
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2-Driven stereochemical inversion of sugars to create thymidine-based polycarbonates by ring-opening polymerisation†
Georgina L.
Gregory
a,
Elizabeth M.
Hierons
a,
Gabriele
Kociok-Köhn
a,
Ram I.
Sharma
b and
Antoine
Buchard
 *a
*a
aDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
bDepartment of Chemical Engineering, University of Bath, Bath BA2 7AY, UK
First published on 13th February 2017
Abstract
The development of biodegradable polymers from renewable resources is vital in addressing the dependence of plastics on petroleum-based feedstocks and growing ocean and landfill waste. Herein, both CO2 and natural sugar diols are utilised as abundant, safe and renewable building blocks for the synthesis of degradable and biocompatible aliphatic polycarbonates. Despite a strong potential for advanced polymer properties, inspired by Nature's supramolecular base-pairing, polycarbonates from the sugar components of DNA, 2′-deoxyribonucleosides have been limited by the inability of phosgene derivatives to form the cyclic carbonate monomers that would allow for controlled ring-opening polymerisation. CO2 insertion at 1 atm pressure into methylated thymidine 2′-deoxyribonucleoside, facilitated by organic base 1,8-diazabicyclo-[5.4.0]-undec-7-ene, affected an intramolecular SN2-like displacement of a tosyl leaving group to yield the cyclic carbonate by stereochemical inversion. Organocatalytic ring-opening polymerisation proceeded rapidly in solution resulting in high monomer conversions of 93% and number-average molecular weights, substantially greater and more controlled than via polycondensation routes. The thermodynamic parameters of the polymerisation (ΔHp = −12.3 ± 0.4 kJ mol−1 and ΔSp = −29 ± 1.1 J mol−1 K−1) were determined from the equilibrium monomer conversions over a temperature range of 0 to 80 °C and pseudo-first order kinetics demonstrated. The amorphous thymidine-based polycarbonates exhibited high glass transition temperatures of 156 °C and were found to be highly degradable to the constituent diol under basic aqueous conditions. Static water contact angle measurements and cell studies with MG-63 cell line indicated slightly hydrophilic and biocompatible materials, promising for tissue-engineering applications. The novel, CO2-driven approach to cyclic carbonate synthesis represents a means of expanding the scope of sugar-based monomers for tailored material properties derived from natural products.
Introduction
Reliance on classical petroleum-derived materials and their lack of biodegradability has instigated a drive towards sustainable bio-based polymers from renewable resources.1 With an ever-growing population, the demand for commodity and engineering plastics is steadily increasing: in 2014, 311 megatonnes of plastics were produced worldwide, a 52% increase since 2002.2 Readily available sugars are a promising inexpensive renewable alternative to fossil-based feedstocks, owing to their natural origin as well as wide structural and stereochemical diversity.3 As such synthetic carbohydrate-based materials can give properties comparable to industrial polymers as well as enhanced biodegradability and biocompatibility characteristics, and have the potential to mimic biological materials and functions.4 For example, the sugar components of DNA, 2′-deoxyribonucleosides are attractive and versatile building blocks, owing to the complementary hydrogen bonding between nucleobase pairs, and have emerged as powerful tools in several fields.5 Furthermore, the metal coordination chemistry of pyrimidine nucleobases such as thymine (T) opens up the potential for unique polymer applications in metal detection and toxic metal removal.6 Subsequently, extensive effort has gone into the integration of nucleobases into the backbone of synthetic polymers7 or as pendent or terminal groups,8 either through direct polymerisation or post-synthetic modification.5b,g,9 Currently, there are several examples, albeit mainly oligomers, of 2′-deoxyribonucleosides bearing carbonate,10 carbamate,11 ester,12 triazole,13 ether14 and silyl ether linkages.15 CO2 is another abundant, safe and renewable building block and its incorporation into polycarbonates constitutes an important research endeavor within the field of CO2 utilisation.16 Moreover, the biodegradability and biocompatibility of aliphatic polycarbonates (APCs) make them a rapidly developing class of polymers for biomedical applications such as tissue engineering scaffolds and drug-delivery carriers.17 Thus, the construction of functionalised sugar-based APCs in the present work was motivated by the use of these two naturally occurring feedstocks and the creation of novel polymers with desirable properties.Polycarbonates have been widely prepared by traditional polycondensation with phosgene, phosgene derivatives or dialkyl carbonates.18 This however overlooks the direct utilisation of CO2, involves hazardous reagents and often presents challenges in molecular weight control. The copolymerisation of epoxides with CO2 has been extensively studied as a greener alternative but can be limited by the substrate scope particularly with regards to carbohydrate feedstocks.19 Enabling larger and well-defined number average molecular weights (Mn) with narrow dispersities (Đ), the controlled ring-opening polymerisation (ROP) of typically 6-membered cyclic carbonates has emerged as an attractive choice in polycarbonate production and often proceeds under mild reaction conditions.20 Both for polymer synthesis and various other applications (e.g. battery electrolytes),21 a growing research effort has focused on the improved synthesis of cyclic carbonates that avoids the use of phosgene or phosgene derivatives and directly incorporates CO2 (ideally at ambient pressure and temperature).22
Currently, sugar-based cyclic carbonates for ROP are limited to those derived from D-glucose,23D-mannose24 and D-xylose25 (Fig. 1). Cyclocarbonation of trans-configured 1,3-diols in 5-membered furanose sugars such as DNA 2′-deoxyribonucleosides has never been achieved to the best of our knowledge. In 1972, Tittensor and Mellish26 reported the attempted synthesis of the 3′,5′-cyclic carbonate of thymidine and suggested the need for an active ester at the 3′-position rather than 5′-hydroxyl group for cyclisation to occur. Subsequently in 2011, Suzuki et al.10b selectively placed a carbonyldimidazole (CDI) component at the 3′-position of thymidine and adenosine nucleosides. However, only polycondensation resulted, leading to thymidine-based polycarbonates of low Mn (4700 g mol−1, Đ 1.86).
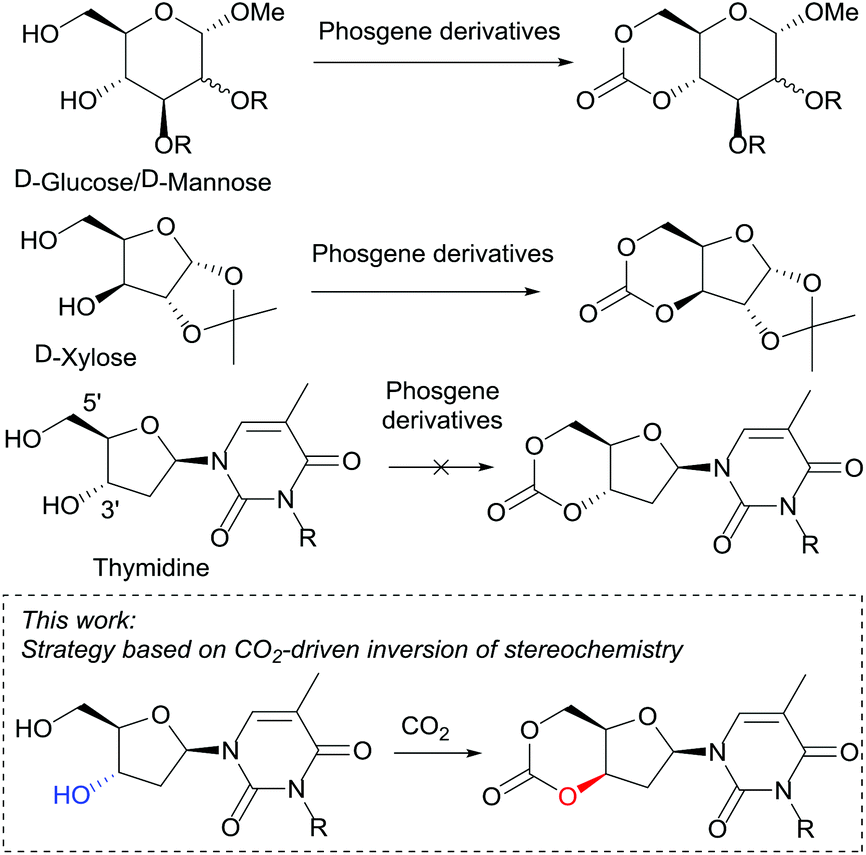 | ||
| Fig. 1 Unlike D-mannose, D-glucose and D-xylose derived monomers, the cyclic carbonate of thymidine cannot be prepared by traditional routes. | ||
Herein, the synthesis and successful ROP of a thymidine-based cyclic carbonate incorporating CO2 at 1 atm pressure is reported. Motivated by the work of Jessop et al.,27 using 1,8-diazabicyclo-[5.4.0]-undec-7-ene (DBU) reagent to capture CO2 into alcohols, we employ the resulting carbonate as a nucleophile to affect cyclisation via an intramolecular SN2-type reaction. We present DFT calculations that rationalise the monomer synthesis and its ability to be polymerised. The thermodynamic and kinetic parameters of the polymerisation are reported, as well as the resulting new polymer microstructure and thermal properties. A preliminary tissue engineering study is finally presented.
