAP) ppm. 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 149.7 (DMAP), 137.8 (–SO2–CHCH2), 129.5 (–SO2–CHCH2), 127.2 (HO–CH2CHCHCH2–O–), 106.7 (DMAP), 86.2 (HO–CH2CCCH2–O–), 82.5 (–O–CH2CCCH2–O–), 80.5 (HO–CH2CCCH2–O–), 63.4 (–O–CH2CCCH2–O–), 58.7 (–SO2–CH2CH2–O–), 54.9 (–SO2–CH2CH2–O–), 50.9 (HO–CH2CCCH2–O–), 39.1 (DMAP) ppm.
1H-NMR (300 MHz, CDCl3, 25 °C): δ 6.82 (dd, 1H, 3JHH = 16.6 Hz, 3JHH = 9.9 Hz, CH), 6.39 (d, 1H, 3JHH = 16.6 Hz, CH2(E)), 6.09 (d, 1H, 3JHH = 9.9 Hz, CH2(Z)), 3.90 (t, 4.9H, –SO2–CH2CH2–O–), 3.64 (s, 39.7H, PEG300), 3.35 (t, 2.9H, –SO2–CH2CH2–O), 3.00 (t, 2.2H, H2CCH–SO2–CH2CH2–O).
To the rest of the reaction mixture, propargylic alcohol (125 μL, 2.162 mmol) was added and the reaction mixture was stirred for further 2 h at 80 °C. An aliquot was removed and investigated by 1H-NMR spectroscopy.
1H-NMR (300 MHz, CDCl3, 25 °C): δ 4.28 (d, 0.36H, HCCCH2OH), 4.20 (d, 1.8H, HCCCH2OCH2CH2–), 4.02–3.85 (m, 7.0H, –SO2–CH2CH2–O–), 3.64 (s, 39.7H, PEG300), 3.46–3.28 (m, 7.1H, –SO2–CH2CH2–O).
1H-NMR (300 MHz, CDCl3, 25 °C): δ 8.18 (DMAP), 6.76 (dd, 1H, 3JHH(E) = 16.6 Hz, 3JHH(Z) = 9.9 Hz, CH), 6.46 (DMAP), 6.38 (d, 1H, 3JHH = 16.6 Hz, CH2(E)), 6.10 (d, 1H, 3JHH = 9.9 Hz, CH2(Z)), 4.53 (s, 16.6H, –O–CH2–C6H4–CH2–O–), 3.89 (s, 119H, –SO2–CH2CH2–O–), 3.63 (s, 105H, –O–CH2CH2–O–), 3.33 (t, 121H, –SO2–CH2CH2–O), 2.98 (DMAP). 13C{1H}-NMR (75 MHz, CDCl3, 25 °C): δ 149.8 (DMAP), 138.0 (–SO2–CHCH2), 137.2 (Ph1,4), 129.1 (–SO2–CHCH2), 128.0 (Ph2,3,5,6), 106.7 (DMAP), 72.7 (HO–CH2CH2–O–), 73.1 (–O–CH2C6H4–CH2–O–), 70.4 (–O–CH2CH2–O–), 70.1 (HO–CH2CH2–O–), 64.7 (–SO2–CH2CH2–O–CH2CH2), 63.9 (–SO2–CH2CH2–O–CH2C6H4–), 55.0 (–SO2–CH2CH2–O–), 39.1 (DMAP) ppm.
