
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-stimuli responsive block copolymers as a smart release platform for a polypyridyl ruthenium complex†
Michael
Appold
a,
Cristina
Mari
 b,
Christina
Lederle
c,
Johannes
Elbert‡
a,
Claudia
Schmidt
d,
Ingo
Ott
d,
Bernd
Stühn
c,
Gilles
Gasser
*e and
Markus
Gallei
*a
b,
Christina
Lederle
c,
Johannes
Elbert‡
a,
Claudia
Schmidt
d,
Ingo
Ott
d,
Bernd
Stühn
c,
Gilles
Gasser
*e and
Markus
Gallei
*a
aErnst-Berl-Institute for Chemical Engineering and Macromolecular Chemistry, Technische Universität Darmstadt, Alarich-Weiss-Straße 4, D-64287 Darmstadt, Germany. E-mail: M.Gallei@mc.tu-darmstadt.de
bDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
cInstitute of Condensed Matter Physics, Technische Universität Darmstadt, Hochschulstraße 8, D-64289 Darmstadt, Germany
dInstitute of Medicinal and Pharmaceutical Chemistry, Technische Universität Braunschweig, Beethovenstr. 55, D-38160, Germany
eChimie ParisTech, PSL Research University, Laboratory for Inorganic Chemical Biology, F-75005 Paris, France. E-mail: gilles.gasser@chimie-paristech.fr
First published on 20th December 2016
Abstract
A variety of applications of amphiphilic block copolymers result from the control of their self-assembled structures. Herein, the synthesis and structure formation of block copolymers (BCPs) consisting of poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) as one segment and poly(methyl methacrylate) (PMMA) or a statistical copolymer (PDMAEMA-co-PMMA) as a second segment, is described. The BCPs provide molar masses between 8.9 kg mol−1 and 35.6 kg mol−1 with low polydispersity index values, Đ = 1.05–1.13. BCPs are synthesized via sequential anionic polymerization strategies while structure formation in water is investigated by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The PDMAEMA-containing micelles in water are loaded with a Ru(II) polypyridyl complex, i.e. [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (bipy = 2,2′-bipyridine; dppz = dipyridophenazine), which was previously shown to act as a potential photosensitizer in photodynamic therapy (PDT). Successful loading of the BCP micelles is evidenced by TEM measurements after dialysis in water. Stimulus-responsive release of the Ru(II) complex from the BCP micelles is shown using ultrasound, change of pH or temperature as external triggers. The quantification and release profiles for the Ru(II) complex are obtained by atomic absorption spectrometry (AAS). As a result, PDMAEMA-b-PMMA is not capable of releasing the Ru(II) complex in a controlled manner after application of, for instance, ultrasound or temperature change as external triggers due to the shielding (stealth effect) of the BCP. On the contrary, micelles made of BCPs featuring PDMAEMA and PDMAEMA-co-PMMA segments reveal excellent Ru(II) complex release profiles due to the tailored molecular composition of the underlying block segments as evidenced by temperature-dependent DLS and AAS measurements. Thus, these smart PDMAEMA-containing BCPs pave the way to a variety of applications for selective triggered release of small molecules.