Results and discussion
Monomer synthesis
Synthesis of the trans-3′,5′-cyclic carbonate of thymidine using our previously reported method24 (tosylation of a DBU-introduced carbonate and cyclisation via an intramolecular nucleophilic addition-elimination mechanism, leading to stereochemical retention), proved unsuccessful (trans-1, Fig. 2).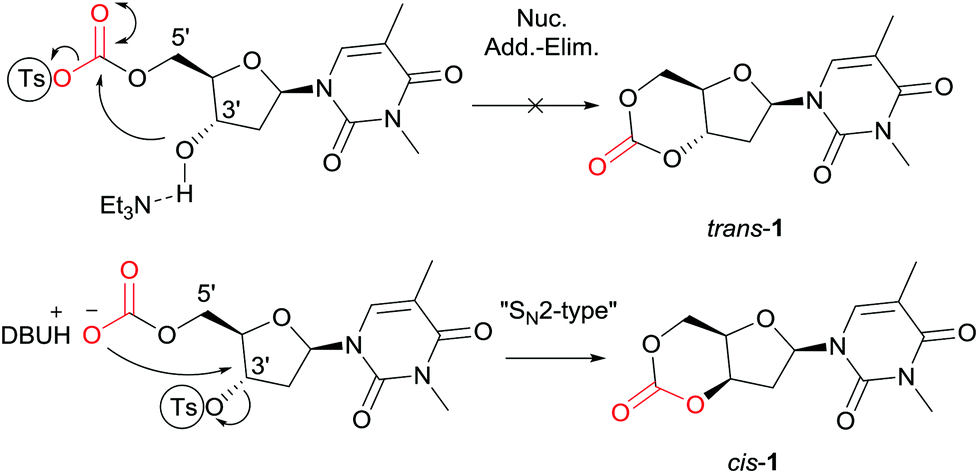 | ||
| Fig. 2 Synthesis of cis-1 by stereochemical inversion at the 3′-position. | ||
However, DFT calculations28 of the ring-closing kinetics (transition state ΔΔG‡ = +13.3 kcal mol−1) and thermodynamic driving force (ΔΔG = −20.8 kcal mol−1) suggested that this pathway should be accessible at ambient reaction conditions (Scheme S2†). Calculations also indicated that the ring-strain of the hypothetical monomer, consisting of a 6-membered cyclic carbonate trans-fused to a furanose sugar ring, was much higher compared to when fused to the 6-membered pyranose ring in reported D-mannose and D-glucose based monomers (Schemes S4 and S5†). For example, ring-opening with methanol is thermodynamically favored with ΔΔG = −6.7 kcal mol−1 compared to −0.9 and −0.2 kcal mol−1 for the mannose and glucose monomers, respectively. The calculated enthalpy of isodesmic ring-opening with dimethyl carbonate (ΔΔH) is also 5.5 kcal mol−1 more favorable for the trans-fused furanose monomer compared to the trans-fused D-mannopyranose cyclic carbonate. The pyranose ring can adopt a chair conformation that can accommodate a trans-fused ring whereas a furanose-cored monomer may be too highly strained to be isolated (especially in the presence of alcohol). The cis-3′,5′-thymidine analogue (cis-1, Fig. 2) however, lies 9.2 kcal mol−1 lower in energy compared to the trans- and shows a similar isodesmic ring-strain and in fact greater thermodynamic driving force for ring-opening compared to the D-xylofuranose derived monomer prepared by Gross and co-workers.25 In that report, cyclocarbonation with ethylchloroformate of the already cis-3,5-diol in D-xylose yielded a cis-fused cyclic carbonate that successfully underwent ROP. Guided by this insight and further DFT calculations, suggesting favorable cyclisation kinetics and thermodynamics (Scheme S3†), we endeavored to invert the sugar stereochemistry of thymidine and prepare the cis-cyclic carbonate by nucleophilic displacement of a tosyl leaving group with a CO2-generated carbonate.
As shown in Scheme 1, selective tosylation of the 3′-hydroxyl position in thymidine was achieved in good yield by a sequential one-pot silylation followed by tosylation reaction. Subsequent deprotection of the 5′-silyl protecting group was carried out under mild reaction conditions with I2 in methanol following the procedure outlined by Vaino and Szarek.29 Unfortunately, protection of the 3-N position of the nucleobase was found to be necessary to avoid undesired side reactions during the cyclisation step and potentially during the ROP. Bases such as DBU are known to lead to formation of 2,3′-anhydrothymidine by intramolecular ring-closing of the nucleobase with 3′-leaving groups.30 As an initial study and proof of concept of the monomer synthesis and ROP potential, the position was methylated in a simple, high yielding step. 3-N-Methyl-3′-tosyl-thymidine 2 was then reacted with DBU under 1 atm CO2 pressure to form a transient amidinium alkyl 5′-carbonate salt, which underwent intramolecular nucleophilic cyclisation with 71% conversion. Subsequent isolation by column chromatography and purification by recrystallisation from toluene gave monomer cis-1 (now referred to as 1 for simplicity), with polymerisation-grade purity, in reasonable yield of 52% (cf. 41% for D-xylose, 25–36% for glucose and 57% for mannose-based cyclic carbonates).