Results and discussion
The reaction of several di- and multifunctional alcohols (Scheme 1) with divinyl sulfone was investigated. Although triphenylphosphine is a good nucleophile for mediating oxa-Michael reactions,9 its solubility in alcohols is generally low. To avoid the use of a solvent in the formulation, better soluble DMAP was used as the nucleophile in this study. Typically a catalytic amount of DMAP was dissolved in the di- or trifunctional alcohol and the corresponding amount of DVS to obtain an equimolar ratio of alcohol- and vinyl-groups was admixed.‡ Instantly an exothermic reaction took place which led to a rapid increase of the viscosity of the reaction mixture. After a reaction time of 24 h an aliquot of the reaction mixture was used for analyses and the rest was placed in an oven preheated to 80 °C for further 24 h. The reaction mixtures were investigated via1H- and 13C{1H}-NMR spectroscopy, gel permeation chromatography (GPC) and matrix assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF).From the 1H-NMR-spectra of all polymers acquired after a reaction time of 24 h at room temperature it is evident that no residual DVS is present in the reaction mixture. A characteristic signal of DVS shows a chemical shift of 6.59 ppm (dd, 3JHH = 16.6 Hz, 3JHH = 9.7 Hz). Instead a resonance at 6.78 appeared (dd, 3JHH = 16.6 Hz, 3JHH = 9.9 Hz) which can be assigned to a vinyl sulfone end-group. The repeating units of the macromolecules of poly1 give rise to three equally intense signals located at 3.91, 3.64 and 3.35 ppm. The alcohol bearing end-group is submersed under the signal at 3.64 ppm which features a slightly higher integral value when compared to the signals located at 3.91 and 3.35 ppm. An apparent degree of polymerisation (n) according to the drawings in Fig. 1 (series A) of 9.5 ± 1 was calculated (using a procedure described in the ESI†). Upon heating of the reaction mixture for further 24 h at 80 °C, n increased to 30.5 ± 2. It is worth mentioning that unreacted DMAP is observed in all samples which were not purified by acidic extraction (see the ESI†). The 13C-NMR spectrum of poly1 features three intense peaks at 70.4, 64.7 and 55.0 ppm representing the repeating unit. GPC of poly1 (after purification as described in the Experimental section) revealed a number average molecular mass lower than 800 g mol−1, which is in contradiction to the apparent degree of polymerization from end-group analysis via NMR. The MALDI-TOF mass spectrum revealed the presence of at least four differently terminated macromolecular series. The heterotelechelic ethylene glycol vinyl sulfone terminated species (Fig. 1 series A) was found as the most abundant species. The presence of the corresponding cyclic species (featuring the same molecular masses) cannot be ruled out at this stage. In fact, the discrepancy of the apparent degree of polymerization and the low molecular masses found from GPC could be explained by the presence of such cyclic products. Homotelechelic vinyl sulfone (Fig. 1 series B) and ethylene glycol terminated macromolecules (Fig. 1 series D) as well as a 4-dimethylamino pyridinium/vinyl sulfone end-capped species (Fig. 1 series C) were found as minor products. MALDI-TOF mass spectra of the polymers prepared from 1,4-butanediol (poly2), (Z)-2-butene-1,4-diol (poly3) and 2-butyne-1,4-diol (poly4) provide very similar information and are available in the ESI†. The only difference is that the anticipated series not observed in the poly1-spectrum, the heterotelechelic 4-dimethylamino pyridinium/alcohol end-capped species, was additionally found in all other cases. The NMR-spectra of poly2–poly4 bear no special features and are given in the Experimental part. Degrees of polymerizations retrieved for samples prepared at room temperature for 24 h were 2.5 ± 0.5 (poly2), 10.6 ± 2 (poly3) and 17.5 ± 2 (poly4). Additional heating for further 24 h at 80 °C increased the apparent n to 7.5 ± 1 (poly2), 17.6 ± 2 (poly3) and 38 ± 2 (poly4), respectively. GPC of poly2 revealed a Mn-value of 1400 while for poly3 a Mn < 800 g mol−1 was retrieved. In contrast, poly4 is characterized by a distinctly higher Mn value of 8000 g mol−1 (polydispersity index 1.8). Also these data suggest, at least in the case of poly2 and poly3, considerable formation of cyclic products. In a first endeavour to gain evidence for cyclic products poly1 was prepared as described and approx. half of the reaction mixture was removed after a reaction time of 30 min at room temperature (DVS and 1 were completely consumed) and subsequently subjected to column chromatography. The first fraction was obtained from eluting with cyclohexane and ethyl acetate (v/v = 1:1) and was identified as the simplest cyclic product 1,4,7-dioxathionane-7,7-dioxide (approx. 