
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vesicles of double hydrophilic pullulan and poly(acrylamide) block copolymers: a combination of synthetic- and bio-derived blocks†
Jochen
Willersinn
,
Anna
Bogomolova
,
Marc Brunet
Cabré
and
Bernhard V. K. J.
Schmidt
 *
*
Max-Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: bernhard.schmidt@mpikg.mpg.de; Fax: (+49) 331-567-9502; Tel: (+49) 331-567-9509
First published on 9th January 2017
Abstract
The formation of vesicular structures with average diameters from 200 to 300 nm consisting of double hydrophilic diblock copolymers pullulan-b-poly(N,N-dimethylacrylamide) (Pull-b-PDMA) and pullulan-b-poly(N-ethylacrylamide) (Pull-b-PEA) in aqueous solution is described. Bio-derived pullulan was depolymerized and functionalized with alkyne endgroups. Furthermore, azide end functionalized acrylamide blocks PDMA and PEA were synthesized via RAFT polymerization. Individual blocks were conjugated via copper catalyzed azide alkyne cycloaddition (CuAAC) to afford defined double hydrophilic block copolymers. Aqueous solutions of the synthesized block copolymers showed formation of completely hydrophilic vesicles that were observed via various techniques including dynamic light scattering (DLS), static light scattering (SLS), laser scanning confocal microscopy (LSCM), and cryogenic scanning electron microscopy (SEM).
Introduction
Block copolymer self-assembly has been an important topic in polymer science in the last few decades, e.g. in bulk or in solution.1,2 In solution micelles or vesicles of amphiphilic block copolymers are commonly studied.3–5 Vesicles formed from amphiphilic block copolymers – the so-called polymersomes – are utilized for various applications e.g. in the biomedical field6–9 or in sensors.10 Moreover, the polymersome interior or the membrane has been utilized frequently as a reaction environment.11,12 Another important direction in polymersome research is the functionalization of the outer surface, which might be useful for molecular recognition in drug-delivery applications.13–15 In addition, significant research focusses on the formation of shape anisotropic polymersomes that might have enhanced biomedical properties.13,16Unlike amphiphilic block copolymers that self-assemble to various structures in aqueous solution due to the insolubility of the hydrophobic block in water,7,17 double hydrophilic block copolymer (DHBC) self-assembly occurs due to a strong difference in hydrophilicity.18,19 In fact, aqueous two-phase systems can be formed from aqueous solutions including two hydrophilic polymers at high concentrations.20,21 In such aqueous multiphase systems the different polymer types occupy one of the water phases, which is a well-known macroscopic demixing process. Therefore, a DHBC should induce phase separation in the respective concentration range as well. As the blocks are covalently bound in a DHBC no macroscopic self-assembly but a microscopic self-assembly occurs, leading to particular structures in aqueous solution, which is driven via the differences in osmotic pressure in the different water-soluble blocks.19,22 In order to achieve efficient self-assembly, the difference in hydrophilicity between the two polymer blocks has to be significant, which was described as a hydrophilic effect by Brosnan et al.18 The first ones who observed the formation of lyotropic mesophases of poly(ethylene oxide)-b-poly(2-methyl-2-oxazoline) block copolymers (PEO-b-PMeOx) in concentrated aqueous solutions were Taubert et al.23 Later, Ryan and coworkers showed the self-assembly of PEO-b-poly(2-(methacryloyloxy)ethyl phosphorylcholine) in concentrated aqueous solutions.22 Furthermore, the investigations of diluted DHBC solutions of PEO-b-poly(N,N-dimethylacrylamide) diblock copolymers (PEO-b-PDMA) by Ke et al.24 showed the formation of loose aggregates at block copolymer concentrations between 0.6 and 2.0 mg mL−1. Among some other publications reporting the presence of aggregates in double hydrophilic block copolymer solutions with and without external triggers,25–29 the most striking discovery was made by Brosnan et al. with the formation of giant vesicles via self-assembly of double hydrophilic polysaccharide-PEO block copolymers pullulan-b-PEO and dextran-b-PEO.18 These so called aquanelles had an average size between 2 and 20 μm and were observed with cryo SEM as well as optical microscopy. The complete hydrophilic nature of DHBC-based self-assemblies leads to expectations of their utilization as drug-delivery vehicles in the future. In general, DHBC self-assembly leads to rather dynamic structures18 that need stabilization to be useful for future applications. Recent research in our group focused on the further improvement of vesicular self-assembly of DHBCs via a PEO-b-poly(N-vinylpyrrolidone-co-N-vinylimidazole) block copolymer that was synthesized via aqueous reversible addition–fragmentation chain transfer/macromolecular design by interchange of xanthate (RAFT/MADIX) techniques starting from a PEO macro chain transfer agent.30 The self-assembled block copolymer could be cross-linked to preserve its spherical shape. However, the self-assembly of PEO-PVP block copolymers was not as efficient as was demonstrated for polysaccharide-b-PEO block copolymers.18,30
In order to further improve DHBC self-assembly and extend the toolbox of purely hydrophilic block copolymer combinations for future research, a modular and more versatile approach towards block copolymer formation would be desirable. A very efficient and prominent route to form complex macromolecular structures in a modular way is the so-called click chemistry approach described by Sharpless et al.31 Taking advantage of the 1,3 dipolar cycloaddition of terminal alkynes and azides first described by Huisgen et al.,32 block copolymer synthesis can be facilitated.33–36 Furthermore, utilization of click chemistry as a tool allowed the formation of other complex macromolecular architectures.37–39 Access to hydrophilic homopolymers with end terminated azides and alkynes can be given by reversible deactivation radical polymerization (RDRP)40–42 or living polymerization techniques.43,44 Moreover, several commercially available homopolymers such as bio-derived dextran and pullulan as well as synthetic PEO can be easily post functionalized to be applicable in copper catalyzed azide alkyne cycloaddition (CuAAC) reactions, for example dextran and pullulan with alkyne moieties45,46 or PEO with either azide or alkyne moieties.47,48 Furthermore, macromolecular ligation via CuAAC features modularity, which easily allows the formation of various block copolymer combinations with respect to polymer type or degree of polymerization.33
Since pullulan block copolymers demonstrated efficient self-assembly behavior with the less hydrophilic PEO, we decided to investigate block copolymers of pullulan and hydrophilic polyacrylamides as a combination of building blocks of biological and synthetic origin. Pullulan is a prominent example of biopolymers in macromolecular science.49–51 Pullulan is formed by the microorganism Aureobasidium pullulans that produces a polysaccharide in order to protect itself from external threats.52 The linear polysaccharide, which consists of maltotriose repeating units that are linked via α-(1,6) glycosidic bonds, has a broad spectrum of applications, such as blood plasma substitutes,53 food manufacturing54 and pharmaceutical applications.49 The second building blocks – polyacrylamides with alkyne or azido functionalities – are facile to synthesize via RDRP techniques starting from a suitable chain transfer agent.42 Functionalized homopolymers such as PDMA were already used in the preparation of macromolecular suprastructures and complex architectures.40,55
In contrast to the DHBC self-assembly reported earlier in the literature, where one polymer block possesses a certain functionality, such as a lower critical solution temperature (LCST) in a biophysical range, ionic functionalities or pH responsive groups that trigger self-assembly upon external changes into the desired fashion,26,56,57 the self-assembly of non-ionic, purely hydrophilic block copolymers occurs through a different, not yet completely understood, pathway. Since only neutral water and no (mostly toxic) selective solvents are used during the self-assembly process, DHBC vesicles are considered to be a more valuable choice for future drug delivery applications. Moreover, it is expected that the formed particles present higher membrane permeabilities compared with traditional polymersomes.
