
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerisation of a terpene-derived lactone: a bio-based alternative to ε-caprolactone†
Helena C.
Quilter
ab,
Marc
Hutchby
b,
Matthew G.
Davidson
b and
Matthew D.
Jones
 *b
*b
aDoctoral Training Centre in Sustainable Chemical Technologies, University of Bath, Bath BA2 7AY, UK
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: mj205@bath.ac.uk
First published on 21st December 2016
Abstract
A high-yielding 4-step process for converting a naturally occurring terpene, β-pinene, into a substituted ε-caprolactone is herein reported. Investigations into the ring-opening polymerisation and copolymerisation of this monomer are also described.
Dwindling fossil fuel supply and growing environmental concerns have increased demand for new renewable, biodegradable plastics.1–4 One such plastic is poly(lactide) (PLA), an aliphatic polyester derived from starchy biomass via ring-opening polymerisation (ROP) of the cyclic monomer lactide (LA).5–8 The physical properties (e.g. rate of degradation, gas permeability) of PLA can be altered by copolymerisation with other cyclic esters, such as petrochemically-derived ε-caprolactone (CL).9 This is an essential method as it widens the usage of PLA in biomedical and engineering applications.10,11 For example, poly(lactide-co-caprolactone) has improved mechanical properties (impact and elongation strengths), and biodegrades more rapidly than PLA alone.12 Renewable routes to ε-caprolactone such as Heeres’ route from HMF (which can be derived from D-fructose)13 or, alternatively, novel bio-derived substituted caprolactones are highly desirable. Caprolactone is an important industrial chemical and is produced on multi-tonne scale via oxidation of cyclohexane.14
A growing source of bio-derived monomers are terpenes and terpenoids.15–17 These naturally occurring molecules are widely utilised in the flavourings and fragrance industries and are the principle component of essential oils in many plants.18 Some of the most commonly occurring terpenes are the cyclic monoterpenes α- and β-pinene (from gum turpentine, crude sulphate turpentine) and D-limonene (from citrus waste), as well as the simplest terpene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :isoprene.15–17,19,20 The abundance of double bonds in terpenes allows for cationic and radical polymerisation, as well as epoxidation as a route to biodegradable oxygenated polymers, as reported for limonene oxide21,22 and α-pinene oxide.23
:isoprene.15–17,19,20 The abundance of double bonds in terpenes allows for cationic and radical polymerisation, as well as epoxidation as a route to biodegradable oxygenated polymers, as reported for limonene oxide21,22 and α-pinene oxide.23
While few examples of terpenoid monomers suitable for ROP are available, Hillmyer and co-workers have reported the use of substituted monomers derived from oxidised menthol24 and carvone25 for the production of functional polyesters and elastomers via block copolymerisation with LA. Building on this work, Winnacker and co-workers reported a method for producing oligoamides from menthone following a similar concept to the synthesis of poly(caprolactam) (nylon 6) from petrochemically-derived cyclohexanone.26 Bio-based polyamide monomers are also highly desirable, with the current scope being somewhat limited, particularly for functionalised polyamides.27
While menthol has shown potential in this area, its annual production of around 19000 tonnes28 is somewhat limiting for development of commodity materials. The production of turpentine, however, is estimated at 350000 tonnes per annum, of which up to almost a third may be β-pinene.29 The work reported herein aims to widen the library of bio-based monomers for ROP available from terpene feedstocks by developing a process of converting abundant, naturally occurring β-pinene into 4-isopropylcaprolactone (4-iPrCL) and its subsequent polymerisation. An intermediate in this process, 4-isopropylcyclohexanone, could also be converted to a lactam via Beckmann rearrangement to prepare a bio-based monomer for functionalised polyamides, similar to nylon. While 4-iPrCL has been previously synthesised as a product in enantioselective Baeyer–Villiger oxidation reactions,30,31 to the best of our knowledge its polymerisation has not been reported nor has it been prepared form a naturally occurring starting material. We herein report the preparation of 4-iPrCL from β-pinene in 4 high yielding steps (see Fig. 1), overall yield 64%.