1% of the fraction, NMR and GC-MS proof, see the ESI†). Further elution with dichloromethane:methanol (v/v = 10:1) released 82% of the fraction, which was identified as poly1 with an apparent n of 3.7 ± 1. The rest of the reaction mixture was heated at 80 °C for 16 h and was then treated as described above. Similar yields for 1,4,7-dioxathionane-7,7-dioxide and poly1 (apparent n of 21 ± 2) were obtained. In the case of both fractions the remaining approx. 25% could not be eluted from the column. From this experiment two points can be made: (a) cyclic products can be formed as exemplified by the formation of 1,4,7-dioxathionane-7,7-dioxide and (b) the thermal stress of the reaction mixture does not increase the yield of 1,4,7-dioxathionane-7,7-dioxide. Unfortunately potentially formed higher cyclic products could not be separated as pure products. Accordingly a second experiment for gaining evidence for the existence of cyclic polymers was set up. Poly1 was prepared using 10 mol% DMAP at 80 °C for 8 h and an aliquot of the reaction mixture was removed and subjected to NMR and MALDI-TOF MS investigations. To the remaining part, propargylic alcohol was added and the reaction mixture was heated for another 8 h at 80 °C and subsequently analysed (for details see the ESI†). The results from MALDI-TOF mass spectrometry are shown in Fig. 2. In the spectrum of poly1 before propargylic alcohol addition, the heterotelechelic ethylene glycol vinyl sulfone terminated species (Fig. 2 series A1) or the isomeric cyclic structure (Fig. 2 series A2) was observed along with the 4-dimethylamino pyridinium/vinyl sulfone end-capped species (Fig. 2 series B). The latter species was not observed in the mass spectrum of poly1 after the reaction with propargyl alcohol. Instead a peak with a 56 Da higher mass was detected, which indicates the formation of the Michael-addition product of B with propargyl alcohol (Fig. 2 series D). In contrast, the peak characteristic for species A1 and A2 is still present after the reaction with propargyl alcohol. Only a very small signal for the propargyl alcohol modified A1 (Fig. 2 series C) could be observed. Moreover, in the 1H-NMR spectrum of the product of the reaction with propargyl alcohol signals characteristic for vinyl sulfone end-groups were not observed. Accordingly it is concluded that the major share of the A series can be regarded as the cyclic isomers A2.
 | ||
| Fig. 1 MALDI TOF-MS of poly1. | ||
 | ||
| Fig. 2 MALDI-TOF mass spectra (details) of poly1 before (blue) and after reaction with propargylic alcohol (black). | ||
The former findings also demonstrated the feasibility of end-group modification of the polymers. The synthesis of a polyethylene glycol 300 (PEG300, 5) DVS polyaddition product poly5 which was subsequently end-group functionalized with propargyl alcohol serves as another example for a complete vinyl sulfone end-group modification. Neither in the 1H-NMR nor in the MALDI-TOF mass spectrum vinyl sulfone terminated species after the reaction with propargyl alcohol could be observed (see the ESI†). The formation of cyclic isomers was not observed in this case. Thus, PEG300 could be oligomerised and subsequently functionalised with a triple bond in a one pot reaction.
Furthermore the possibility to conduct polyaddition reactions with two different diols was investigated. 1,4-Phenylenedimethanol (6) is a solid diol which is neither soluble in neat DVS nor in neat 1 at room temperature. Nevertheless when heating a mixture of 1 and 6 in a molar ratio of 5:1 and the according amount of DVS to 60 °C a solution formed which was cured upon addition of 5 mol% DMAP. As evidenced by NMR spectroscopy of the reaction mixture both diols, 1 and 6, were fully incorporated into the macromolecules yielding poly6 with an apparent n of 29 ± 2. GPC revealed a Mn value < 800 g mol−1.
In the next step, the trifunctional alcohols propane-1,2,3-triol (7, glycerol) or 2-ethyl-2-(hydroxymethyl)propane-1,3-diol (8) were employed to prepare duroplastic materials. Polyol 7 is liquid at room temperature and miscible with DVS. The formulation was composed of 1.5 equiv. of DVS and 1 equiv. of 7. Upon addition of DMAP (2, 5 or 10 mol% according to DVS) the reaction mixture became hot and its viscosity was immediately increased allowing for a pot life of approx. 20 sec. During this time the formulation was transferred to moulds and was then cured at 80 °C for 16 h yielding solid specimens of poly7. Solid polyol 8 is insoluble in DVS at room temperature but soluble at 60 °C. Accordingly 1 equiv. of 8 was dissolved in 1.5 equiv. DVS at 60 °C and subsequently DMAP (2, 5 or 10 mol% according to DVS) was added. The formulation was immediately transferred into moulds. In contrast to poly7, poly8 prepared with 2 mol% DVS yielded no proper specimens, while curing with 5 and 10 mol% DVS yielded specimens suitable for DMA measurements.