Introduction
In the last decade stimuli-responsive polymers have proven their utility for a range of important applications.1–3 These types of polymers can be designed to feature certain chemical functionalities, which can be changed by invasive or non-invasive external triggers. They are capable of changing their conformation, solubility, or even of breaking or forming covalent bonds upon, for instance, a change of temperature or pH value, light irradiation, electrochemical stimuli or the presence of an electrical field, or combinations thereof.4–11 Polymer-based nanocontainers filled with payloads are promising systems for selective and gated transport in the fields of, e.g., biomedicine,12–14 or anticorrosion.15,16 The preparation of block copolymers (BCPs), i.e., polymers consisting of two or more homogeneous polymer fragments that are covalently connected, feature the intrinsic capability for microphase separation yielding fascinating structures in the bulk state or in selective solvents. Encapsulation of molecules and oligomers has already been carried out in BCP micelles, vesicles or BCP capsules featuring even more complex substructures.17–20 For instance, multicompartment micelles were successfully prepared from triblock terpolymers and their fascinating self-assembled structures were studied in different media.21–24 Furthermore, in our first attempts, the selective release of two different payloads out of such complex BCP micelles featuring different compartments has been studied.25 Amongst all potential external triggers for selective release of payloads, thermal and pH value changes have been intensively investigated owing to their important role in physiological environments. Within the field of stimuli-responsive polymers, poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) is one major player due to its capability for rapid conformational changes during the change of temperature, pH (or the presence of CO2), and ionic strength.26–28 As another external trigger ultrasound is reported to be a potential stimulus for drug release from polymer micelles.29 The release can occur by diffusion of the payload or by degradation triggered by ultrasound. For example, the release of doxorubicin from Pluronic micelles could be achieved by insonation with low frequency ultrasound as reported by Husseini et al.30 or Marin et al.31Like other cationic polymers, PDMAEMA is reported to be slightly cytotoxic and changes regarding the molecular architecture (for example copolymers with hydrophobic or hydrophilic monomers, branched system, chain ends) as well as the overall molar mass can significantly influence the cytotoxicity.32–35 Moreover, it is reported that the masking effects of PDMAEMA complexes play a crucial role in the cytotoxicity.34 To the best of our knowledge, the use of PDMAEMA as a multi-stimuli responsive polymer (and BCP thereof) has not been reported for the selective release of photodynamic therapy (PDT) photosensitizers. PDT is a medical method used to treat certain types of cancer and other skin conditions.36,37 A photodynamic effect is achieved due to the synergistic action of light, molecular oxygen normally present in tissues (3O2) and a photosensitizer. In detail, the first step is usually the intravenous administration of the photosensitizer to the patient. After 1–3 days, when the photosensitizer has localized in cancer cells, the photosensitizer is excited by light irradiation. The photosensitizer reaches its excited state, which has a triplet