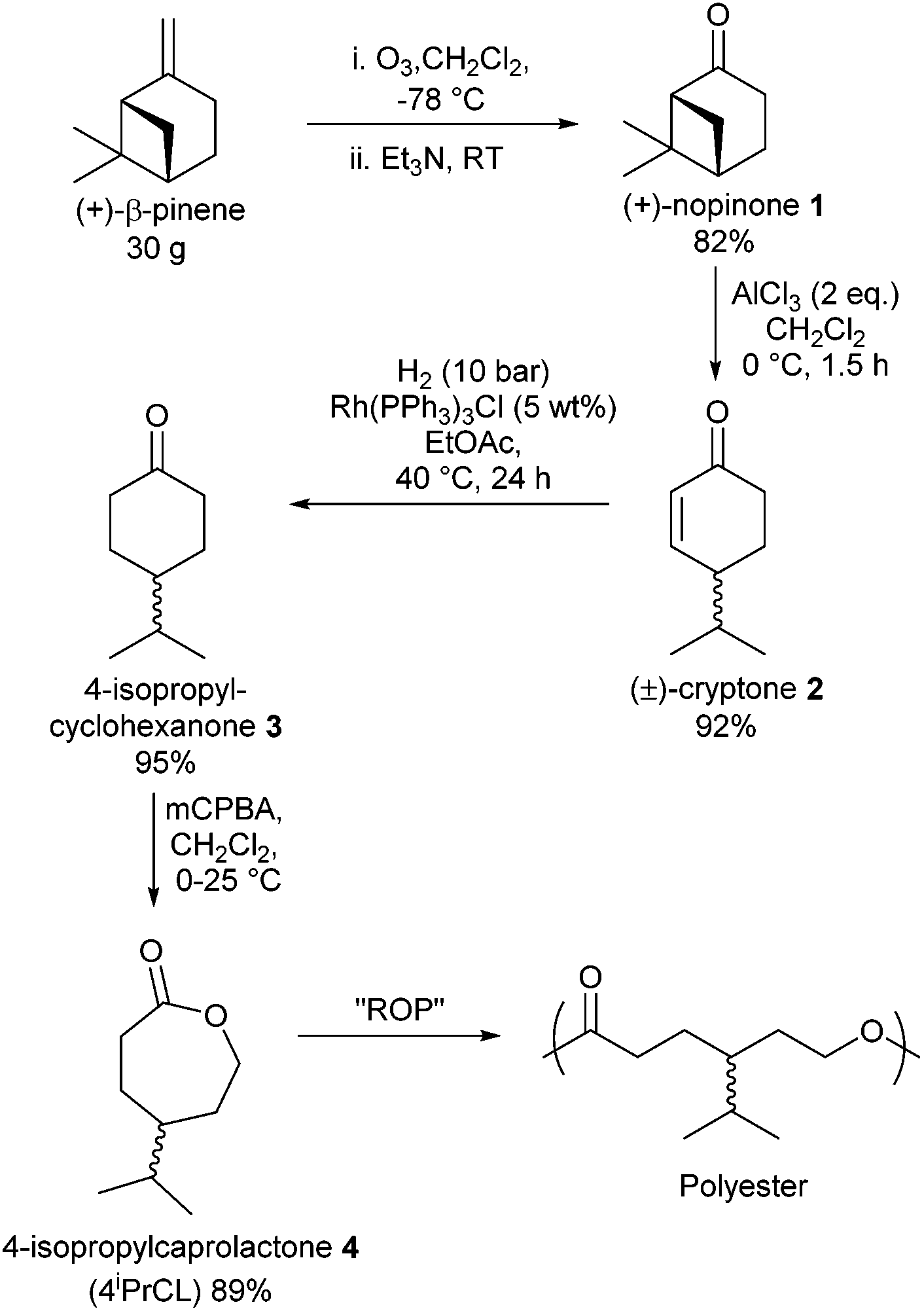 | ||
| Fig. 1 Synthesis of 4-isopropylcaprolactone from β-pinene in 4 steps and its subsequent polymerisation to poly(4-ispropylcaprolactone). | ||
Ozonolysis of β-pinene afforded the terpenoid (+)-nopinone (1) on a 30 g scale, with characterisation matching literature data.32 Lewis acid promoted isomerisation of 133 was found to cleave the strained cis-cyclobutane ring to produce exclusively the monocyclic product (±)-cryptone (2) in high yield. No isomeric by-products were observed on a 10 g scale, evident by the absence of unconjugated alkene resonances in the 1H NMR spectrum. This contrasts with the mixture of alkene products observed in this rearrangement by Mori, who additionally reported a 6-step synthesis of enantiopure (+)-cryptone from another terpenoid:perillyl alcohol.33 Hydrogenation, with Wilkinson's catalyst with H2 at 10 bar and 40 °C in EtOAc gave quantitative conversion of the alkene to the desired saturated ketone 3. Baeyer–Villiger oxidation of 3 or commercially available 4-isopropylcyclohexanone (Fluorochem) with meta-chloroperoxybenzoic acid (m-CPBA) gave the desired lactone 4 in good yields after purification via column chromatography, and was characterised by NMR (1H and 13C{1H}), GC-MS and ESI-MS.† We have also shown it is possible to prepare the oxime from 3 (produced from β-pinene) and the subsequent Beckmann rearrangement yields the lactam, thus this methodology could have benefits for polyamide production.†
This route represents a laboratory scale proof of principle to establish the feasibility of utilization of abundant terpenes for monomer and polyester synthesis. However, it is encouraging that all four steps to achieve monomer, 4, are common transformations that are amenable to scale-up through standard process development methodologies.