The resulting specimens of poly7 and poly8 were investigated by DMA, TGA and DSC. Results are presented in Table 1. Poly7 is characterized by a storage modulus (G′) in the range of 2–4 GPa, while the G′ of poly8 is between 1.8 and 1.9 GPa. The glass transition temperatures (Tg) as determined from the peak of the loss modulus were around 38 °C for poly7 and around 33 °C for poly8. While the G′ and Tg of poly8 are well reproducible, the same values show a rather broad distribution in the case of poly7 (Table 1 and the ESI†). The reason for that is not entirely clear yet. A possible explanation is an inhomogeneous distribution of DMAP due to solubility issues leading to less perfect test specimens. DSC gave Tgs around 30 °C for poly7 and 21 °C for poly8. TGA measurements revealed thermal stability under inert conditions up to about 300 °C (as revealed from the temperature for 5% mass loss, see the ESI†). Thermal stability decreased with an increasing amount of DMAP. Since DMAP was not removed from the specimens in the case of poly7 and poly8 and its boiling point is 195 °C (melting point: 109–111 °C) evaporation of DMAP might be an explanation for this finding. Furthermore, it cannot be ruled out that DMAP catalyses the retro-Michael reaction at such elevated temperatures. The samples of poly8 prepared with 5 and 10 mol% DMAP were further investigated by extraction with dichloromethane and acetone. The specimens slightly swelled in both solvents. After two times extraction with the respective solvent for 24 h at room temperature and subsequent removal of the solvent, 10 ± 2 wt% residue was obtained in all cases (see the ESI†). This residue contained DMAP and (oligomeric) poly8 according to 1H-NMR spectroscopy. The ratio DMAP:poly8 was approx. 1:2 in the case of the reaction with 5 mol% DMAP and 2:1 in the case of the reaction with 10 mol% DMAP. Neither unreacted 8 nor unreacted DVS could be observed in all cases. These results suggest that at least more than 90% of the monomers are incorporated in the insoluble part and that this share becomes higher with higher DMAP loading. Moreover, it is evident that only a small amount of DMAP is actually incorporated in the network.
| poly7 | poly8 | ||||
|---|---|---|---|---|---|
| DMAPb [mol%] | 2 | 5 | 10 | 5 | 10 |
| a Proper test specimens from the formulation cured by 2 mol% DMAP could not be prepared. b DMAP loading with respect to DVS. c Storage modulus. d Glass transition temperature (Tg) as determined from the peak of the loss modulus. e As determined from DSC measurements. f As determined from STA measurements. | |||||
| G′ [GPa]c | 3.7 ± 1.4 | 2.4 ± 0.6 | 3.3 ± 0.7 | 1.8 ± 0.1 | 1.9 ± 0.1 |
| T g from DMA [°C]d | 39 ± 1 | 40 ± 1 | 37 ± 2.5 | 33.5 ± 0.6 | 32.6 ± 0.3 |
| T g from DSC [°C]e | 32.0 | 27.3 | 30.6 | 21.5 | 20.5 |
| 5% mass loss [°C]f | 298 | 260 | 193 | 283 | 201 |
| 50% mass loss [°C]f | 364 | 369 | 354 | 368 | 364 |
Based on the studies of the related thiol-Michael reaction12 a general mechanistic rational is sketched in Scheme 2 on the left side. First DMAP undergoes a conjugate addition to DVS forming the zwitterionic species I, which is transformed upon protonation by the alcohol to the ion pair II. The latter step is supposed to be the rate determining step.13 The generated alkoxide then reacts with a vinyl-sulfone group forming the ion pair III. Finally, the catalytic cycle is closed by protonation of the carbanion in III by an alcohol generating ion pair II and the addition product. The proposed mechanism is supported by two findings. First, cations similar to that depicted in ion pairs II and III were detected by the MALDI-TOF MS (cf.Fig. 1, 2 and the ESI†) as well as by NMR (ESI†).§ Second, performing the reaction of DVS and 1 in CDCl3 using 100 mol% DMAP resulted in partial deuteration of the oxa-Michael product in the α-position to the sulfone group (ESI†), suggesting the formation of a strong base (e.g. the carbanion in ion pair III) capable of detracting deuterium from CDCl3.¶ In the same experiment, species featuring vinyl sulfone groups partially deuterated at the internal vinylic position were identified (ESI†), suggesting that the Michael addition of either DMAP or the alcohol is reversible under these particular conditions.