Herein, we present the synthesis of two novel double hydrophilic block copolymers, namely Pull-b-PDMA and pullulan-b-poly(N-ethylacrylamide) (Pull-b-PEA), which show the ability to form vesicular structures in highly diluted purely aqueous solutions without the involvement of external stimuli (Scheme 1). The bio-derived pullulan block was obtained via depolymerization. The polyacrylamide homopolymer blocks were synthesized via RAFT polymerization and conjugated to the respective pullulan building blocks via CuAAC. The presence of formed vesicular structures was investigated via cryogenic electron microscopy (cryo SEM), laser confocal scanning microscopy (LSCM), static light scattering (SLS) and dynamic light scattering (DLS) measurements.
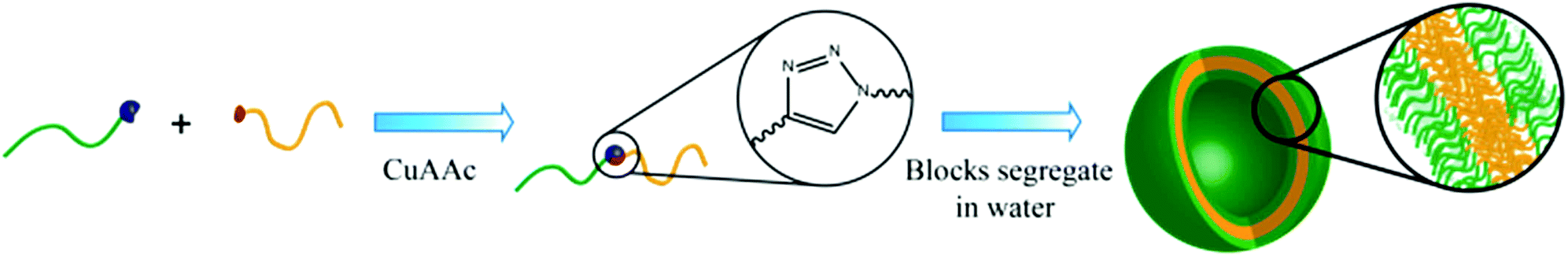 | ||
| Scheme 1 Conjugation and self-assembly scheme of double hydrophilic block copolymers in water. | ||
Experimental
Materials
Azobis(isobutyronitrile) (AIBN, 99%, Sigma Aldrich) was recrystallized twice from methanol. Dichloromethane (DCM, 99.9%, Acros Organics) and acetone (99%, J.T. Baker) were stored over molecular sieves (3 Å) prior to use. N,N-Dimethylacrylamide (DMA, 99%, TCI) was passed over neutral aluminium oxide prior to use. N-Ethylacrylamide (EA, 99%, TCI) was distilled under vacuum prior to use. Millipore water was obtained from an Integra UV plus pure water system by SG Water (Germany). Ammonium chloride (99%, Roth KG), ascorbic acid (98%, Alfa Aesar), 2-bromo-2-methylpropanoic acid (98%, Sigma Aldrich), 2-bromopropionyl bromide (97%, Sigma Aldrich), 3-bromo-1-propanol (97%, Sigma Aldrich), carbon disulfide (CS2, anhydrous 99%, Sigma Aldrich), chloromethyl polystyrene resin (2.4 mmol g−1, TCI), copper sulfate (CuSO4, 99%, Carl Roth), N,N′-dicyclohexylcarbodiimide (DCC, 99%, Sigma Aldrich), diethylether (analytical grade, Sigma Aldrich), N,N-dimethylaminopyridine (DMAP, 99%, Sigma Aldrich), dimethylsulfoxide (DMSO, VWR Chemicals), dodecanethiol (98%, Alfa Aesar), ethyl acetate (EtOAc, analytical grade, Chem Solute), hexane (analytical grade, Fluka), hexylamine (>99%, Fluka), hydrochloric acid (fuming, Carl Roth), magnesium sulfate (dried, Fisher Scientific), methanol (MeOH, analytical grade, Fisher Scientific), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 98%, Sigma Aldrich), potassium phosphate (K3PO4, Sigma Aldrich), propargylamine (98%, Sigma Aldrich), pullulan (Pull, pure, TCI), Rhodamine B (RhB, 99%, Sigma Aldrich), Rhodamine B isothiocyanate (RITC, 99%, Sigma Aldrich), sodium azide (>99.5%, Fluka), sodium bicarbonate (>99%, Fluka), sodium cyanoborohydride (NaCNBH3, 95%, Sigma Aldrich), sodium iodide (Acros Organics, anhydrous), tetrahydrofuran (THF, extra dry, Acros Organics) and triethylamine (99.5%, Sigma Aldrich) were used as received. Azido functionalized PS-resin, azido terminated PDMA, azido terminated PEA, dodecylthiocarbonylthio-2-methylpropanoic acid 3′-azidopropylester and pullulan alkyne were synthesized according to the literature (refer to the ESI† for details).45,58,59Methods
1H- and 13C-NMR spectra were recorded at ambient temperature at 400 MHz for 1H and 100 MHz for 13C with a Bruker Ascend400. Dynamic light scattering (DLS) and static light scattering (SLS) were performed using an ALV-7004 Multiple Tau Digital Correlator in combination with a CGS-3 Compact Goniometer and a HeNe laser (Polytec, 34 mW, λ = 633 nm at θ = 90° setup for DLS and 30° to 150° with steps of 10° for SLS). Sample temperatures were adjusted to 25 °C. Toluene was used as immersion liquid. Apparent hydrodynamic radii (Rapp) have been determined from fitting autocorrelation functions by using REPES algorithms. Radii of gyration (Rg) were determined via SLS with an ALV Stat ALV-5000 using a Guinier plot. Cryogenic scanning electronic microscopy (cryo SEM) was performed on a Jeol JSM 7500 F and the cryo-chamber from Gatan (Alto 2500). Size exclusion chromatography (SEC) for polyacrylamides was conducted in NMP (Fluka, GC grade) with 0.05 mol L−1 LiBr and BSME as internal standard at 70 °C using a column system with a PSS GRAM 100/1000 column (8 × 300 mm, 7 μm particle size) and a PSS GRAM precolumn (8 × 50 mm) and a Shodex RI-71 detector and a PS calibration with standards from PSS. Pullulan samples were analyzed in acetate buffer containing 20% MeOH at 25 °C using a PSS NOVEMA Max analytical system XL (pre column size 50 mm × 8 mm – 10 μm, main column size 300 mm × 8 mm – 10 μm) using a pullulan calibration with standards from PSS. Laser scanning confocal microscopy (LSCM) measurements were conducted with a Leica TCS SP5 (Wetzlar, Germany) confocal microscope, using a 63× (1.2 NA) water immersion objective. The dye stained samples were excited with a diode pumped solid-state laser at 561 nm. The emission bands were collected at 640 nm. Turbidimetry measurements to obtain the lower critical solution temperature (LCST) were conducted with a T70+ UV/Vis Spectrometer (PG Instruments Ltd) at a wavelength of 660 nm and a temperature control system consisting of a Peltier Temperature Controller PTC-2 and a Manson Switching Mode Power Supply 1-36VDC-10A. Typically, 0.5 wt% solutions were investigated with a heating rate of 1 K min−1 and the transmission values were detected within a 5 second interval.Synthesis procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1, pullulan standard in acetate buffer with 20% MeOH, Đ = 1.9) as a white powder.
500 g mol−1, pullulan standard in acetate buffer with 20% MeOH, Đ = 1.6) as a white powder.
600 g mol−1, pullulan standard in acetate buffer with 20% MeOH, Đ = 2.7) as a purple powder.
500 g mol−1, pullulan standard in acetate buffer with 20% MeOH, Đ = 1.9) as a white powder.
500 g mol−1, pullulan standard in acetate buffer with 20% MeOH, Đ = 1.6) as a white powder.
600 g mol−1, pullulan standard in acetate buffer with 20% MeOH, Đ = 2.7) as a purple powder.