character. The excited photosensitizer can then interact with 3O2. Upon energy transfer from the excited state of the photosensitizer to the ground state of 3O2, a very reactive singlet oxygen species (1O2) is formed. This toxic form of oxygen is able to rapidly interact and damage the surrounding biological substrates, inducing a biological cascade of events that will eventually lead to cell death. PDT has a very localized toxic effect since 1O2 is produced just at the site of light irradiation. Moreover, since its lifetime is very short in a biological environment (40 ns), 1O2 will exert its toxic activity just in the proximity of its site of generation.38 The temporally and spatially defined formation of these toxic species is responsible for the low side effects of PDT compared to those that cancer patients have to usually go through during chemotherapeutic treatment. The characteristics of the photosensitizer are of crucial importance for the medical outcomes. Nowadays, most of the clinically approved photosensitizers have a tetrapyrrolic structure (i.e. porphyrins, chlorins).39,40 This class of molecules is known for their very high 1O2 production, long wavelength excitation and high cancer tissue accumulation. However, some drawbacks associated with these systems (i.e. non-trivial synthesis and purification, scarce water solubility, systemic accumulation) highlight the importance for the development of alternative photosensitizers. A great effort has been made in the last few decades to develop novel photosensitizers with a non-porphyrin based structure. Among the different types of compounds investigated, Ru(II) polypyridyl complexes were found to be extremely promising.37,41–45 Encouragingly, such a complex will soon enter into clinical trials as a photosensitizer for the treatment of non-muscle invasive bladder cancer.46 At our end, we have focused our attention, over the past few years, in the application of metal complexes as photosensitizers in PDT for the treatment of cancer47–49 and bacterial infections (aPDT)50 as well as in photoactivated chemotherapy (PACT).51,52 In particular, some Ru(II) polypyridyl complexes showed very promising results with, among others, an efficient cellular uptake and very high phototoxic indexes (PI, the ratio of dark toxicity over phototoxicity).37 Another important drawback of the tetrapyrrolic compounds used as PDT photosensitizers is related to their poor bioavailability. Porphyrins and porphyrin molecules are generally characterized by low water solubility and high lipophilicity, generating problems with regard to medicinal/biological applications. To overcome these issues, the use of delivery systems was envisaged to improve the pharmacokinetic properties of the photosensitizers. As mentioned above for other payloads, a wide variety of carriers for photosensitizers have been applied, ranging from liposomes to micelles or organic or inorganic nanoparticles or polymers.53,54 As an example, FOSLIP®, a water-soluble liposomal formulation of the photosensitizer FOSCAN® ((m-tetra(hydroxyphenyl) chlorin), a PDT photosensitizer approved in Europe) currently in preclinical tests, displayed rapid biodistribution and clearance from the bloodstream, together with selective tumor accumulation and reduced side effects.54–56 Worth mentioning, although not related to cancer therapy, is the case of Vysudine®, in which the photosensitizer verteporfin is formulated as a green liposome powder. It was approved by the FDA as a photosensitizer for the treatment of age-related macular degeneration in 2000 and for the treatment of pathological myopia in 2001.54 Of note, Lemercier et al. reported the encapsulation of two Ru(II)-based photosensitizers in poly(D,L-lactide-co-glycolide) nanoparticles. The free complexes were characterized by very high dark toxicity, which was strongly reduced upon encapsulation.57