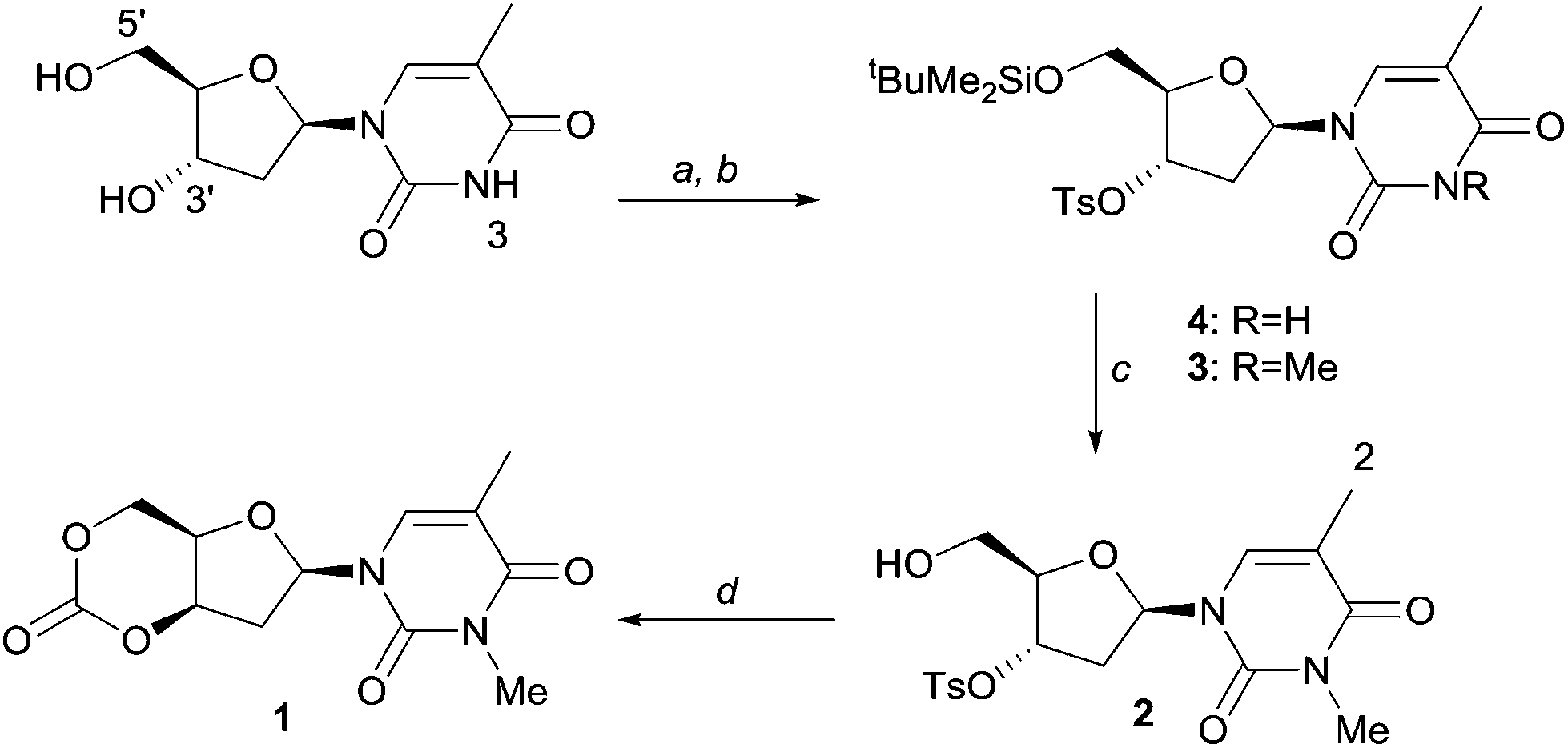 | ||
| Scheme 1 Synthesis of 1 (a) i: tBuMe2SiCl, 4-dimethylaminopyridine (DMAP), pyridine, rt, 12 h; ii: TsCl, rt, 24 h, 84%; (b) MeI, K2CO3, acetone, 12 h, 93%; (c) 1 wt% I2 in MeOH, reflux, 2 h, 95%; (d) DBU, CO2 (1 atm), MeCN, 0–40 °C, 24 h, 52%. | ||
The strategy represents a generally applicable method to cyclic carbonate synthesis from natural furanose sugars bearing a trans-1,3-diol. It extends the range of sugar-based cyclic carbonate monomers beyond those possible with known sugars and classical reagents such as phosgene and its derivatives, which require a nucleophilic addition-elimination mechanism. Although the need to protect the 3-N position of the nucleobase is somewhat limiting, the functionalisation potential of the pyrimidine arm is tremendous, ranging from Re(CO)3 complexes31 to alkyne functionalities for click chemistry.5c,32
As a further demonstration, we also prepared the 3-N-benzoyl derivative of 1 (1-Bz). Benzoylation could be carried out in tandem with the silylation and tosylation, reducing the number of overall synthetic steps (Scheme S1 and Fig. S12–S14†). Literature precedence has furthermore shown post-polymerisation removal of benzoyl protecting groups.10b
The structure of 1 was confirmed by 1H NMR (Fig. 6), 13C{1H} NMR and FTIR spectroscopies as well as electrospray ionisation mass spectrometry (ESI-MS), elemental analysis and single crystal X-ray diffraction. Full NMR assignment was aided by DEPT135, HSQC and COSY experiments (Fig. S1–S5†). In the 13C NMR spectrum, further to the carbonyl environments of the nucleobase, an additional C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O resonance was observed at 147 ppm, typical of a 6-membered cyclic carbonate, and a vicinal 3J3′–4′ coupling constant in the 1H NMR spectrum of just 3.9 Hz was consistent with a cis-configuration. X-ray analysis of crystals of 1 grown from toluene (Fig. 3) further corroborated stereochemical inversion at the 3′-carbon to form the 3′,5′-cis-cyclic carbonate in which the furanose ring adopts a strained 4′-endo-3′-exo twist (4T3) conformation. This is likely retained in solution as the ring-methylene H-2′ protons display very different chemical shifts and J-coupling constants consistent with the Karplus equation. In contrast the unconstrained furanose ring in the preceding trans-3′,5′-diol 2 adopts a 2-endo (2E) envelope conformation (see crystal structure in ESI†).
O resonance was observed at 147 ppm, typical of a 6-membered cyclic carbonate, and a vicinal 3J3′–4′ coupling constant in the 1H NMR spectrum of just 3.9 Hz was consistent with a cis-configuration. X-ray analysis of crystals of 1 grown from toluene (Fig. 3) further corroborated stereochemical inversion at the 3′-carbon to form the 3′,5′-cis-cyclic carbonate in which the furanose ring adopts a strained 4′-endo-3′-exo twist (4T3) conformation. This is likely retained in solution as the ring-methylene H-2′ protons display very different chemical shifts and J-coupling constants consistent with the Karplus equation. In contrast the unconstrained furanose ring in the preceding trans-3′,5′-diol 2 adopts a 2-endo (2E) envelope conformation (see crystal structure in ESI†).