A range of four initiators were investigated for the polymerisations (Table 1). Tin(II) 2-ethylhexanoate {Sn(oct)2} and diethylzinc/benzyl alcohol systems, favoured by Hillmyer and Tolman,24,25 were chosen for their industrial relevance. Two zirconium complexes (Fig. 2), which have previously been shown to be stereoselective in the ROP of rac-LA, were chosen to investigate the potential for stereoselective enchainment of the racemic monomer 4. A zirconium amine(trisphenolate) complex, {Zr(tris)(OiPr)}, which shows heterotactic bias with rac-LA34 and a zirconium bipyrollidine-based salan complex, {Zr(bis)(OiPr)2}, which exhibits an isotactic bias with rac-LA.35
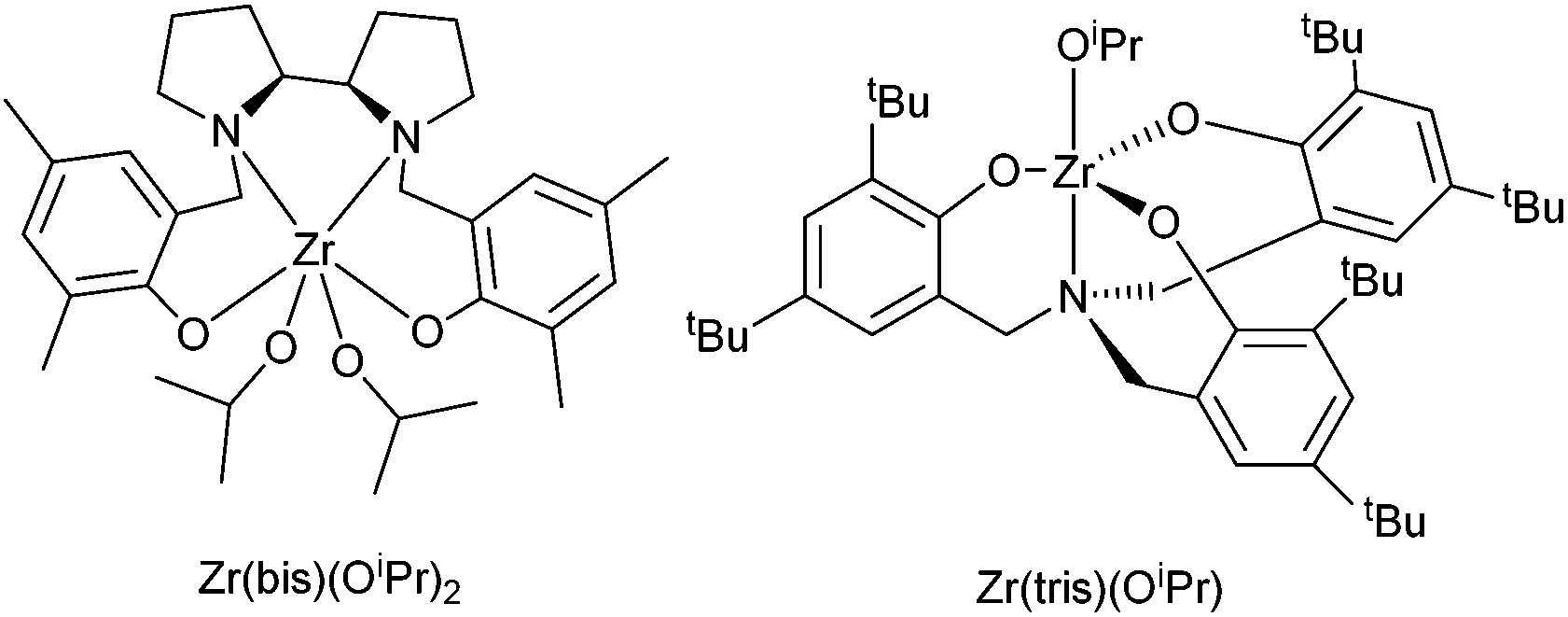 | ||
| Fig. 2 Structures of zirconium initiators used in this study. | ||
| Entry | Initiator | Time/h | Temp./°C | Con.a/% |
M
nb |
PDIb |
|---|---|---|---|---|---|---|
| All polymerisations performed solvent free except entries 4, 6 and 7 which were conducted in toluene. For all entries [M]:[I] = 100.a Determined from the crude 1H NMR spectrum.b Determined by GPC (THF), using polystyrene standards.c Monomer produced from β-pinene. The predicted molecular weight can be calculated by the following equation: (156 × Meq × conversion)/Ieq + end group. |
||||||
| 1 | ZnEt2/BnOH | 2 | 100 | 91 | 22500 |
1.34 |
| 2 | ZnEt2/BnOH | 24 | 80 | 44 | 16700 |
1.19 |
| 3 | ZnEt2/BnOH | 24 | 40 | 14 | 16300 |
1.35 |
| 4 | Sn(oct)2 | 4 | 100 | 74 | 32000 |
1.55 |
| 5 | Zr(bis)(OiPr)2 | 2 | 100 | 99 | 11500 |
1.46 |
| 6 | Zr(bis)(OiPr)2 | 24 | 80 | 88 | 5700 | 1.18 |
| 7 | Zr(tris)(OiPr) | 24 | 100 | 96 | 11400 |
1.42 |