Results and discussion
Synthesis of alkyne functionalized pullulan
Being a polysaccharide which is produced from starch by the fungus Aureobasidium pullulans, commercially available pullulan generally possesses a broad molecular mass distribution with high average molecular masses. In order to afford pullulan with a sufficient Đ below 2.0 and molecular masses in the intended region, high molecular mass pullulan has to be depolymerized to a certain degree. Since pullulan predominantly consists of maltotriose units that are linked via α-(1,6) glycosidic bonds, the easiest access to depolymerized pullulan is a cleavage between the maltotriose units. Ilic et al. described a facile method to achieve a controlled depolymerization of pullulan, using 0.025 M hydrochloric acid solution and elevated temperatures in order to cleave α-(1,6) glycosidic bonds.58 Following that procedure commercial pullulan was depolymerized to a Mn of 14000 g mol−1 with a Đ of 1.8. Attempts to further decrease the polydispersity of the obtained pullulan were not successful with the employed starting material without further decreasing the molecular mass, due to the broad molecular mass distribution of the starting material. Since efficient self-assembly of pullulan based block copolymers with Đ values around 1.8 was shown earlier by Brosnan et al.,18 further optimization was not attempted.
The alkyne functionalization of the depolymerized pullulan was conducted in acetate buffer at 50 °C (Fig. 1a) via reductive amination. In order to afford full conversion of the terminal aldehyde group of the ω-glucose unit, a 100 fold excess of propargylamine and NaCNBH3 was used. Additionally, acetate buffer and a high excess of reactants were utilized to ensure the desired reaction pathway towards reductive amination and diminish further depolymerization of pullulan. The presence of the alkyne could not be directly detected by 1H-NMR due to an overlap of propargyl proton signals with the signals corresponding to pullulan. Nevertheless, the full disappearance of the anomeric proton peaks (α-centered at 6.7 ppm and β-centered at 6.3 ppm) is a strong indication of the complete conversion of the terminal group (Fig. 1b and S1†). Regarding the SEC elution curves of the alkyne functionalized pullulan (Fig. 1c and Table S1†), only a very slight decrease in molecular weight was observed. Therefore, it can be stated that there is predominant reductive amination of the α-1-aldehyde at the terminal glucose unit and no significant depolymerization. Thus, the first part of the block copolymer – the bio-derived alkyne containing block – was synthesized.
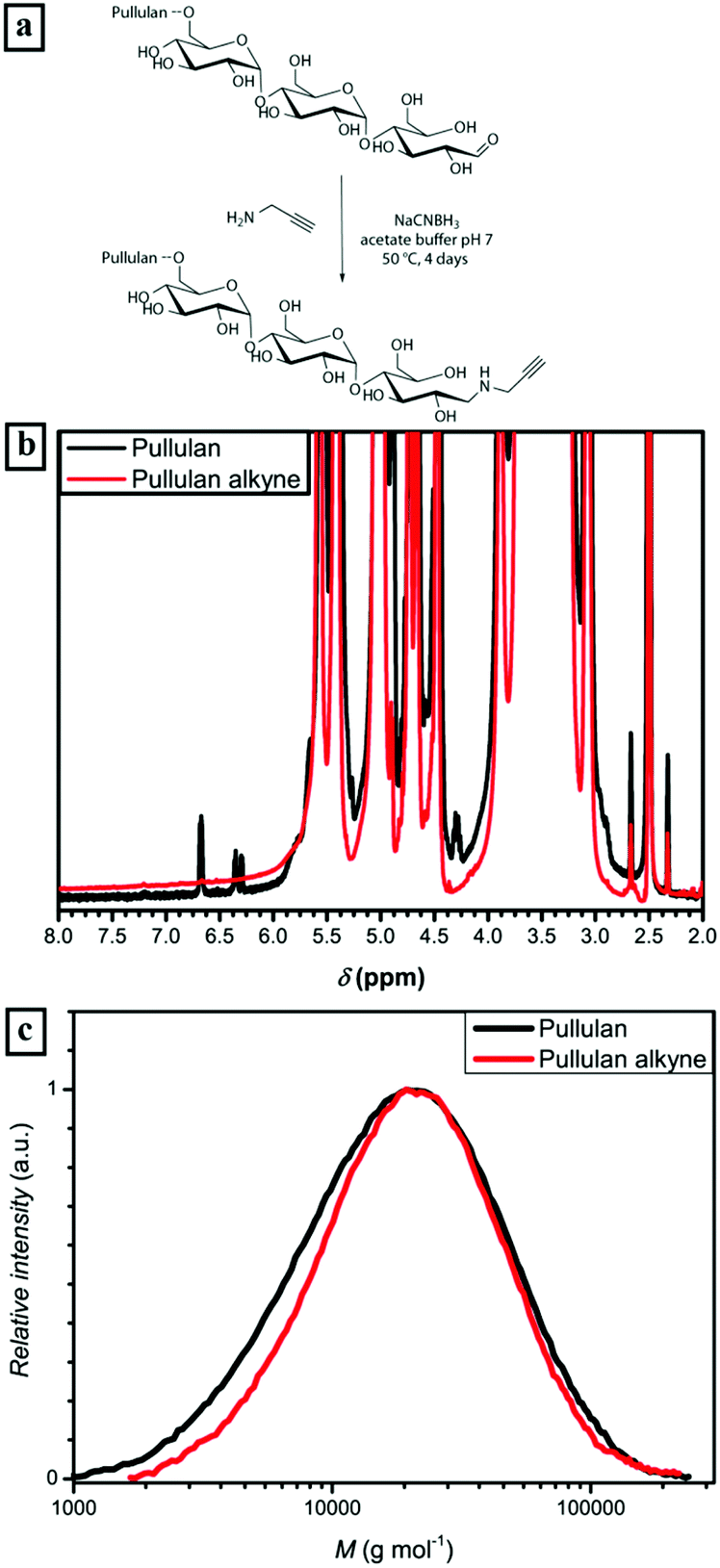 | ||
| Fig. 1 (a) Synthesis scheme of pullulan-alkyne; (b) 1H-NMR comparison of anomeric proton area of pullulan and pullulan alkyne recorded at 400 MHz in DMSO-d6; (c) corresponding molecular mass distribution curves determined via SEC in acetate buffer solution against pullulan standards. | ||
Synthesis of azide terminated acrylamide homopolymers
Azido terminated PDMA and PEA are easily accessible via RAFT polymerization. An azido functionalized trithiocarbonate, namely dodecylthiocarbonylthio-2-methylpropanoic acid 3′-azidopropylester,42 was used as chain transfer agent according to a known procedure (Scheme 2).42,59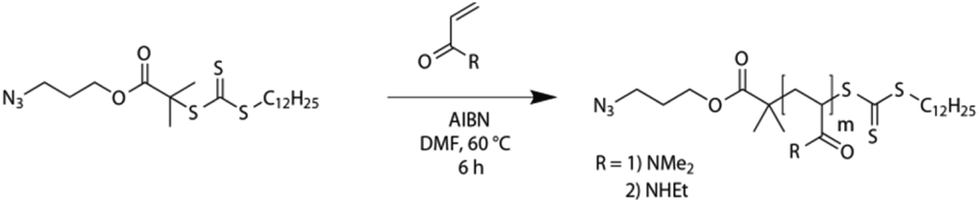 | ||
| Scheme 2 RAFT polymerization procedure for the synthesis of azido terminated polyacrylamides. | ||
The afforded homopolymers PEA-N3 and PDMA-N3 possessed a narrow size distribution (Đ values from 1.26 to 1.37) and molecular masses of 16800 g mol−1 (PDMA-N3) and 13900 g mol−1 (PEA-N3) (Fig. S3, S5 and Table S1†). Moreover, the polymers were characterized via1H-NMR (Fig. S2 and S4†). The synthesized polyacrylamide homopolymers were well soluble in water even without removal of the RAFT endgroup. However, the presence of a certain amount of homopolymer aggregation was observed for all blocks as shown via DLS (Fig. S6†). Turbidimetry measurements proved their solubility up to 60 °C in water (Fig. S7†) as well. Within the double hydrophilic block copolymer systems of interest, PDMA and PEA are considered to be the less hydrophilic part of the block copolymers, whereas pullulan is the more hydrophilic block, respectively.