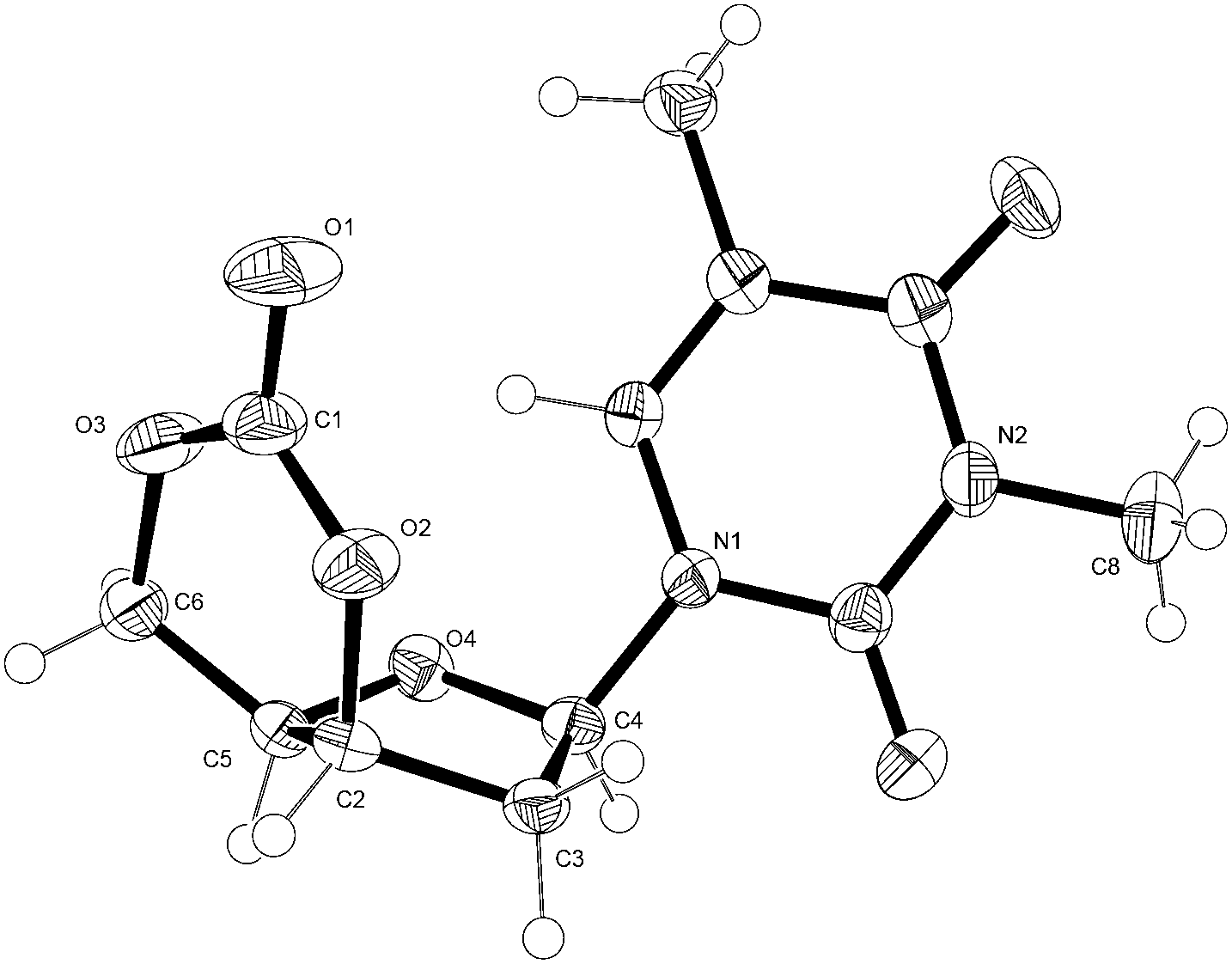 | ||
| Fig. 3 ORTEP33 drawing of the crystal structure of 1 with thermal ellipsoids at the 50% probability level (see ESI†). Selected bond lengths (Å) and dihedral angles (°): C(2)–O(2) 1.466 (2), O(2)–C(1) 1.326 (3), O(1)–C(1) 1.202 (3), C(1)–O(3) 1.326 (4), O(3)–C(6) 1.456 (3); C(4)–C(3)–C(2)–C(5) −29.61 (19), C(3)–C(2)–C(5)–O(4) −36.87 (19), C(2)–C(5)–O(4)–C(4) 30.01 (2), C(5)–O(4)–C(4)–C(3) 10.5 (2), O(4)–C(4)–C(3)–C(2) 12.6 (2). | ||
Ring-opening polymerisation (ROP)
The ROP of 1 was initially explored with bifunctional organocatalyst, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 4-methyl benzyl alcohol initiator. Polymerisation proceeded rapidly at 25 °C in CH2Cl2, reaching a plateau at 80% monomer conversion (determined by 1H NMR spectroscopy) after 1 h for a monomer-to-initiator feed ratio of 100 and 1 mol L−1 initial monomer concentration, [M]0. Further addition of monomer after 24 h led to establishment of a new equilibrium monomer conversion indicating no catalyst deactivation and a concentration dependent equilibrium polymerisation. A higher monomer conversion of 90% was achieved at a higher [M]0 of 2.5 mol L−1 (Fig. 4).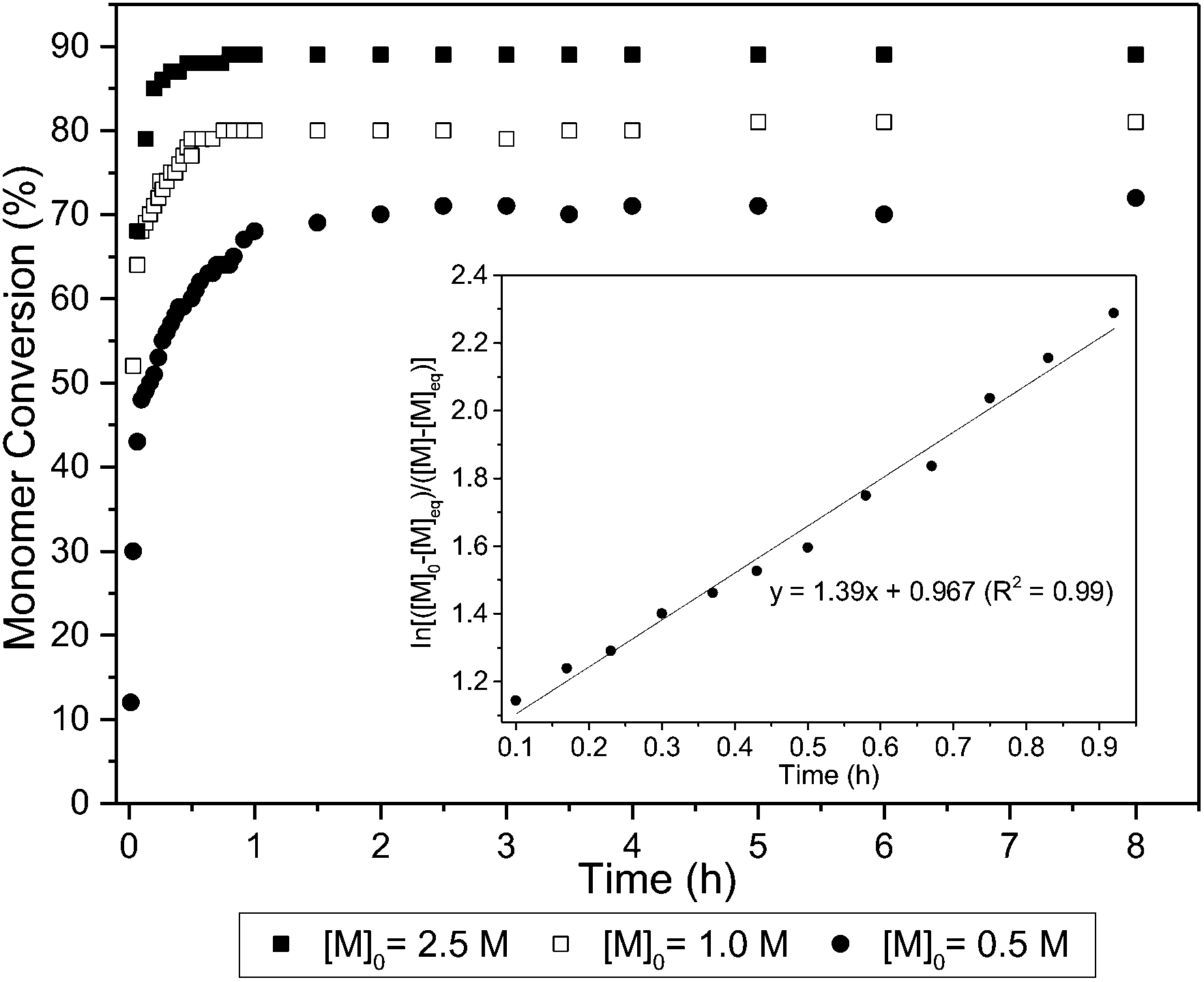 | ||
Fig. 4 ROP of 1 with 4-MeBnOH and TBD catalyst: conversion versus time for different [M]0. Carried out at 25 °C in CH2Cl2 for [M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TBD]0:[4-MeBnOH]0 ratio of 100:1:1. Monomer conversion was determined by 1H NMR spectroscopy by relative integration of the anomeric H-1′ proton. (inset) Kinetic plot carried out under the same conditions for [M]0 = 0.5 mol L−1. :[TBD]0:[4-MeBnOH]0 ratio of 100:1:1. Monomer conversion was determined by 1H NMR spectroscopy by relative integration of the anomeric H-1′ proton. (inset) Kinetic plot carried out under the same conditions for [M]0 = 0.5 mol L−1. | ||
A linear kinetic plot is consistent with pseudo-first order kinetics, typical for ROP, from which a kapp of 1.39 ± 0.005 h−1 was determined (inset Fig. 4 and S15†). The high melting point of the monomer (204–205 °C) and low thermal stability (178–205 °C, 88% mass loss, Tinf 248 °C) prevented polymerisations from being carried out in the melt. The temperature dependence of the equilibrium polymerization was also investigated at fixed [M]0, catalyst loading and initiator concentration in 1,2-dichloroethane solvent to access a wider temperature range of 0–80 °C. A plot of ln[M]eqversus the reciprocal of the absolute temperature gave an estimate of the polymerisation thermodynamic parameters (Fig. 5): ΔHp = −12.3 ± 0.4 kJ mol−1 and ΔSp = −29 ± 1.1 J mol−1 K−1. For comparison, Endo and co-workers34 reported an enthalpic driving force of −26.4 kJ mol−1 for the anionic polymerisation in THF of unsubstituted trimethylene carbonate and accompanying −44.8 J mol−1 K−1 entropy decrease ([M]0 = 1 mol L−1). A decrease in ΔHp with increased steric bulk of substituted derivatives was attributed to distortion of the polymer backbone as opposed to a conformational effect on the monomer. Interestingly, for polymer recyclability, the polymerisation mixture at 0 °C, which showed a 72% conversion to polymer at equilibrium, resulted in near quantitative recovery of the monomer upon heating to 80 °C.