| 8 | Zr(tris)(OiPr) | 24 | 80 | 66 | 3200 | 1.29 |
| 9c | ZnEt2/BnOH | 2 | 100 | 95 | 4900 | 1.54 |
| 10c | Zr(bis)(OiPr)2 | 2 | 100 | 95 | 4900 | 1.52 |
| 11c | Zr(tris)(OiPr) | 24 | 100 | 96 | 3400 | 1.57 |
Generally, polymerisations were performed solvent free at 100 °C or in toluene at 80 °C, with a monomer (M) to initiator (I) ratio of 100:1. Benzyl alcohol was added as co-initiator if required to generate the necessary metal alkoxide for ROP. Polymers were found to be thick, colourless gels with low glass transition temperatures (Tg ≈ −50 °C), see ESI Fig. S18.†1H NMR spectra are consistent with the expected polyester resulting from the ROP of 4, with the structure confirmed by MALDI-TOF analysis (Table 1, entry 6), showing repeat units of 156.20 Da, as expected. A series was observed which was capped by an isopropoxide group via insertion from the initiator. A second series was also evident, corresponding to cyclic oligomers – the product of intramolecular transesterification, a common competing side-reaction in ROP.
The ROP of 4 (prepared with commercial 3) with ZnEt2/BnOH was found to proceed rapidly at 100 °C, with quantitative conversion to polymer within 2 h. {Sn(oct)2} afforded a conversion of 74% conversion after 4 hours although a broad PDI and exaggerated Mn could be indicative of transesterification. Kinetics with ZnEt2/BnOH were investigated by varying the time intervals for polymerisation, giving a first order rate constant, kapp of 1.7 × 10−2 min−1 (ESI, Fig. S13†). The polymerisation was found to be poorly controlled in terms of molecular weight and polydispersity, which led to studies with the Zr(IV) complexes.