Conjugation of block copolymers via copper catalyzed azide alkyne cycloaddition
The CuAAC reaction of homopolymers bearing terminal alkynes and azides is a versatile and easy tool to form block copolymers (Schemes 1 and 3).34 In order to conjugate two hydrophilic polymers to a double hydrophilic block copolymer, cycloaddition was conducted in a mixture of water and DMSO to ensure complete solubility of all reagents. Since the absolute molecular masses of the starting materials were not determined, the ratio between the homopolymers for a full conversion of both polymer blocks can only be assessed via multiple conjugation experiments. Thus, a different route was utilized. A 1.2 molar excess of the alkyne terminated pullulan was used to ensure a full conversion of the acrylamide blocks to the conjugated block copolymers. The excess of pullulan alkyne was removed after a sufficient reaction time by employing an azidomethyl polystyrene resin (Fig. S8†), which was added to the reaction mixture. The resin particles with a mesh size of 100 to 200 can be easily removed after the cycloaddition reaction by filtration, which facilitates the purification of the block copolymers. The conjugated block copolymers were analysed with 1H-NMR and SEC in aqueous acetate buffer solution and compared with mixtures of the corresponding homopolymers (Table S1†).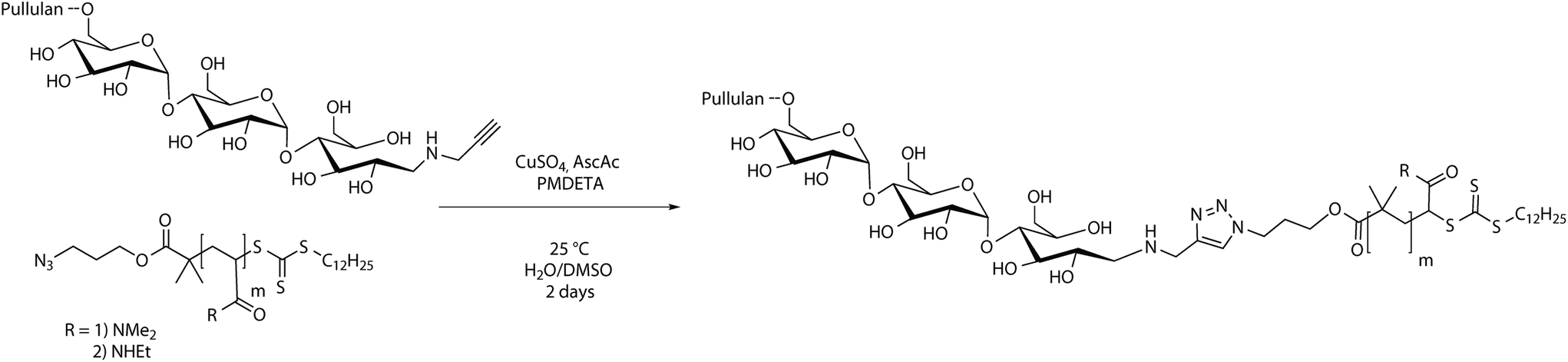 | ||
| Scheme 3 CuAAC conjugation reaction scheme of pullulan alkyne and azide terminated acrylamides. | ||
300 g mol−1 to 21500 g mol−1. Since an equimolar amount of resin was applied to theoretically remove all alkyne blocks, a full conversion of PDMA to the block copolymer can be assumed. The 1H-NMR spectrum of the conjugated block copolymer displaying the presence of the pullulan as well as the PDMA block underlines this assumption, e.g. the signals of anomeric protons in the pullulan block between 5.0 and 5.3 ppm and the signals of the methyl sidegroups in the PDMA block between 2.7 and 3.1 ppm (Fig. 2c) are clearly visible.
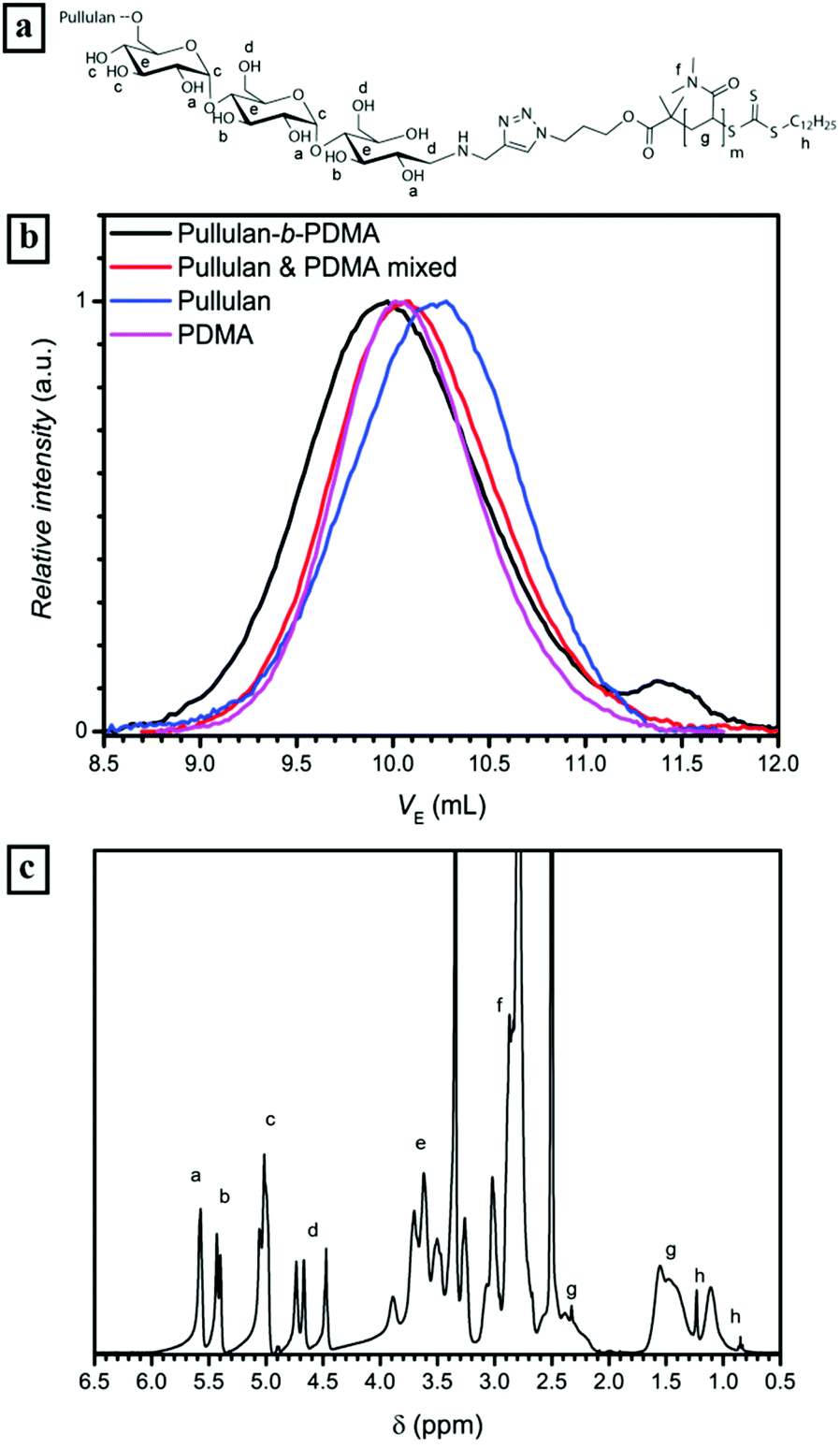 | ||
| Fig. 2 (a) Chemical structure of Pull-b-PDMA, (b) SEC traces of conjugated Pull-b-PDMA, a homopolymer mixture and the homopolymers in acetate buffer and (c) corresponding 1H-NMR spectrum of the block copolymer in DMSO-d6 at 25 °C. | ||
500 g mol−1 and Đ = 1.6, Pull-b-PEA stands in clear contrast to the mixture with a high polydispersity of 3.5 and an apparent Mn of 6000 g mol−1. The 1H-NMR spectrum of the block copolymer displays the presence of both blocks as well, e.g. the signals of anomeric protons in the pullulan block between 5.0 and 5.3 ppm and the signals of the methyl sidegroups in the PEA block between 0.9 and 1.1 ppm (Fig. 3c). A full conversion of PEA to the block copolymer can be stated here as well as for Pull-b-PDMA.