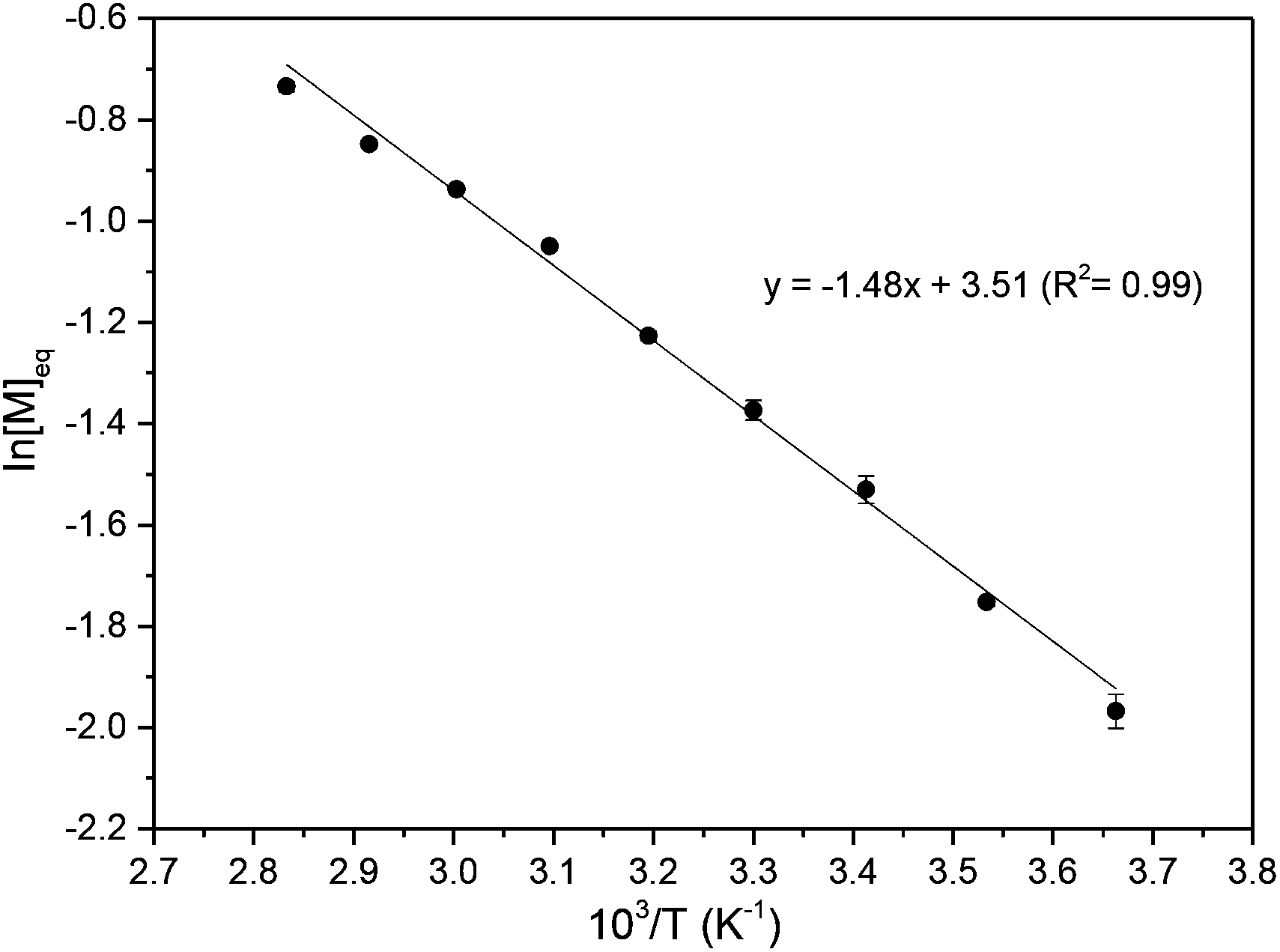 | ||
| Fig. 5 ROP of 1 with 4-MeBnOH and TBD catalyst: calculation of the thermodynamic parameters from a plot of ln[M]eq as a function of 1/T, where T is the absolute temperature. Carried out in 1,2-dichloroethane, over a temperature range of 0–80 °C for [M]0 = 0.5 mol L−1 and [M]0:[TBD]0:[4-MeBnOH]0 = 100:1:1. Polymerisations were repeated three times at each temperature and an average taken. | ||
A linear increase in Mn with percentage monomer conversion whilst maintaining narrow dispersities indicated that the polymerisation proceeded in a controlled fashion up to 60% monomer conversion (Fig. 6). Initially excellent agreement was observed between the calculated Mn and those estimated by size exclusion chromatography (SEC) versus polystyrene standards with CHCl3 eluent. For example, the polymer at 54% monomer conversion had a Mn of 7210 g mol−1 (Đ 1.12) as estimated by SEC (Fig. S17†), only slightly lower than the predicted35 7740 g mol−1. Once the thermodynamic equilibrium was reached and with longer reaction times, a broadening of the polymer distribution was observed due to transesterification reactions.
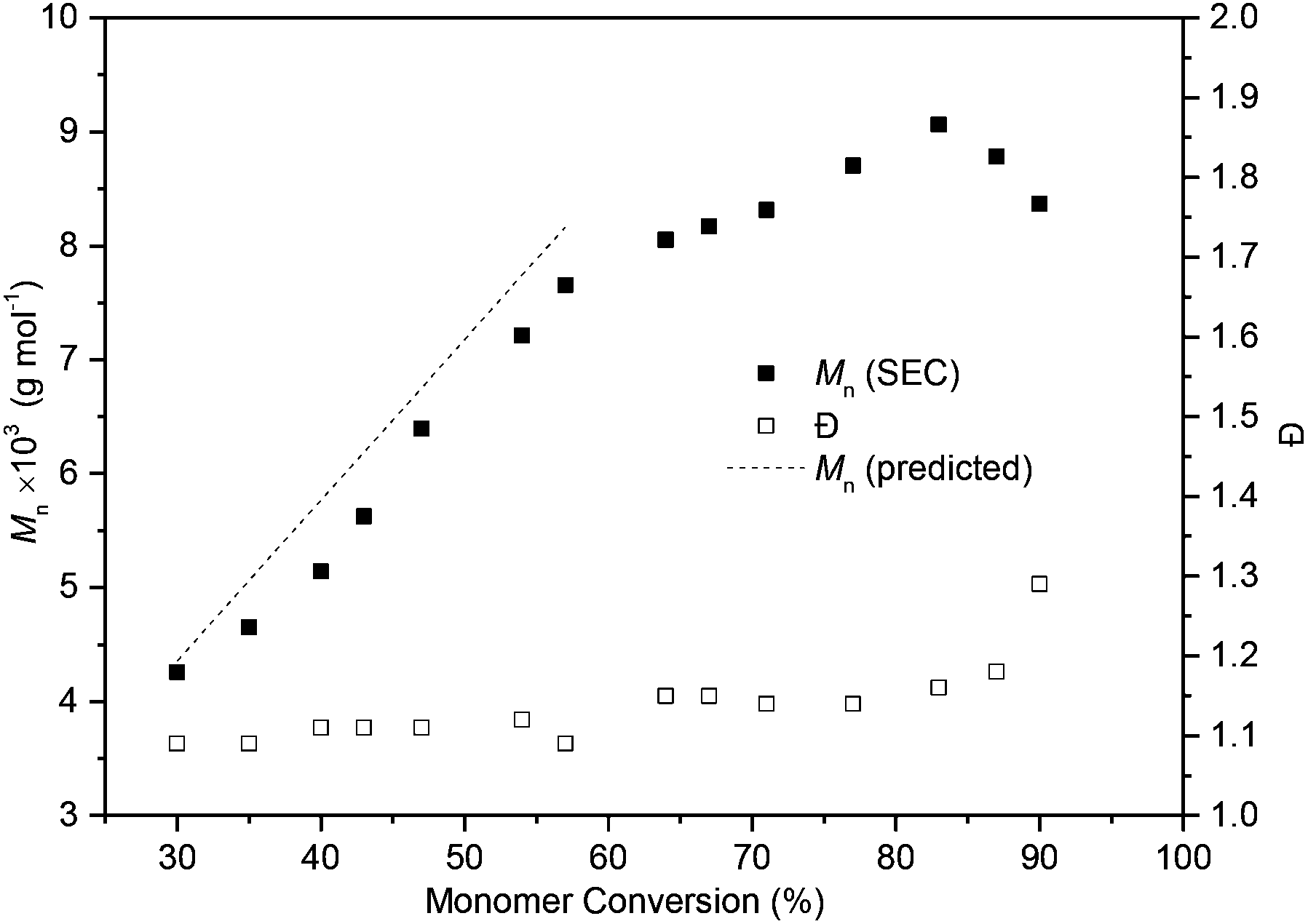 | ||
| Fig. 6 Plot of Mn and Đ versus monomer conversion for a polymerisation carried out at 0 °C with [M]0 = 2.5 mol L−1 and 50:1:1 [M]0:[TBD]0:[I]0. Conversion was determined by 1H NMR spectroscopy of aliquots quenched with excess benzoic acid. Mn and Đ were estimated by SEC versus polystyrene standards using a refractive index (RI) detector and CHCl3 eluent. | ||
Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-ToF) mass spectrometry of this polymer showed a major linear polymer series of Mn 7120 g mol−1 and expected 4-methyl benzyl alcohol and O–H end groups (Fig. 7 and S19†). A minor (19%) cyclic species with m/z consistent with no end-groups and an integer number of repeat units (m/z ∼282.25), was also observed due to backbiting of the polymer chain. As the polymer chain grows and monomer concentration decreases, a greater rate of chain backbiting relative to chain propagation may account for deviations observed between the calculated and SEC estimated Mn at higher monomer conversions. Dilution of the reaction conditions to 0.5 mol L−1 in an attempt to favour macrocycle formation resulted in lower Mn (Table 1, entry 1) and in more cyclic species observed by MALDI-ToF analysis (Fig. S21†). The lack of chain end in macrocycles should prevent reversion of the polymer to the monomer at elevated temperatures, however, difficulty in separating the cyclic and linear species prevented us from rigorously investigating the issue. Polymeric macrocycles are also interesting in their own right, including those derived from nucleosides, e.g. as supramolecular complexes with metal coordination characteristics.12
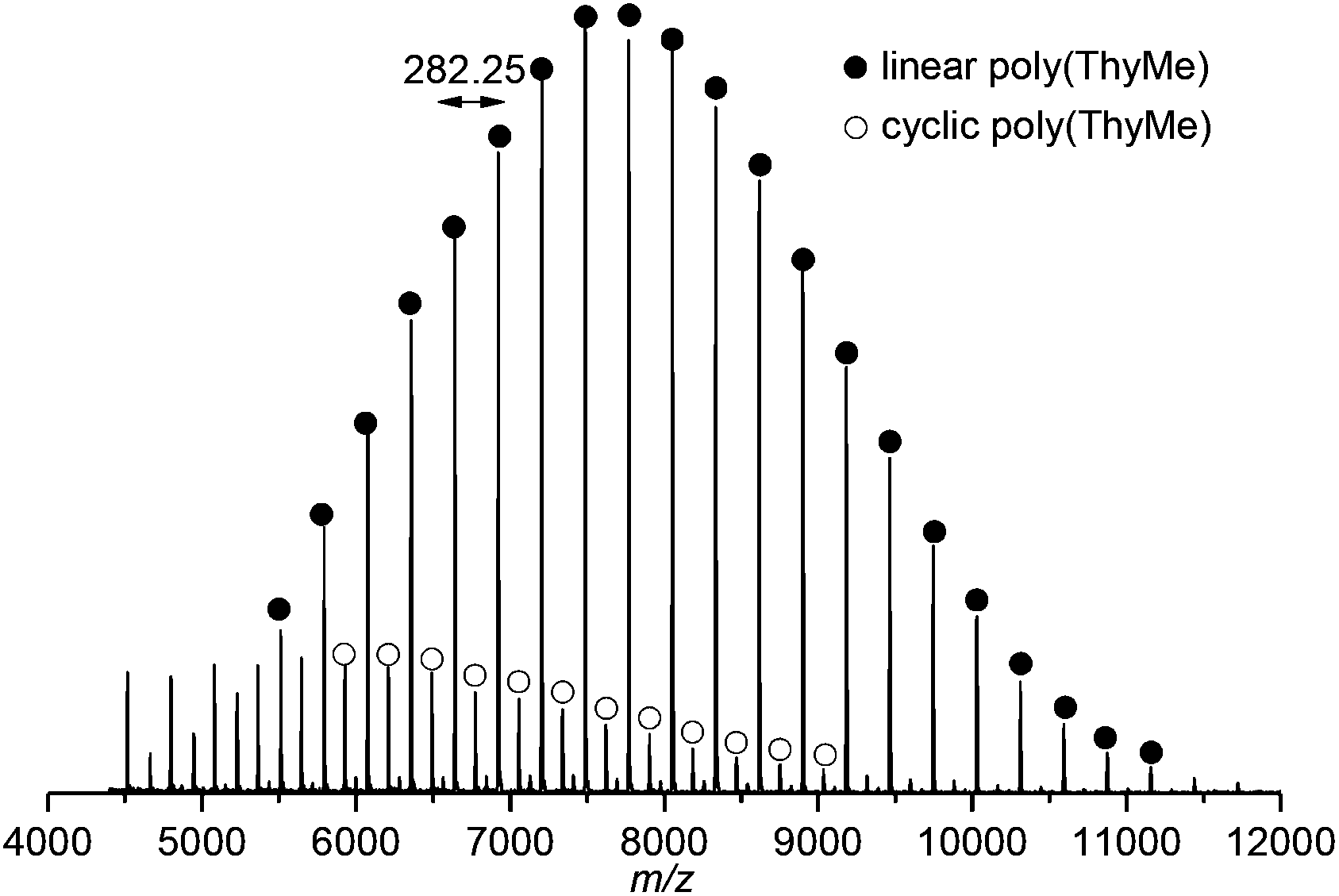 | ||
| Fig. 7 MALDI-ToF mass spectrum of polymer (Mn,SEC 7210 g mol−1, Đ 1.12) showing major linear and minor cyclic polymeric species (distribution assigned based on molecular masses and corresponding formulas, see Fig. S19† for details). | ||
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Temp. (°C) | Cat. | [M]0:[C]0:[I]0 |
Conv.b (%) | Time (h) |
M