Herein, we present the encapsulation of a Ru(II) polypyridyl complex, i.e., [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (bipy = 2,2′-bipyridine; dppz = dipyridophenazine), which was previously shown to be a promising PDT photosensitizer by some of us.47 PDMAEMA-based block copolymers with either poly(methyl methacrylate) (PMMA) or a statistical copolymer (PMMA-co-PDMAEMA) as a second block segment were prepared by sequential anionic polymerization strategies. The thermo-responsiveness of PDMAEMA-containing block copolymer (BCP) micelles was investigated in detail by temperature-dependent dynamic light scattering (DLS) measurements in water. The micelles were loaded with the Ru(II) polypyridyl complex and the release was triggered by ultrasound, pH and temperature variations. The Ru(II)complex-loaded and empty BCP micelles were characterized with respect to structure formation by transmission electron microscopy (TEM) and DLS, while the release profiles were obtained after dialysis via atomic absorption spectrometry (AAS).
Results and discussion
Polymer synthesis and characterization
For encapsulation of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2, block copolymers consisting of PMMA and poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) were investigated. The stimuli-responsive block copolymers PMMA-b-PDMAEMA 4 were synthesized via anionic polymerization in THF with diphenylhexyllithium (DPHLi) as an initiator – prepared by the nucleophilic addition of n-butyl lithium to 1,1-diphenylethylene (DPE) – at low temperatures (see the Experimental section). The synthesis is depicted in Fig. 1 (top).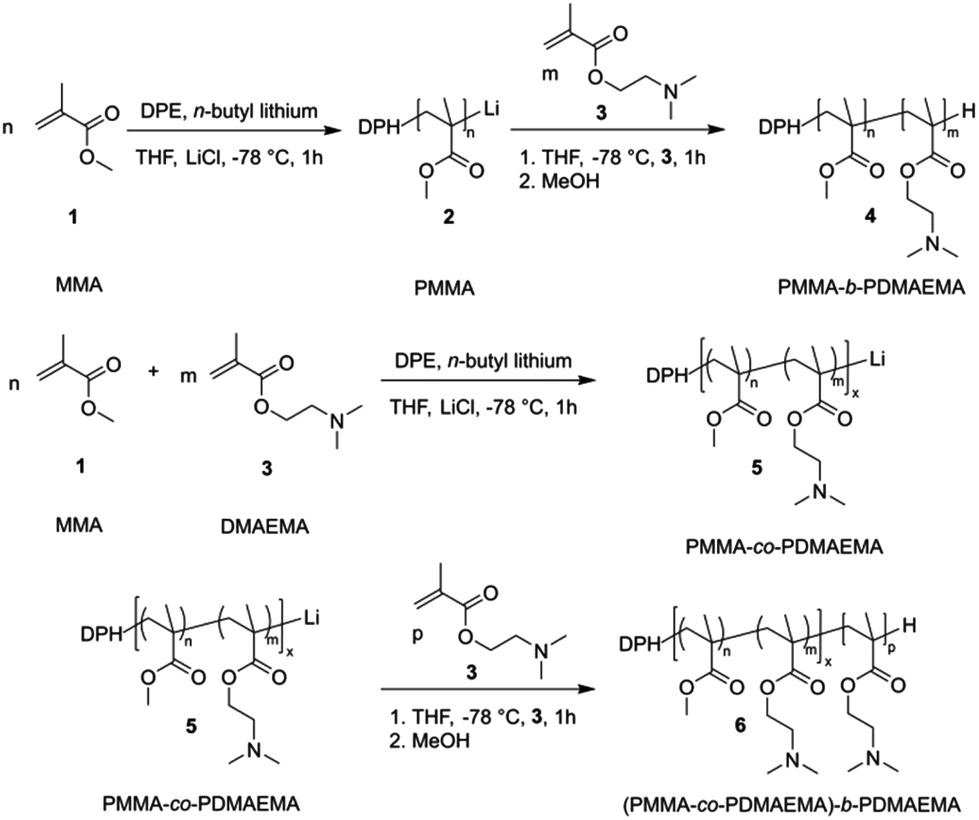 | ||
| Fig. 1 Top: Anionic block copolymerization of methyl methacrylate 1 (MMA) and N,N-dimethylaminoethyl methacrylate 3 (DMAEMA) in THF in the presence of LiCl at low temperature to yield PMMA-b-PDMAEMA 4. Bottom: MMA and DMAEMA were copolymerized by means of anionic polymerization followed by the addition of DMAEMA to generate the corresponding PMMA-co-PDMAEMA-b-PDMAEMA 6. | ||
The corresponding PMMA-b-PDMAEMA BCPs 4 were analyzed by size exclusion chromatography (SEC) in dimethylformamide (DMF) and 1H NMR spectroscopy. Exemplarily, the results on the sequential anionic polymerization of DMAEMA and MMA for the preparation of PMMA54-b-PDMAEMA95 are given. In Fig. 2, the molar mass distribution of the PMMA macro precursor (black) and the corresponding PMMA-b-PDMAEMA is shown, evidencing the successful BCP formation without significant termination of the PMMA anionic macro initiator. Additionally, the corresponding 1H NMR spectrum for the final BCP with signal assignments is given in Fig. 2 (bottom).
 | ||
| Fig. 2 Molar mass distributions (top) obtained by SEC measurements vs. PMMA standards in DMF obtained for the PMMA54 precursor (black line) and PMMA54-b-PDMAEMA95 (red line). The 1H NMR spectrum of PMMA54-b-PDMAEMA95 in CDCl3 (bottom). | ||
All obtained data for the PMMA homopolymers and block copolymers – with PDMAEMA or PDMAEMA-co-PMMA as a second block segment – investigated in this study comprising Mn, Mw, and Đ are compiled in Table 1.