 | ||
| Scheme 2 Left, boxed in: general mechanistic rational; right: mechanistic explanation for the formation of cyclic products. | ||
To test for the reversibility under the conditions used for polymerisations, poly1 was dissolved in propargylic alcohol in the presence of DMAP. After stirring the reaction mixture for 3 days at 80 °C no reaction occurred. Subsequently, DVS was added to the reaction mixture. Now the di-adduct of propargylic alcohol and DVS formed. No evidence for a mixed (ethylene glycol/propargylic alcohol) adduct could be retrieved. In a similar experiment the di-adduct of propargylic alcohol and DVS9 was mixed with benzylic alcohol and DVS, and DMAP was added. After heating this reaction mixture for 16 h at 80 °C the di-adduct of benzylic alcohol almost quantitatively formed and the vast majority of the propargylic alcohol adduct remained unchanged. However a small amount of free propargylic alcohol was detected (see the ESI† for details). The amount of free propargylic alcohol did not change upon heating for further 6 d at 80 °C. Accordingly it can be assumed that the reversibility of ether formation is negligible under the reaction conditions used.
The formation of cyclic products deserves special attention. Scheme 2 on the right side summarizes a plausible mechanism accounting for the cyclisation. Based on previous studies a prearrangement of the Michael donor, acceptor and the nucleophile is necessary to achieve the initiation of the reaction.12,13 This prearrangement could involve an interaction of the pyridinium based cation with the lone pair of the alcohol, resulting in an increased acidity of the alcohol's proton (Scheme 2, right, I). Subsequently, the carbanion present in I is protonated leading to the ion pair II featuring the generated alkoxide in proximity to the pyridinium-cation. The attack of the alkoxide on the nearby Michael acceptor leads to a situation (III) similar to I; again the alcohol group stabilizes the cation and the carbanion leading to the protonation of the carbanion and the formation of the alkoxide/pyridinium zwitterion II′. This species further reacts with DVS leading to the zwitter-ion III′, which upon further protonation by an alcohol and attack of the generated alkoxide is converted into the zwitterion II′′. Repetition of these steps leads to propagation, and nucleophilic substitution of the pyridinium moiety upon intramolecular attack of the alkoxide leads to termination and the formation of the corresponding cyclic polyaddition product. Another termination reaction, i.e. the nucleophilic substitution of the pyridinium moiety by the carbanion present e.g. in III′, is also conceivable but seems to be of less importance.|| In order to obtain cyclic macromolecules, propagation must be fast relative to termination, i.e. cyclisation. This situation prevails in principle since DVS and the di- or polyol are completely consumed in the first minutes of the polyaddition reaction under the conditions used and small rings (e.g. 1,4,7-dioxathionane-7,7-dioxide) are only formed in negligible amounts. Upon longer reaction times and elevated temperatures the apparent degree of polymerisation determined by end-group analysis then increases (but not molecular masses determined by GPC) suggesting that cyclisation occurs predominantly in this latter step. This is not surprising since alkylated DMAP derivatives are known to be relatively poor leaving groups.14 What is surprising is that a high share of cyclic macromolecules forms although the reaction is performed in a solvent free manner, conditions counterintuitive for conducting macrocyclisation reactions.15 Likewise, the mechanistically related zwitterionic ring opening polymerisation of cyclic esters (ZROP) is typically performed in diluted solution.16
Conclusions
In summary, an oxa-Michael polyaddition reaction with divinyl sulfone as a Michael-acceptor and alcohols as Michael-donors has been described. The reaction proceeds via the nucleophile-mediated mechanism using 4-dimethylaminopyridine as the mediator under solvent-less conditions. Upon addition of DMAP to the formulation containing divinyl sulfone and the di- or multifunctional alcohol a fast and exothermic reaction is triggered which completely consumes both reactants within minutes. Upon prolonged curing predominantly macrocyclic products are formed with difunctional alcohols such as ethylene glycol or 1,4-butanediol. The use of trifunctional alcohols such as glycerol or 2-ethyl-2(hydroxymethyl)propane-1,3-diol leads to duroplastic polymers. Furthermore, endgroup functionalization of the vinyl sulfone terminus with monofunctional alcohols such as propargylic alcohol is feasible in a one-pot procedure.Acknowledgements
The authors thank David Fast and Karin Bartl for MS measurements, Josefine Hobisch for TGA and DSC measurements and Petra Kaschnitz for NMR measurements.Notes and references
- (a) W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS; (b) S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185–3200 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- (a) A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC; (b) D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- (a) B. H. Northrop, S. H. Frayne and U. Choudhary, Polym. Chem., 2015, 6, 3415–3430 RSC; (b) G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddelton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- (a) S. Chatani, D. P. Nair and C. N. Bowman, Polym. Chem., 2013, 4, 1048–1055 RSC; (b) J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS; (c) W. X. Xi, C. Wang, C. J. Kloxin and C. N. Bowman, ACS Macro Lett., 2012, 1, 811–814 CrossRef CAS.