The polymerisation of β-pinene-derived 4 was performed under analogous conditions (Table 1, entries 9–11). The conversion was observed to be similar to the monomer from commercial 4-iPrCL, although the molecular weights of the polymers were reduced and broader polydispersities were observed. The ROP of 4 with {Zr(tris)(OiPr)} (100 °C, solvent-free, [4]:[Zr] = 100) was further investigated, from commercial 3. Reasonable control over the polymerisation was observed (Fig. 3). The Mn was found to increase in a linear fashion with conversion, while the PDI remained fairly constant, although some control was lost at high conversions. Near quantitative conversion (96%) was observed after 24 h with [M]:[I] = 100, with rate constant, kapp = 2.4 × 10−3 min−1 (see ESI, Fig. S14†). Solution-state kinetics were also investigated on an NMR scale with this system in d8-toluene at 80 °C, with the kapp found to be slower by an order of magnitude than for solvent-free (kapp = 2.0 × 10−4 min−1, ESI Fig. S15†). The controlled nature of the polymerisation was further investigated by varying [M]:[I]. While generally the Mn was observed to increase relative to the concentration of monomer, at lower initiator loadings, the polymerisation did not reach completion even after 72 h. DSC analysis of these polymer products showed slight variation in the Tg with the molecular weight of the polymer (ESI, Table S1†). The rate of polymerisation with {Zr(bis)(OiPr)2} was investigated in d8-toluene at 60 °C and found to proceed readily (kapp = 29.6 × 10−3 min, see ESI Fig. S16†), polymer from commercial 3.†
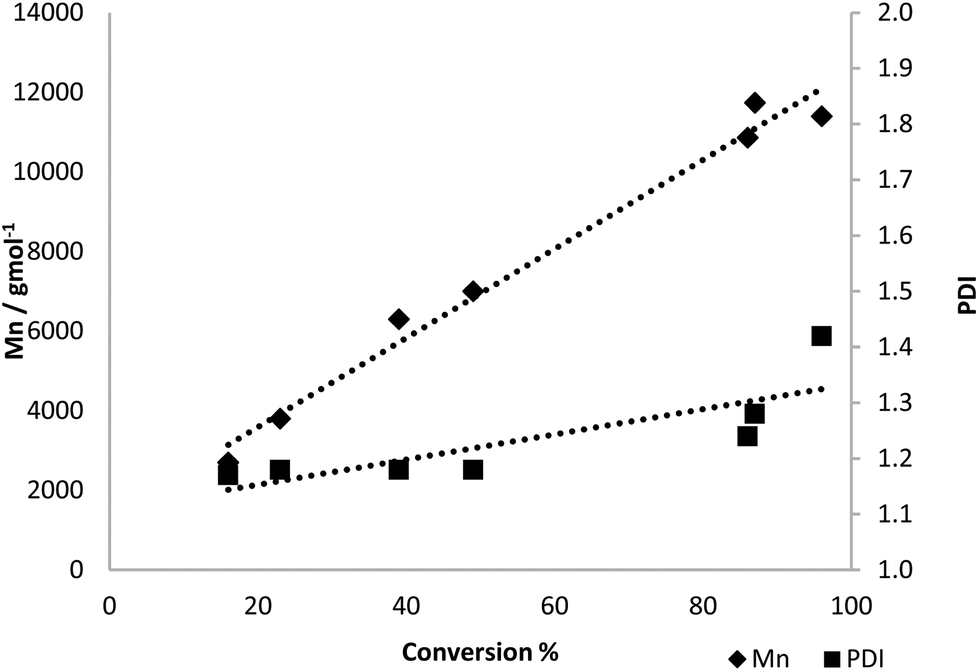 | ||
| Fig. 3 Graph showing the relationship between conversion and Mn or PDI. Conditions = [4]:[{Zr(tris)(OiPr)}] = 100, 100 °C, solvent-free, 0–24 h. Using 4 derived from commercial 3. | ||
Copolymerisation studies with L- and rac-LA were also conducted, with the aim of preparing copolymers with different ratios of each monomer via “one-pot” or sequential addition respectively (see Table 2). Investigations were initially conducted with L-LA to produce purely isotactic LA regions in polymer products. In all cases the conversion of L-LA was higher than for 4, particularly for entries with higher [LA]:[4], indicating that the relative rate of enchainment for lactide monomers is faster than for the substituted caprolactone. Polymer products were generally found to be low molecular weight. Copolymerisations were then attempted with {Zr(bis)(OiPr)2} – both neat and in solution (Table 2, entries 5–10) with 4-iPrCL derived from β-pinene. In all cases a high conversion of both LA and 4 was observed, and DOSY analysis of a random (“one-pot”) copolymer (Table 2, entry 14) was consistent with copolymer formation (see ESI, Fig. S25†). Attempts to sequentially form block copolymers (Table 2, entries 11–13) were performed with both zirconium-based initiators. Again, higher conversion of both monomers was observed with {Zr(bis)(OiPr)2}. When monomer 4 was added first followed by the addition of LA after 24 h (entry 12) very low conversion of both monomers was observed. DSC analysis (Table 2, entries 11 & 13) were found to have Tm = 137.9 °C and 139.3 °C, slightly lower than expected for pure PLLA. However, DOSY analysis of entry 13 (see ESI, Fig. S28†) showed the polymer product to be a mixture of poly(4) and poly(lactide), rather than a block copolymer.