| Polymer |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kg mol−1) a (kg mol−1) |
M
wa (kg mol−1) |
Đ |
|---|---|---|---|
| a Molecular weight determined by SEC in kg mol−1(PMMA standards, DMF). b Molecular weight in kg mol−1 determined by SEC in DMF and the relative composition for PDMAEMA and PMMA determined by 1H NMR spectroscopy in CDCl3. | |||
| PMMA63a |
6.3 | 6.4 | 1.02 |
| PMMA63-b-PDMAEMA25a |
8.5 | 8.9 | 1.15 |
| PMMA54a |
5.4 | 5.8 | 1.08 |
| PMMA54-b-PDMAEMA95a |
10.0 | 10.4 | 1.03 |
| PMMA19-co-PDMAEMA22b |
5.4 | 6.0 | 1.11 |
| (PMMA19-co-PDMAEMA22)-b-PDMAEMA191b |
31.8 | 35.8 | 1.13 |
| PMMA13-co-PDMAEMA22b |
4.7 | 5.3 | 1.11 |
| (PMMA13-co-PDMAEMA22)-b-PDMAEMA132b |
26.9 | 29.6 | 1.10 |
As described in the ensuing sections, structure formation in water as well as capability for the stimuli-responsive release of payloads were initially carried out with PMMA-b-PDMAEMA BCPs. Based on these results, the usability of PDMAEMA BCPs having a PDMAMEA-co-PMMA segment was taken into account. For this purpose, the mixtures of MMA and DMAEMA were used prior to the formation of the PDMAEMA block to yield polymer 6 (see the Experimental section and Fig. 1). An exemplary molar mass distribution of (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 is given as Fig. S1 in the ESI,† while the corresponding 1H NMR spectrum is given as Fig. S2.† Data on SEC and 1H NMR spectroscopy proved the successful synthesis of the PDMAEMA-containing BCP (see also Table 1), which will be investigated in the following sections. Additionally, thermal phase behavior of the BCPs was investigated by DSC measurements revealing glass transition temperatures of approx. 30 °C and 90–100 °C which could be assigned to PDMAEMA or PDMAEMA-co-PMMA segments, respectively.58,59 All data on SEC, NMR spectroscopy and DSC measurements for the investigated BCPs and BCPs with a statistical segment are given in the ESI as Fig. S3–S10.†
As the thermo-responsiveness of the PDMAEMA-containing segment was of major interest, the ratio of MMA and DMAEMA was varied in order to tailor the LCSTs. For this purpose, the weight content of MMA was varied from 4.5 wt%, over 17 wt% to 26.9 wt% and UV-Vis spectroscopy measurements were carried out to determine the LCST (Fig. S11†). As can be concluded from these experiments, the LCST for the statistical PDMAEMA-co-PMMA could be tailored in the range of 17 °C to approximately 35 °C. A rough trend for the relationship of the LCST with the increasing MMA content is given as Fig. S12 in the ESI.† All data on glass transition temperatures and LCSTs for the investigated BCPs in this study are compiled in Table S1 of the ESI.†
Structure formation of PDMAEMA-containing block copolymers in water
Before the stimulus-induced release of a PDT agent payload is studied in more detail, structure formation of the PDMAEMA-based diblock copolymers in a block-selective solvent for PDMAEMA, i.e. water, was investigated. In general, due to their amphiphilic nature, the BCPs are capable of self-assembling into micelles in aqueous solution.60–62 We investigated the micellation in the presence or absence of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 complex 7 as depicted in Scheme 1 and as described in the Experimental section. In brief, BCPs (and complex 7) were diluted in a small amount of acetone followed by dropwise addition of water (Milli-Q) and dialysis. In Fig. S13,† a photograph of the BCP micelles in water is given. In the case of BCP micelles loaded with [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 complex 7, a slightly yellow to orange color could be observed after dialysis, while in the case of the unloaded micelles, no turbidity or color was observed. This was a first hint that entrapment of Ru(II) complex 7 was successful. | ||
| Scheme 1 Simplified illustration of BCP micelle formation for PMMA-b-PDMAEMA and (PMMA-co-PDMAEMA)-b-PDMAEMA BCPs in water. The micelles were directly compared by using TEM measurements in the presence of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 complex 7 (see the text). | ||
In order to confirm these findings, comparative TEM measurements of the empty BCP micelles, which were formed by the polymer (PMMA13-co-PDMAEMA22)-b-PDMAEMA132, and BCP micelles loaded with Ru(II) complex 7 were carried out (Fig. 3). For TEM measurements, one drop of the resulting micellar solution was deposited on a carbon-coated copper grid, and the solvent was removed in vacuo. Interestingly, the empty BCP micelles could not be observed during TEM measurements, potentially due to the weak electron contrast of the micelles compared to the carbon background of the coated TEM copper grids. However, after staining with iodine the empty micelles of the statistical block copolymer (PMMA13-co-PDMAEMA22)-b-PDMAEMA132, which will be important for the thorough DLS measurements in the ensuing section, could be observed using TEM (Fig. S14†). The average diameter of the stained spherical micelles was determined to be 8–9 nm. Compared to this, the corresponding TEM images for the loaded micelles of PMMA54-b-PDMAEMA95 (Fig. 3A and B) and both BCPs featuring a statistical PDMAEMA-co-PMMA as the second block segment, i.e., (PMMA19-co-PDMAEMA22)-b-PDMAEMA191 (Fig. 3C and D) and (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 (Fig. 3E and F) are shown. In this case, for the loaded BCP micelles, no further treatment was necessary to increase the contrast due to the high electron density of Ru(II) complex 7.