nc [Đ] |
| a All polymerisations were carried out at [M]0 = 2.5 mol L−1 in CH2Cl2 unless stated otherwise. b Determined by 1H NMR spectroscopy in CDCl3. c g mol−1, calculated by SEC with CHCl3 eluent versus polystyrene standards (RI detector). d [M]0 = 0.25 mol L−1. e L1 = 2,4-di-tert-butyl-6-{[(2′-dimethylaminoethyl)methylamino]-methyl}phenolate. f BDI-1 = 2-((2,6-diisopropylphenyl)amido)-4-((2,6-diisopropylphenyl)-imino)-2-pentene. | ||||||
| 1d | 0 | TBD | 100:1:1 |
68 | 4 | 4260 [1.09] |
| 2 | 0 | TBD | 150:1:1 |
90 | 3 | 17000 [1.30] |
| 3e | 25 | L1ZnOEt | 100:1 |
71 | 20 | 7250 [1.22] |
| 4f | 25 | (BDI-1) | ||||
| ZnEt | 100:1:1 |
60 | 24 | 5240 [1.15] | ||
| 5 | 25 | TBD | 100:1:1 |
90 | 3 | 11000 [1.27] |
| 6 | 0 | TBD | 100:1:1 |
93 | 3 | 15400 [1.28] |
The largest molecular weights, up to 17000 g mol−1 (Đ 1.3) were achieved at 0 °C and [M]0 of 2.5 mol L−1 (Table 1, entry 2). A greater disparity between the theoretical Mn and those determined by SEC was observed at higher [M]0/[I]0 ratios, most likely due to chain back-biting reactions. To explore the influence of the catalytic system on the control of Mn, metal complexes were tested for the ROP of 1. Sn(Oct)2 catalyst with 4-MeBnOH initiator at 120 °C in toluene did not yield homopolymer. Y(OiPr)3 or Al(OTf)3 with BnOH initiator resulted in no control with only low Mn (1800–3910 g mol−1, Đ 1.35–1.39) in DCM, dioxane and toluene solvents at 0, 25, 70 and 120 °C. Although highly active for lactide ROP in CH2Cl2 at rt, the zinc(II) alkoxide catalyst (L1ZnOEt) reported by Williams et al.36 was slower compared to TBD for the ROP of 1, as was the (BDI-1)ZnEt catalyst37 with 4-MeBnOH initiator (Table 1, entries 3 and 4).
Polycarbonate properties
Initial insight into the structure of the thymidine-based polycarbonates was gained by NMR spectroscopy. Shown in Fig. 8, coalescence of the H-5′ and H-4′ proton environments in the 1H NMR spectrum of the polymer compared to the monomer was consistent with loss of constraint upon ring-opening of the cyclic carbonate moiety. In addition, retention of the distinct chemical environments of the furanose ring 2′-methylene protons suggest that the sugar may adopt a similar puckered conformation in the polymer backbone to that observed in the monomer. 13C{1H} NMR analysis revealed three carbonate environments centered at 154.4, 153.6 and 152.8 ppm, consistent with tail–tail, head–tail and head–head linkages that would arise from the cleavage at either side of the asymmetric carbonate by either a free secondary or primary propagating alcohol chain. Relative assignments were suggested based on previous work,23a,24,25 and by the 1:2:1 integration ratio observed, which further suggested a regiorandom propagation of the chain (Fig. S9†). Further splitting observed within these three distinct environments is attributed to long range sequence effects as also reported for regiorandom glucose23a and xylose25 derived polycarbonates. DFT calculations28 of the initiation step in the ROP of 1 with 4-MeBnOH and TBD catalyst support this lack of preference for ring-opening to expose either a free primary (ΔΔG = −5.6 kcal mol−1) or secondary hydroxyl group (ΔΔG = −6.6 kcal mol−1) for chain propagation (Fig. S38†).