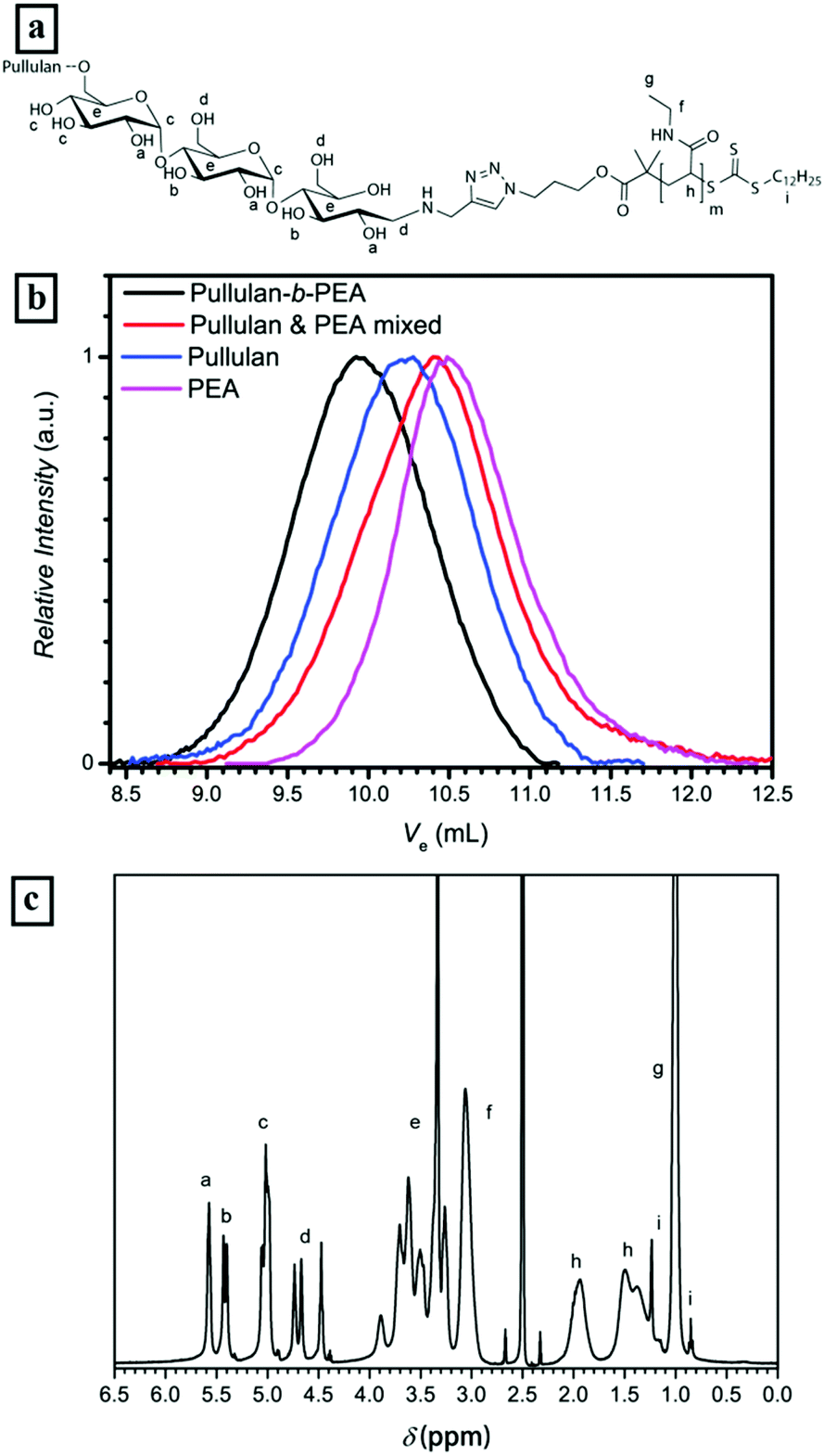 | ||
| Fig. 3 (a) Chemical structure of Pull-b-PEA, (b) SEC traces of conjugated Pull-b-PEA, a homopolymer mixture and the homopolymers in acetate buffer and (c) corresponding 1H-NMR spectrum of the block copolymer in DMSO-d6 at 25 °C. | ||
Aqueous self-assembly of pullulan-b-PDMA and pullulan-b-PEA
DHBC self-assembly without the influence of external stimuli such as pH or temperature requires certain properties of the block copolymer composition. In previous reports a strong difference in hydrophilicity,18,60i.e. the interaction of the polymer with water molecules, was regarded as the key role for successful self-assembly. Regarding this assumption, a comparison of the second virial coefficient A2 of the homopolymers should be further taken into account. The A2 factor, which can be determined via SLS can be seen as a quantitative measure for solvent solute interactions.61 The second virial coefficients for pullulan and PDMA in the molecular weight range of interest were already studied in the literature and determined to be 3.2 × 10−4 mol cm3 g−2 for pullulan,62 and 8.0 × 10−4 mol cm3 g−2 for PDMA, respectively.63 When comparing these two values, PDMA appears to possess a stronger interaction with the solvent water than pullulan. Since PEA is a quite similar polymer, its A2 factor should be in the same range as that of PDMA. (It is assumed that phase separation of the different hydrophilic polymer blocks occurs, when the difference in hydrophilicity is large enough.) Furthermore, chain rigidity is assumed to influence self-assembly behaviour, too. Hydrogen bonding and the corresponding thermoresponsivity as well as phase separation of the backbone as was observed for homopolymer self-assembly play only minor roles.64,65 Furthermore, the functional groups of pullulan and both polyacrylamide polymer blocks are not known to show any thermoresponsive behavior (Fig. S6†) in the investigated temperature range, which allows the formation of a pure hydrophilic self-assembly. Nevertheless, PEA is known for its LCST behavior at elevated temperatures above 70 °C.66 In fact, PEA was chosen as a block due to this fact since the comparison between a block without LCST and a block with LCST at elevated temperatures should give some insights into the fundamentals of DHBC aggregate formation. First, since the mechanism of DHBC self-assembly has not been completely understood yet, investigations of the dissolved DHBCs via DLS were conducted.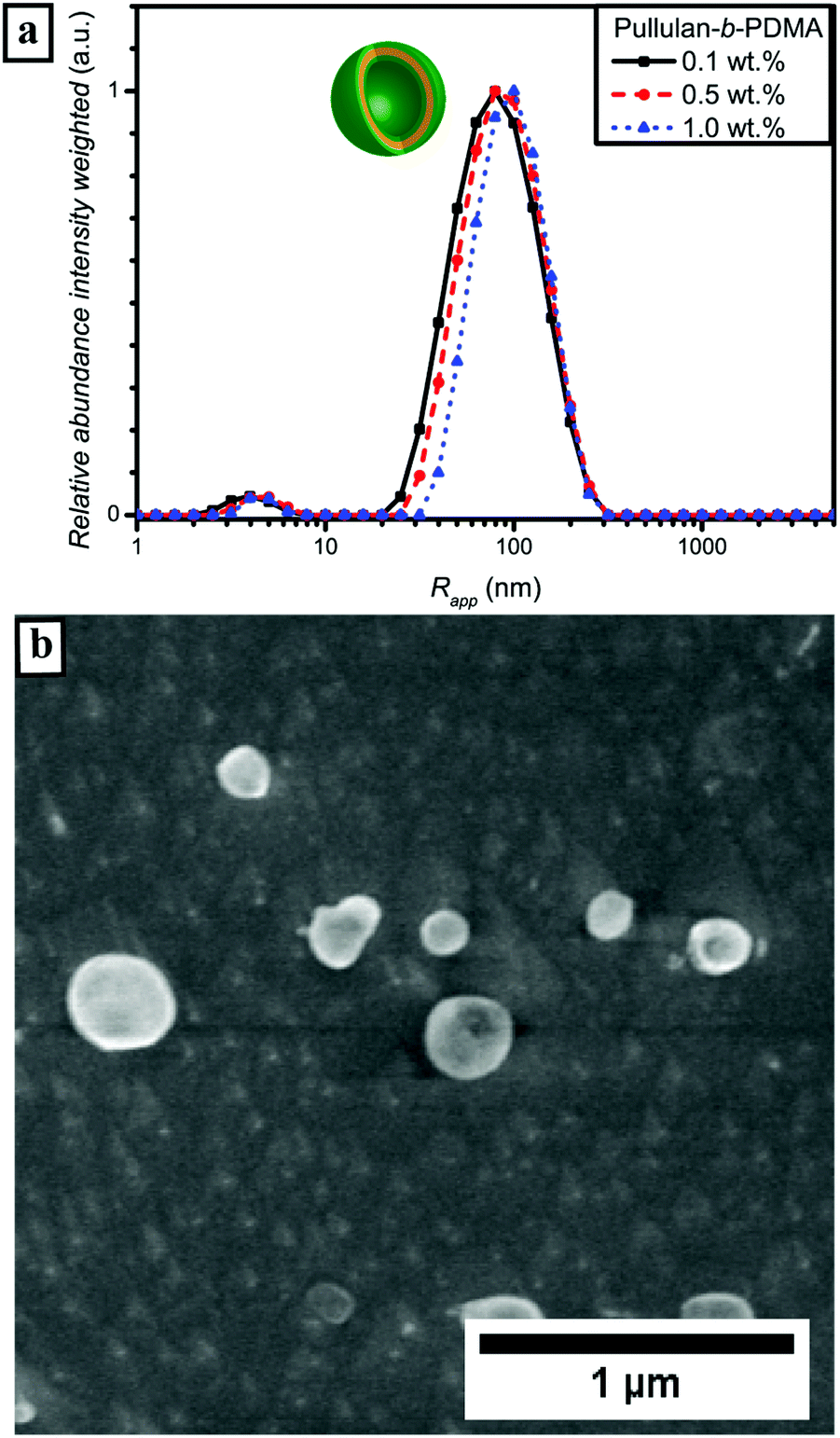 | ||
| Fig. 4 (a) Intensity weighted particle size distributions of Pull-b-PDMA in water measured via DLS at 25 °C; (b) cryo SEM micrograph of self-assembled spherical particles at a concentration of 0.5 wt%. | ||
In order to exclude an effect of the aliphatic RAFT-agent backbone C12 unit on self-assembly behavior, a pullulan-b-PDMA block copolymer was synthesized after oxidative removal of the RAFT chain transfer group (Fig. S11†).40 As visible from the particle size distribution curves in Fig. S15,† the self-assembly behavior did not change drastically apart from a slight increase in the hydrodynamic radius (Fig. S12†). For that reason, an influence of the RAFT chain transfer group towards enhancing the self-assembly behavior of Pull-b-PDMA significantly can be excluded. Moreover, DLS measurements at pH values of 5 and 8 did not show significant differences as well (Fig. S13†). In addition the effect of filtration with cellulose acetate filters was probed via DLS (Fig. S14†). No significant differences were found for filters with 0.45, 0.8 and 1.2 μm pore sizes.