 | ||
| Fig. 3 TEM images of Ru(II) complex 7 loaded BCP micelles PMMA54-b-PDMAEMA95 (A and B), (PMMA19-co-PDMAEMA22)-b-PDMAEMA191 (C and D) and (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 (E and F) obtained after dialysis, drop-casting on carbon-coated copper grids and without staining. | ||
The TEM images for PMMA54-b-PDMAEMA95 clearly revealed a core–shell structure for the BCP micelles. The dark appearing core was attributed to the presence of Ru(II) complex 7 (Fig. 3A and B). Compared to this, both BCPs with a statistical PDMAEMA-co-PMMA segment did not feature an observable core–shell structure and homogeneous micelles could be observed (Fig. 3C–F). Summarizing the results of TEM measurements on the loaded BCP micelles, the herein investigated PDMAEMA-containing BCPs are capable of encapsulating the photosensitizer in the interior of the micellar structure in an aqueous environment. Before we turn to the Ru(II) complex 7 release, temperature-dependent dynamic light scattering (DLS) experiments are performed and discussed below.
Thermoresponsive behaviour of BCP micelles by dynamic light scattering (DLS) measurements
The temperature-responsiveness of the BCP micelles in water was followed using temperature-dependent dynamic light scattering (DLS) measurements. It is worth mentioning that the focus was on investigating the polymer (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 as the model system, which has the highest ratio of PDMAEMA in the statistical block segment (as compared for all polymers in Table S2†) in this study. As will be described in the ensuing section, the polymer PMMA54-b-PDMAEMA95 was not able to release a significant amount of Ru(II) complex 7 after 90 min of ultrasound, while (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 featured a reasonable leakage of the Ru(II) complex already at room temperature. Compared to this, the polymer (PMMA19-co-PDMAEMA22)-b-PDMAEMA191 having a lower ratio of PDMAEMA in the statistical block segment (as compared for all polymers in Table S2†) featured the best properties with respect to the payload release applying external triggers, i.e., by applying