| Entry | Initiator | LA | [LA]:[4] |
Time/h | Temp./°C | Con.a/% LA | Con.a/% 4 |
M
nb |
PDIb |
|---|---|---|---|---|---|---|---|---|---|
| All polymerisations performed neat except entries 8, 9 and 10 which were conducted in toluene.a Determined from analysis of the crude 1H NMR spectrum.b As determined by GPC (THF), using polystyrene standards.c LA added first, addition of 4-iPrCL after 24 h.d 4-iPrCL added first.e 4-iPrCL monomer prepared from β-pinene. | |||||||||
| 1 | Zr(tris)(OiPr) | L-LA | 25:75 |
48 | 100 | 99 | 90 | 4540 | 1.42 |
| 2 | Zr(tris)(OiPr) | L-LA | 50:50 |
48 | 100 | 99 | 90 | 3800 | 1.46 |
| 3 | Zr(tris)(OiPr) | L-LA | 75:25 |
48 | 100 | 85 | 45 | 4570 | 1.23 |
| 4 | Zr(tris)(OiPr) | L-LA | 85:15 |
48 | 100 | 85 | 44 | 6870 | 1.32 |
| 5e | Zr(bis)(OiPr)2 | L-LA | 50:50 |
2 | 100 | 96 | 89 | 4900 | 2.00 |
| 6e | Zr(bis)(OiPr)2 | L-LA | 25:75 |
2 | 100 | 98 | 91 | 4600 | 1.79 |
| 7e | Zr(bis)(OiPr)2 | L-LA | 75:25 |
2 | 100 | 98 | 87 | 7500 | 1.67 |
| 8e | Zr(bis)(OiPr)2 | L-LA | 50:50 |
6 | 80 | 96 | 84 | 4600 | 1.42 |
| 9e | Zr(bis)(OiPr)2 | L-LA | 25:75 |
6 | 80 | 96 | 89 | 3350 | 1.51 |
| 10e | Zr(bis)(OiPr)2 | L-LA | 75:25 |
6 | 80 | 96 | 80 | 6860 | 1.39 |
| 11c | Zr(tris)(OiPr) | L-LA | 50:50 |
24 + 24 | 80 | 99 | 31 | 7900 | 1.15 |
| 12d | Zr(tris)(OiPr) | L-LA | 50:50 |
24 + 24 | 80 | 22 | 37 | 2020 | 2.32 |
| 13c | Zr(bis)(OiPr)2 | rac-LA | 50:50 |
24 + 4 | 80 | 93 | 91 | 6220 | 1.19 |
| 14 | Zr(bis)(OiPr)2 | rac-LA | 50:50 |
24 | 80 | 92 | 74 | 9160 | 1.72 |
We have reported the preparation of 4-isopropyl-ε-caprolactone as a new monomer for ROP. This monomer can be prepared on a multigram scale from an abundant, cheap, renewable feedstock: β-pinene. In this paper we have used 4 derived directly from β-pinene or from commercially available 3. The polymer fully produced via the β-pinene route has a lower Mn, this may well be due to small contaminants from the ozonolysis step, which then act as chain transfer agents. Work is on-going to investigate the nature of this apparent difference.