Subsequently, microscopy was utilized to image the formed particles. The corresponding micrographs of the performed cryo SEM investigations of the 0.5 wt% solution of Pull-b-PDMA (Fig. 4b) display spherical particles with diameters in the range between 80 nm and 250 nm. Since there are no ruptured particles visible in the micrographs, the particle's morphology cannot be assessed via cryo SEM only. In order to gain deeper insight into the particle morphology, a 2.5 wt% block copolymer solution in water was stained with Rhodamine B to examine the spherical structures with LSCM techniques. The solution of self-assembled Pull-b-PDMA was directly stained with 10 μL of an aqueous 0.08 mM Rhodamine B solution. As visible from the LSCM micrographs in Fig. 5a and b, Rhodamine B is homogeneously distributed in the solution. An increased concentration of dye was observed inside spherical micron sized structures.
 | ||
| Fig. 5 (a) LSCM micrographs of a 2.5 wt% Pull-b-PDMA solution stained with 0.08 mM Rhodamine B solution; (b) magnification of dye stained spherical particles; (c) LSCM micrographs of a 2.5 wt% Pull-b-PDMA/Pull-b-PDMA-RhB solution and (d) magnification of Pull-b-PDMA/Pull-b-PDMA-RhB based particles. | ||
The enrichment of Rhodamine B inside these structures indicates a certain permeability of the membrane towards the dye to diffuse inside the particle. Since the spherical particles self-assembled before the addition of Rhodamine B, permeation of the dye into the spherical structures is quite likely. However, interactions of the block copolymer membrane with Rhodamine B seem to prevent diffusion back to the external media. Therefore, the observed enrichment inside the particles may occur. As visible from the LSCM micrographs, the diameter of the observed spherical particles exceeds the one determined from DLS and cryo-SEM by almost five fold. The increase in the diameter can be attributed to the particle motion inside the solution, resulting in an uncertainty of the image. Furthermore, the lower resolution level of LSCM instruments within the range of the determined particle size prevents a clear resolution and broadens the signal. Therefore, the size of the depicted spherical particles has to be handled with care. Moreover, the resolution of the LSCM instrument prevents imaging of particles with smaller size. Besides the spherical structures within the micrometer size, smaller spherical particles without increased Rhodamine B concentration are visible in Fig. 5a. These particles were observed quite frequently but could not be resolved efficiently due to the fast particle motion inside the solution. In order to further investigate the interactions of dye and block copolymer via exclusion of non-specific interactions, a PDMA-N3 homopolymer was labelled with RITC and conjugated to a pullulan alkyne via CuAAC that was analyzed via1H-NMR and DLS (Fig. S15 and S16†). The direct attachment of fluorescent dye to the block copolymer should on the one hand ensure that only spherical structures self-assembled from the DHBC were investigated. On the other hand, the proposed membrane structure should be further investigated, since the dye can only be present in areas where the block copolymer is expected to concentrate. The corresponding Rhodamine B labelled block copolymer was added to a 2.5 wt% solution of Pull-b-PDMA and investigated via LSCM. As visible from the micrographs in Fig. 5c and d, the amount of fluorescent material is far lower in comparison with the stained samples as expected. Furthermore, spherical structures with a vesicular morphology and average diameters of 1 to 2 μm could be observed, where the fluorescent membrane can be clearly attributed to the labelled Pull-b-PDMA block copolymer. This is a strong indication that the predicted vesicular structure is present in the case of larger particles. However, the particle motion and resolution limit of LSCM prevent a closer look at submicron structures. Structures in the submicron-size range were observed but could only be resolved as fluorescent spheres because the volume excited by the laser is too small to be resolved by the detector. In conclusion, it can be stated that Pull-b-PDMA forms particular self-assembled structures in aqueous solution with high abundance. Moreover, SLS and LSCM with RhB labelled block copolymers indicate that hollow structures are formed.