- (a) I. Ugur, A. Marion, S. Parant, J. H. Jensen and G. Monard, J. Chem. Inf. Model., 2014, 54, 2200–2213 CrossRef CAS PubMed; (b) S. Takahashi, L. A. Cohen, H. K. Miller and E. G. Peake, J. Org. Chem., 1971, 36, 1205–1209 CrossRef CAS.
- C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988–999 RSC.
- (a) Y. Yu and Y. Chau, Biomacromolecules, 2012, 13, 937–942 CrossRef CAS PubMed; (b) S.-i. Matsuoka, S. Namera and M. Suzuki, Polym. Chem., 2015, 6, 294–301 RSC; (c) T. Saegusa, S. Kobayashi and Y. Kimura, Macromolecules, 1975, 8, 950–952 CrossRef CAS; (d) M. Gibas and A. Korytkowska-Walach, Polymer, 2003, 44, 3811–3816 CrossRef CAS.
- S. Strasser and C. Slugovc, Catal. Sci. Technol., 2015, 5, 5091–5094 CAS.
- A. C. Spivey and S. Arseniyadis, Angew. Chem., Int. Ed., 2004, 43, 5436–5441 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- (a) S. Chatani, D. P. Nair and C. N. Bowman, Polym. Chem., 2013, 4, 1048–1055 RSC; (b) J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS; (c) W. X. Xi, C. Wang, C. J. Kloxin and C. N. Bowman, ACS Macro Lett., 2012, 1, 811–814 CrossRef CAS; (d) C. Wang and C. Qi, Tetrahedron, 2013, 69, 5348–5354 CrossRef CAS.
- A. V. Salin, A. R. Faktkhutdinov, A. V. Il'in, V. I. Galkin and F. G. Shamsutdinova, Heteroat. Chem., 2014, 25, 205–216 CrossRef CAS.
- H. R. Kricheldorf, M. Garaleh and G. Schwarz, J. Macromol. Sci., Pure Appl. Chem., 2005, A42, 139–148 CrossRef CAS.
- (a) Z. Jia and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2085–2097 CrossRef CAS; (b) H. Kammiyada, M. Ouchi and M. Sawamoto, Polym. Chem., 2016, 7, 69911–66917 RSC.
- H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Comprehensive characterization data (NMR spectra, DSC and TGA measurements, MALDI-TOF mass spectra) and protocols for all experiments. See DOI: 10.1039/c7py00152e |
| ‡ In the case of solid alcohols the alcohol was dissolved in the corresponding amount of DVS, and DMAP was added as the last component. |
| § As evidenced by NMR-spectroscopy only a minor amount of DMAP participates in the reaction since 4-dimethylaminopyridinium species are, if at all, only detected in very small amounts. |
| ¶ The conjugated base CCl3− is then supposed to react with an alcohol to give the corresponding alkoxide and CHCl3. Indeed an increase of CHCl3 over the reaction time was verified by 1H-NMR spectroscopy. |
| || In some MALDI-TOF mass spectra peak-series with the according masses but low abundance were found, see the ESI.† |
| This journal is © The Royal Society of Chemistry 2017 |