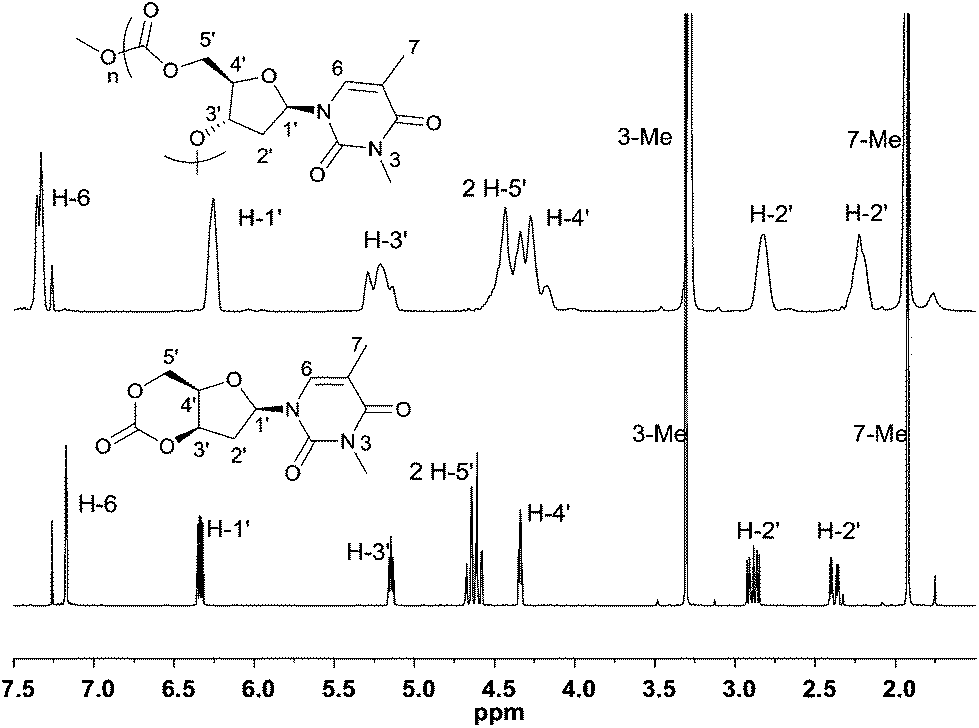 | ||
| Fig. 8 Comparison of the 1H NMR spectra (400 MHz, CDCl3) of monomer 1 (bottom) and its polymer (top, Table 1, entry 6). | ||
The thermal properties of the polycarbonates were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). The onset of thermal degradation occurred at relatively low temperatures of ∼170 °C, reaching a maximum rate of mass loss (Tinf) around 246 °C for polymers of Mn 11000 and 15400 g mol−1 (Table 1, entries 5 and 6). High mass losses of 91 and 94%, respectively were achieved by ∼300 °C (Fig. S22†). Analysis of the gases evolved by tandem mass spectrometry confirmed the presence of m/z 44 attributed to the loss of CO2+ (Fig. S24†). The polymers exhibited high glass transition temperatures (Fig. S25–S27†); a Tg of 156 °C was measured for polycarbonate of Mn 15400 g mol−1 and is attributed to restricted rotation about the furanose ring backbone and to the bulky thymine group. For comparison, furanose-cored and isopropylidene-protected D-xylose polycarbonate exhibited a Tg of 128 °C (for Mn = 13200 g mol−1).25 Pyranose backboned polycarbonates derived from isopropylidene-protected D-mannose and methyl protected D-glucose showed Tg of 152 °C (13600 g mol−1)23 and 122 °C (14700 g mol−1),24 respectively. No transitions associated with a melting domain were observed implying an amorphous polymer, which was further confirmed by powder X-ray diffraction (Fig. S28†).
The hydrolytic stability of the water insoluble polymer was investigated at 0.25 wt% at rt in water, 1 mol L−1 HCl and 1 mol L−1 NaOH, as well as in PBS buffer at 37 °C in presence of Candida antartica lipase B. After 1 week, no dissolution or loss of mass by SEC was observed in the case of water and 1 mol L−1 HCl. NMR analysis of the supernatant in D2O revealed no environments. Under the alkali conditions, the polymer was observed to dissolve after 4 hours. 1H NMR spectroscopy in D2O after 24 h at 4 wt% revealed proton environments consistent with the cis-diol of 3-N-methyl thymidine (Fig. S29 and S30†). Mass spectrometry (HR-MS ESI) after 24 h further confirmed the presence of the diol suggesting complete hydrolytic degradation of the polymer to its constitute components. In the case of lipase B, unknown degradation products were detected in solution after one week. Static water contact angle measurements of polymer thin films drop cast onto glass slides revealed an average contact angle of 48 ± 1°. This was more hydrophobic than the monomer but less hydrophilic compared to the glass control (Fig. S32†).
Cell attachment studies were carried out with a human osteoblast cancer cell line, MG-63, on polymer films drop cast onto glass slides. Cell attachment after a 1 h incubation period (37 °C, 5% CO2) was comparable to the empty tissue culture polystyrene well-plate and glass controls, and indicated the possibility of thymidine-based materials to support cells (Fig. S33–S35†). After a longer 24 h incubation period, cell attachment dropped compared to the empty well-plate control suggesting further modifications to the polymer surface are required for it to serve as an effective tissue engineering scaffold. Nevertheless, the choice of the 3-N protecting group and general applicability of the strategy present wide-scope and future opportunities for novel biomaterials with applications in the tissue engineering and regenerative medicine fields. Other degradable polymers attractive in this area, such as PLA and PLGA, are notoriously difficult to functionalise.38 Therefore, the successful preliminary ROP of benzoyl-protected monomer (1-Bz, see ESI†) offers promise for more elaborate functionality as well as post-polymerisation modifications, including self-assembly with base-paired nucleoside polycarbonates.
Conclusions
In summary, a novel strategy for the synthesis of challenging sugar-based cyclic carbonate monomers is presented that utilises CO2 as a C1 resource at 1 atm pressure. Sugar-based resources previously inaccessible for ROP such as 2′-deoxyribonucleosides can thus be efficiently converted to cyclic carbonate monomer. Here, novel polycarbonates derived from thymidine are reported. DBU-facilitated CO2 insertion into the 5′-hydroxyl group of 3-N-protected thymidine 2′-deoxyribonucleoside sugar introduces a carbonate nucleophile to affect an intramolecular displacement of a 3′-tosyl leaving group, resulting in stereochemical inversion. The isolated cis-configured 3′,5′-cyclic carbonate undergoes ROP under mild reaction conditions with organocatalyst TBD and 4-MeBnOH alcohol initiator to yield novel thymidine-based polycarbonates. The kinetic and thermodynamic parameters of the equilibrium polymerisation were established and MALDI-ToF MS revealed thymidine-based polycarbonates with both linear and cyclic topologies. Thermal analysis revealed high glass transition temperatures and a low onset of thermal degradation. Static water contact angle measurements were carried out and cell attachment studies with MG-63 cell line indicated promising biocompatible materials. Work is ongoing to explore the functionalisation of the 3-N position of the thymine nucleobase to impart specific properties for targeted applications.Acknowledgements
The EPSRC (EP/N022793/1; EP/L016354/1/CDT in Sustainable Chemical Technologies, studentship to GLG), Roger and Sue Whorrod (fellowship to AB) and the Royal Society (RG/150538) are acknowledged for research funding. We thank the University of Bath HPC for computing resources.Notes and references
- (a) A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2015, 116, 1637–1669 CrossRef PubMed; (b) C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS; (c) A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS PubMed; (d) A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS; (e) A. Pellis, E. Herrero Acero, L. Gardossi, V. Ferrario and G. M. Guebitz, Polym. Int., 2016, 65, 861–871 CrossRef CAS; (f) M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC; (g) T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 2–8 CrossRef PubMed.
- http://www.plasticseurope.org/information-centre/publications.aspx , (accessed 08/12/2016).