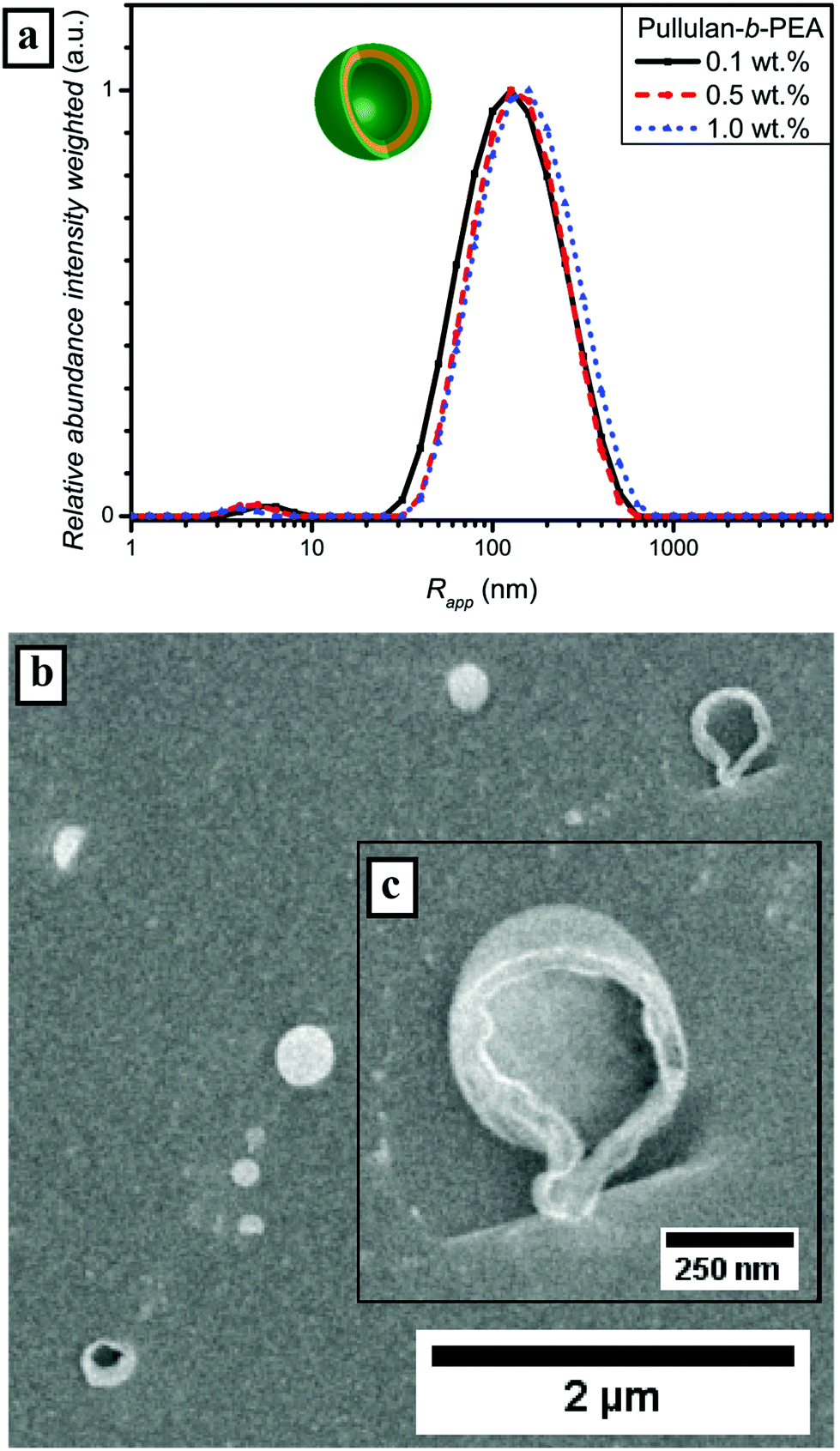 | ||
| Fig. 6 (a) Intensity weighted particle size distributions of Pull-b-PEA in water measured via DLS at 25 °C; (b) cryo SEM micrograph of self-assembled spherical particles at a concentration of 0.5 wt% and (c) a magnification of a ruptured vesicular structure. | ||
The enhanced self-assembly behavior of Pull-b-PEA leads to the conclusion that the difference in hydrophilicity of pullulan and PEA is even higher compared with pullulan and PDMA. The secondary amide of the N-ethylacrylamide unit interacts differently with water than the N,N-dimethylacrylamide unit of PDMA. The difference in interaction can be seen as well in the DLS particle size distributions of the homopolymers (Fig. S6†). It seems that the incorporation of a block that features LCST behavior at elevated temperatures – even if the LCST is significant above the self-assembly temperature, resulting in less hydrophilicity of the acrylamide-derivative block – improves self-assembly. The value of Rg determined via SLS (Fig. S17 and Table S4†) for a concentration of 0.5 wt% is 129 nm and the corresponding quotient of Rg and Rapp is 1.03 and in the same range for hollow spheres as is the case for Pull-b-PDMA. Again, a strong indication for vesicular structures is present. A more outstanding difference can be seen from the cryo SEM micrographs in Fig. 6b. Along with spherical particles with an average diameter of up to 300 nm, several larger particles with a ruptured membrane were observed (Fig. 6c). These ruptured particles strongly indicate that a vesicular structure is present. The vesicular structures appear to be hollow, which is consistent with cryo SEM observations of dextran-b-PEO block copolymer vesicles displayed by Brosnan et al.18 The fact that such small vesicular structures could be afforded with Pull-b-PEA is quite astonishing.
LSCM measurements of Pull-b-PEA block copolymer solutions stained with Rhodamine B display spherical vesicular particles with an average diameter of 1 μm. The vesicular structures were observed either attached on the glass surface (Fig. 7a and b) or in motion in the solution. The motion of the vesicular structures caused a distortion of the particles when a micrograph was recorded as seen in Fig. 7c. The magnification of a vesicular structure (Fig. 7d) shows that Rhodamine B concentrates inside the structures similarly to the observed pullulan-b-PMA structures. In conclusion, self-assembled particles from Pull-b-PEA in aqueous solution are observed. Investigations via SLS and cryo SEM imaging strongly indicate formation of hollow vesicular structures.
 | ||
| Fig. 7 (a) LSCM overview of Rhodamine B stained Pull-b-PEA vesicles in water. (b) Magnification displaying several spherical particles. (d) A magnification of a vesicular structure. The distortion origins from the particle motion through the solution. (c) Magnified spherical particle. | ||
Conclusions
In summary, we were able to extend the principle of double hydrophilic self-assembly to novel polysaccharide–polyacrylamide block copolymers. The blocks were separately synthesized either via depolymerisation of bio-derived pullulan or via RAFT polymerization of acrylamide-derivatives. Subsequently, the blocks were conjugated via CuAAC to afford the corresponding DHBCs in an efficient manner. The block copolymers were characterized via SEC and 1H NMR techniques. Furthermore, it was demonstrated that such block copolymers perform self-assembly in water to vesicular structures with average diameters of 200 nm to 500 nm with high efficiency. The vesicular structure was visualized via cryo SEM. In addition LSCM was utilized using Rhodamine B staining or Rhodamine B labelled block copolymers. Interesting applications of the novel vesicular structures are expected, e.g. in the field of drug-delivery.Acknowledgements
The authors thank the Max Planck Society for funding. The authors acknowledge Marlies Gräwert for SEC measurements, Heike Runge for assistance with cryogenic SEM measurements, and Dr Tom Robinson as well as Carmen Remde for assistance with LSCM measurements. Open Access funding provided by the Max Planck Society.Notes and references
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- R. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- G. Yu, W. Yu, L. Shao, Z. Zhang, X. Chi, Z. Mao, C. Gao and F. Huang, Adv. Funct. Mater., 2016, 26, 8999–9008 CrossRef CAS.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- M. Antonietti and S. Forster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, J. Mater. Chem., 2011, 21, 12777–12783 RSC.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262–15275 CrossRef CAS PubMed.