Investigation of its polymerisation to a low-Tg polymer highlights its potential application as a bio-based alternative to petrochemically-derived ε-caprolactone. Promising copolymerisation data with rac-LA and L-LA suggest that 4 provides a means of enhancing the range of available renewable aliphatic polyester materials. Work is ongoing to further investigate the potential for copolymerisation with this monomer and to optimise a synthetic route to facilitate the scale-up of the process from, with particular emphasis on the Baeyer–Villiger oxidation step. The polymers reported herein complement and expand the existing body of literature concerning the utilisation of terpenes. Recent work has focused on the preparation of epoxides or anhydrides from terpenes as a route to producing polycarbonates or polyesters.36–40 In these reports the thermal properties of the polymer have been modified by the incorporation of a renewable aliphatic moiety. This research is generally focused on production of high Tg materials, whereas the work reported herein also demonstrates their utility as drop in replacements for low Tg polymers. For terpene based monomers to fulfil their potential several strands of research are necessary.
We wish to thank the EPSRC for funding (i) EP/L016354/1, CDT in sustainable chemical technologies (HQ and MGD); (ii) EP/K014889/1, terpene-based manufacturing for sustainable chemical feedstocks (MH, MDJ, MGD); (iii) the mass spectrometry service in Swansea for MALDI-ToF facilities.
Notes and references
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS PubMed.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- M. Hiljanen-Vainio, T. Karjalainen and J. Seppälä, J. Appl. Polym. Sci., 1996, 59, 1281–1288 CrossRef CAS.
- R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS.
- L. Wang, Z. Zhang, H. Chen, S. Zhang and C. Xiong, J. Polym. Res., 2010, 17, 77–82 CrossRef CAS.
- J. M. Becker and A. P. Dove, in Green Polymerization Methods, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 201–220 Search PubMed.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Gröger, A. Liese, H.-G. Herz and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2015, 54, 2784–2787 CrossRef CAS PubMed.
- M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS PubMed.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- J. M. Bolton, M. A. Hillmyer and T. R. Hoye, ACS Macro Lett., 2014, 3, 717–720 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- J. Zhao and H. Schlaad, in Bio-synthetic Polymer Conjugates, Springer, Berlin, Heidelberg, 2012, pp. 151–190 Search PubMed.
- M. Golets, S. Ajaikumar and J.-P. Mikkola, Chem. Rev., 2015, 115, 3141–3169 CrossRef CAS PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo and G. W. Coates, Macromolecules, 2015, 48, 2534–2550 CrossRef CAS.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef PubMed.
- D. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS PubMed.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- M. Winnacker, S. Vagin, V. Auer and B. Rieger, Macromol. Chem. Phys., 2014, 215, 1654–1660 CrossRef CAS.
- M. Winnacker and B. Rieger, Macromol. Rapid Commun., 2016, 37, 1391–1413 CrossRef CAS PubMed.
- J.-G. Yin, G.-C. Xu, G.-W. Zheng and J.-H. Xu, Appl. Biochem. Biotechnol., 2015, 176, 1102–1113 CrossRef CAS PubMed.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1–39 CrossRef CAS.
- L. Zhou, X. Liu, J. Ji, Y. Zhang, X. Hu, L. Lin and X. Feng, J. Am. Chem. Soc., 2012, 134, 17023–17026 CrossRef CAS PubMed.
- B. G. Kyte, P. Rouvière, Q. Cheng and J. D. Stewart, J. Org. Chem., 2004, 69, 12–17 CrossRef CAS PubMed.
- T. Szuppa, A. Stolle, B. Ondruschka and W. Hopfe, ChemSusChem, 2010, 3, 1181–1191 CrossRef CAS PubMed.
- K. Mori, Tetrahedron: Asymmetry, 2006, 17, 2133–2142 CrossRef CAS.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC.
- M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schäfer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- C. Martín and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 CrossRef.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 CrossRef CAS PubMed.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- M. Reiter, S. Vagin, A. Kronast, C. Jandl and B. Rieger, Chem. Sci., 2017, 43, 6618–6639 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterisation of monomer and polymer. See DOI: 10.1039/c6py02033j |
| This journal is © The Royal Society of Chemistry 2017 |