- (a) J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2015, 116, 1600–1636 CrossRef PubMed; (b) J. A. Galbis and M. G. García-Martín, Top. Curr. Chem., 2010, 295, 147–176 CrossRef CAS PubMed; (c) J. A. Galbis and M. G. García-Martín, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Oxford, 2008, pp. 89–114 Search PubMed; (d) Biobased Monomers, Polymers, and Materials, ed. P. B. Smith and R. A. Gross, American Chemical Society, 2012 Search PubMed; (e) Y. Tokiwa and M. Kitagawa, in Polymer Biocatalysis and Biomaterials II, American Chemical Society, 2008, vol. 999, pp. 379–410 Search PubMed; (f) G. L. Gregory, E. M. Lopez-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- (a) A. S. Carlini, L. Adamiak and N. C. Gianneschi, Macromolecules, 2016, 49, 4379–4394 CrossRef CAS PubMed; (b) N. G. Ricapito, C. Ghobril, H. Zhang, M. W. Grinstaff and D. Putnam, Chem. Rev., 2016, 116, 2664–2704 CrossRef CAS PubMed.
- (a) J. Shelton, X. Lu, J. A. Hollenbaugh, J. H. Cho, F. Amblard and R. F. Schinazi, Chem. Rev., 2016, 116, 14379–14455 CrossRef CAS PubMed; (b) R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS; (c) A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC; (d) S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef CAS; (e) A. Poma, H. Brahmbhatt, J. K. Watts and N. W. Turner, Macromolecules, 2014, 47, 6322–6330 CrossRef CAS; (f) P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed; (g) H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef CAS PubMed.
- (a) B. Lippert and P. J. Sanz Miguel, Acc. Chem. Res., 2016, 49, 1537–1545 CrossRef CAS PubMed; (b) C. Xing, H. Yuan, S. Xu, H. An, R. Niu and Y. Zhan, ACS Appl. Mater. Interfaces, 2014, 6, 9601–9607 CAS.
- G. S. Collier, L. A. Brown, E. S. Boone, B. K. Long and S. M. Kilbey, ACS Macro Lett., 2016, 5, 682–687 CrossRef CAS.
- A. S. Karikari, B. D. Mather and T. E. Long, Biomacromolecules, 2007, 8, 302–308 CrossRef CAS PubMed.
- (a) I. H. Lin, C.-C. Cheng, C.-W. Huang, M.-C. Liang, J.-K. Chen, F.-H. Ko, C.-W. Chu, C.-F. Huang and F.-C. Chang, RSC Adv., 2013, 3, 12598–12603 RSC; (b) B. D. Mather, M. B. Baker, F. L. Beyer, M. A. G. Berg, M. D. Green and T. E. Long, Macromolecules, 2007, 40, 6834–6845 CrossRef CAS; (c) A. Jatsch, A. Kopyshev, E. Mena-Osteritz and P. Bäuerle, Org. Lett., 2008, 10, 961–964 CrossRef CAS PubMed; (d) I. H. Lin, C.-C. Cheng, Y.-C. Yen and F.-C. Chang, Macromolecules, 2010, 43, 1245–1252 CrossRef CAS; (e) H. J. Spijker, F. L. van Delft and J. C. M. van Hest, Macromolecules, 2007, 40, 12–18 CrossRef CAS.
- (a) J. R. Tittensor, J. Chem. Soc. C, 1971, 2656–2662 RSC; (b) M. Suzuki, T. Sekido, S.-i. Matsuoka and K. Takagi, Biomacromolecules, 2011, 12, 1449–1459 CrossRef CAS PubMed.
- J. M. Coull, D. V. Carlson and H. L. Weith, Tetrahedron Lett., 1987, 28, 745–748 CrossRef CAS.
- N. Calcerrada-Muñoz, I. O'Neil and R. Cosstick, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1347–1350 Search PubMed.
- H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Org. Lett., 2008, 10, 3729–3732 CrossRef CAS PubMed.
- J. Sengupta and A. Bhattacharjya, J. Org. Chem., 2008, 73, 6860–6863 CrossRef CAS PubMed.
- K. K. Ogilvie and J. F. Cormier, Tetrahedron Lett., 1985, 26, 4159–4162 CrossRef CAS.
- (a) W. N. R. Wan Isahak, Z. A. Che Ramli, M. W. Mohamed Hisham and M. A. Yarmo, Renewable Sustainable Energy Rev., 2015, 47, 93–106 CrossRef CAS; (b) A. Dibenedetto, A. Angelini and P. Stufano, J. Chem. Technol. Biotechnol., 2014, 89, 334–353 CrossRef CAS; (c) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed; (d) Y. Qin, X. Sheng, S. Liu, G. Ren, X. Wang and F. Wang, J. CO2 Util., 2015, 11, 3–9 CrossRef CAS; (e) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (f) S. M. Guillaume and L. Mespouille, J. Appl. Polym. Sci., 2014, 131, 40081 CrossRef.
- (a) S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC; (b) J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822–39838 Search PubMed; (c) J. Feng, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS.
- E. Leino, P. Mäki-Arvela, V. Eta, D. Y. Murzin, T. Salmi and J. P. Mikkola, Appl. Catal., A, 2010, 383, 1–13 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- G. Rokicki and P. G. Parzuchowski, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 247–308 Search PubMed.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Bardé, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- (a) M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 1893–1896 CrossRef CAS; (b) M. Honda, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2830–2845 RSC; (c) F. D. Bobbink, W. Gruszka, M. Hulla, S. Das and P. J. Dyson, Chem. Commun., 2016, 52, 10787–10790 RSC.
- (a) K. Mikami, A. T. Lonnecker, T. P. Gustafson, N. F. Zinnel, P. J. Pai, D. H. Russell and K. L. Wooley, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed; (b) R. Kumar, W. Gao and R. A. Gross, Macromolecules, 2002, 35, 6835–6844 CrossRef CAS; (c) O. Haba, H. Tomizuka and T. Endo, Macromolecules, 2005, 38, 3562–3563 CrossRef CAS.
- G. L. Gregory, L. M. Jenisch, B. Charles, G. Kociok-Köhn and A. Buchard, Macromolecules, 2016, 49, 7165–7169 CrossRef CAS.
- Y. Shen, X. Chen and R. A. Gross, Macromolecules, 1999, 32, 2799–2802 CrossRef CAS.
- J. R. Tittensor and P. Mellish, Carbohydr. Res., 1972, 25, 531–534 CrossRef CAS.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102–1102 CrossRef CAS PubMed.
- DFT calculations of the ring-closing kinetics and thermodynamics were carried out using the following protocol: ωB97XD/6-31+g(d)/SCRF=(cpcm,solvent=acetonitrile) at a temperature of 298 K. The protocol includes an dispersion term shown to reproduce reaction barriers effectively. DFT calculations of monomer ring-strain were carried out the ωB97XD/6-311++g(2d,p)/SCRF=(cpcm,solvent=dichloromethane)/298 K level of theory. DFT computations of the initiation step in the ROP of 1 were carrried at the following level of theory: rωB97XD/6-311+g(d,p)/6-31+g(d)/cpcm=dichloromethane/298 K.
- A. R. Vaino and W. A. Szarek, Chem. Commun., 1996, 2351–2352 RSC.
- W. Hua, C. Changmei, Z. Hongchao, L. Xiaohong, F. Chang, X. Shanshan and Z. Yufen, Chem. Lett., 2005, 34, 432–433 CrossRef.
- M. D. Bartholoma, A. R. Vortherms, S. Hillier, J. Joyal, J. Babich, R. P. Doyle and J. Zubieta, Dalton Trans., 2011, 40, 6216–6225 RSC.
- K. Liu, L. Zheng, Q. Liu, J. W. de Vries, J. Y. Gerasimov and A. Herrmann, J. Am. Chem. Soc., 2014, 136, 14255–14262 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- J. Matsuo, K. Aoki, F. Sanda and T. Endo, Macromolecules, 1998, 31, 4432–4438 CrossRef CAS.
- Calculated as Mr(4-MeBnOH) + (Mr(1) × [1]0/[4-MeBnOH]0 × conv./100%).
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed; (b) M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- G. L. Fiore, F. Jing, J. V. G. Young, C. J. Cramer and M. A. Hillmyer, Polym. Chem., 2010, 1, 870–877 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details; spectroscopic and crystallographic data for 1 and 2; SEC traces, MALDI-ToF spectra, spectroscopic and thermal (TGA, DSC) data for polymers; DFT calculations data and associated digital repositories; tissue engineering protocol and results. CCDC 1523960 and 1523961. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7py00118e |
| This journal is © The Royal Society of Chemistry 2017 |