- K.-K. Huang, Y.-K. Fang, J.-C. Hsu, C.-C. Kuo, W.-H. Chang and W.-C. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 147–155 CrossRef CAS.
- H. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- M. C. M. van Oers, W. S. Veldmate, J. C. M. van Hest and F. P. J. T. Rutjes, Polym. Chem., 2015, 6, 5358–5361 RSC.
- L. K. E. A. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef PubMed.
- B. Iyisan, J. Kluge, P. Formanek, B. Voit and D. Appelhans, Chem. Mater., 2016, 28, 1513–1525 CrossRef CAS.
- L. Ruiz-Pérez, L. Messager, J. Gaitzsch, A. Joseph, L. Sutto, F. L. Gervasio and G. Battaglia, Sci. Adv., 2016, 2, e1500948 Search PubMed.
- R. S. M. Rikken, H. Engelkamp, R. J. M. Nolte, J. C. Maan, J. C. M. van Hest, D. A. Wilson and P. C. M. Christianen, Nat. Commun., 2016, 7, 12606 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- S. M. Brosnan, H. Schlaad and M. Antonietti, Angew. Chem., Int. Ed., 2015, 54, 9715–9718 CrossRef CAS PubMed.
- O. Casse, A. Shkilnyy, J. Linders, C. Mayer, D. Haussinger, A. Volkel, A. F. Thunemann, R. Dimova, H. Colfen, W. Meier, H. Schlaad and A. Taubert, Macromolecules, 2012, 45, 4772–4777 CrossRef CAS.
- R. Dimova and R. Lipowsky, Adv. Mater. Interfaces, 2016, 4, 1600451 CrossRef.
- C. D. Keating, Acc. Chem. Res., 2012, 45, 2114–2124 CrossRef CAS PubMed.
- A. Blanazs, N. J. Warren, A. L. Lewis, S. P. Armes and A. J. Ryan, Soft Matter, 2011, 7, 6399–6403 RSC.
- A. Taubert, E. Furrer and W. Meier, Chem. Commun., 2004, 2170–2171 RSC.
- F. Y. Ke, X. L. Mo, R. M. Yang, Y. M. Wang and D. H. Liang, Macromolecules, 2009, 42, 5339–5344 CrossRef CAS.
- W. Agut, A. Brûlet, C. Schatz, D. Taton and S. Lecommandoux, Langmuir, 2010, 26, 10546–10554 CrossRef CAS PubMed.
- M. Bathfield, J. Warnant, C. Gerardin and P. Lacroix-Desmazes, Polym. Chem., 2015, 6, 1339–1349 RSC.
- N. Chan, S. Y. An, N. Yee and J. K. Oh, Macromol. Rapid Commun., 2014, 35, 752–757 CrossRef CAS PubMed.
- H. Park, S. Walta, R. R. Rosencrantz, A. Korner, C. Schulte, L. Elling, W. Richtering and A. Boker, Polym. Chem., 2016, 7, 878–886 RSC.
- T. Rudolph, S. Crotty, M. von der Luehe, D. Pretzel, U. S. Schubert and F. H. Schacher, Polymers, 2013, 5, 1081–1101 CrossRef.
- J. Willersinn, M. Drechsler, M. Antonietti and B. V. K. J. Schmidt, Macromolecules, 2016, 49, 5331–5341 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC.
- W. Agut, R. Agnaou, S. Lecommandoux and D. Taton, Macromol. Rapid Commun., 2008, 29, 1147–1155 CrossRef CAS.
- S. R. S. Ting, A. M. Granville, D. Quémener, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Aust. J. Chem., 2007, 60, 405–409 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS PubMed.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Polym. Chem., 2014, 5, 2461–2472 RSC.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960–4965 CrossRef CAS.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474–481 CrossRef CAS.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- M. W. M. Fijten, C. Haensch, B. M. van Lankvelt, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 1887–1895 CrossRef CAS.
- C. Schatz, S. Louguet, J. F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS PubMed.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2845–2857 CrossRef CAS.
- H. F. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS PubMed.
- C. Hua, S. M. Peng and C. M. Dong, Macromolecules, 2008, 41, 6686–6695 CrossRef CAS.
- K.-C. Cheng, A. Demirci and J. M. Catchmark, Appl. Microbiol. Biotechnol., 2011, 92, 29–44 CrossRef CAS PubMed.
- R. K. Shukla and A. Tiwari, Carbohydr. Polym., 2012, 88, 399–416 CrossRef CAS.
- R. S. Singh, N. Kaur and J. F. Kennedy, Carbohydr. Polym., 2015, 123, 190–207 CrossRef CAS PubMed.
- I. W. Sutherland, Surface carbohydrates of the prokaryotic cell, Academic Press, 1977 Search PubMed.
- W. M. Kulicke and T. Heinze, Macromol. Symp., 2005, 231, 47–59 CrossRef.
- M. E. Gounga, S. Y. Xu, Z. Wang and W. G. Yang, J. Food Sci., 2008, 73, E155–E161 CrossRef CAS PubMed.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Macromolecules, 2013, 46, 1054–1065 CrossRef CAS.
- A. E. Smith, X. W. Xu, S. E. Kirkland-York, D. A. Savin and C. L. McCormick, Macromolecules, 2010, 43, 1210–1217 CrossRef CAS.
- V. A. Vasantha, S. Jana, S. S. C. Lee, C. S. Lim, S. L. M. Teo, A. Parthiban and J. G. Vancso, Polym. Chem., 2015, 6, 599–606 RSC.
- L. Ilic, K. Jeremic and S. Jovanovic, Eur. Polym. J., 1991, 27, 1227–1229 CrossRef CAS.
- B. V. K. J. Schmidt, M. Hetzer, H. Ritter and C. Barner-Kowollik, Polym. Chem., 2012, 3, 3064–3067 RSC.
- J. Wu, Z. Wang, Y. Yin, R. Jiang, B. Li and A.-C. Shi, Macromolecules, 2015, 48, 8897–8906 CrossRef CAS.
- W. Schärtl, in Light Scattering from Polymer Solutions and Nanoparticle Dispersions, Springer, Berlin, Heidelberg, 2007, ch. 1, pp. 1–24 Search PubMed.
- T. Kato, T. Okamoto, T. Tokuya and A. Takahashi, Biopolymers, 1982, 21, 1623–1633 CrossRef CAS.
- R. H. Lambeth, S. Ramakrishnan, R. Mueller, J. P. Poziemski, G. S. Miguel, L. J. Markoski, C. F. Zukoski and J. S. Moore, Langmuir, 2006, 22, 6352–6360 CrossRef CAS PubMed.
- M. Sedlák, J. Phys. Chem. B, 2012, 116, 2356–2364 CrossRef PubMed.
- Y. Zhu, L. Liu and J. Du, Macromolecules, 2013, 46, 194–203 CrossRef CAS.
- J. S. Lowe, B. Z. Chowdhry, J. R. Parsonage and M. J. Snowden, Polymer, 1998, 39, 1207–1212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic procedures, NMR, SEC, DLS and SLS data. See DOI: 10.1039/c6py02212j |
| This journal is © The Royal Society of Chemistry 2017 |