ultrasound, changing the pH value and temperature variation. We started studying the thermo-responsive behaviour of (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 in more detail in order to clarify the leakage occurring already at room temperature. All detailed measurement conditions for the DLS experiments are given in Table S3 of the ESI.†Fig. 4 illustrates two examples of the measured intensity autocorrelation functions for the micelles at 20 °C and 6 °C. Note that the decay of the correlation functions shows clearly two steps. The faster process corresponds to the translation dynamics of uniform micelles formed from (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 chains in water, while the slower process belongs to the dynamics of some micellar aggregates. For a direct comparison, the drop-cast and dried micelles were investigated by TEM as already described in the section above (Fig. S13†). From the TEM images it was concluded that the spherical micelles featured an average diameter of approximately 8–9 nm. For DLS measurements, both processes are well separated on the time scale and it is therefore possible to analyse these two dynamics in detail. To analyse the temperature-responsiveness of the hydrodynamic radii of the micelles it is necessary to calculate the correlation time τ for every process.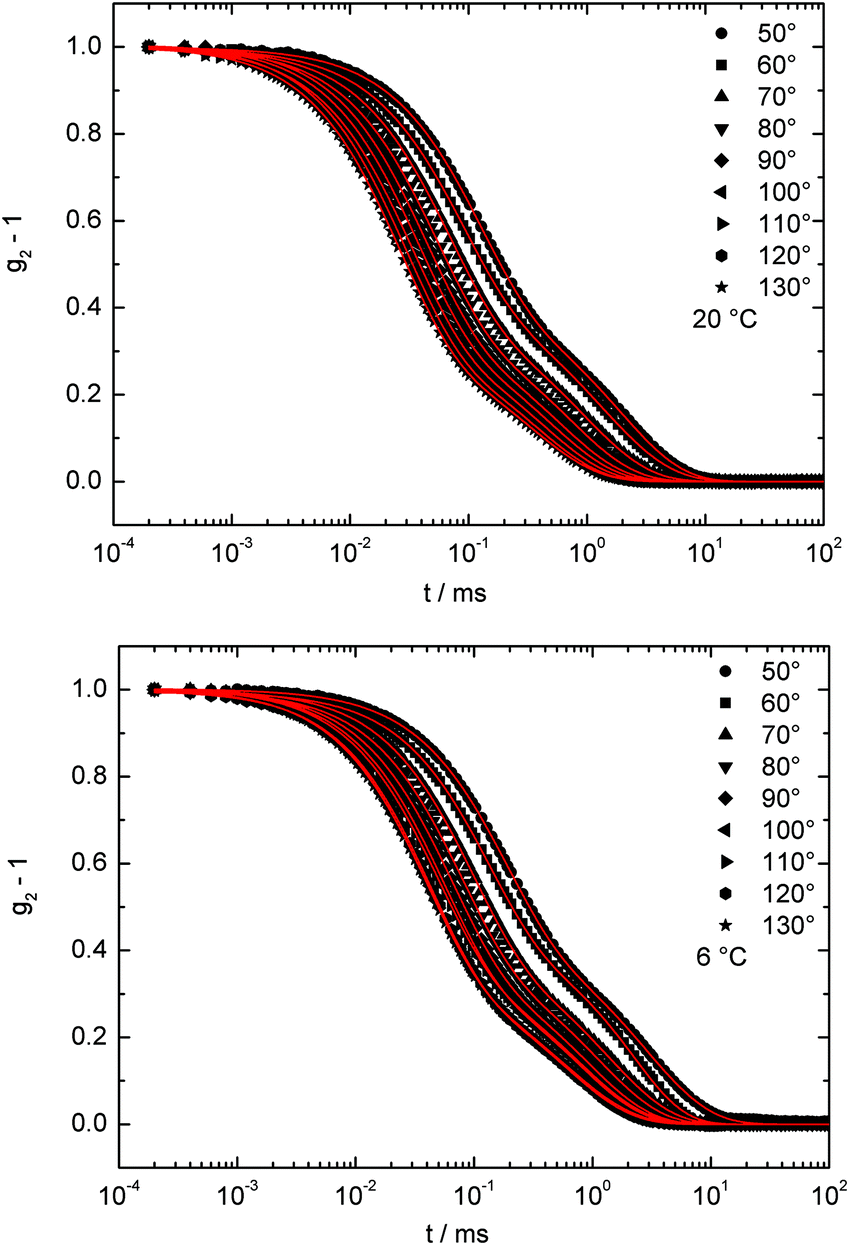 | ||
| Fig. 4 Intensity autocorrelation functions for BCP micelles formed from (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 in water at 2θ: 50°–130°, measured at 20 °C (top) and 6 °C (bottom). The solid lines represent a fit using a squared sum of two exponential decays. | ||
The intensity autocorrelation functions can be described by a squared sum of two exponential decays (see Fig. 4):
| g2 − 1 = (A1exp(−(t/τ1)β1) + A2exp(−(t/τ2)β2))2. |
A
1 and A2 describe the amplitudes of the processes, with A1 + A2 = 1, τ1 and τ2 are the correlation times and β1 and β2 are stretching parameters. These parameters correspond to the polydispersity of the micelles and aggregates. For example if β is equal to 1, the micelles are ideally monodisperse. This is the case for the faster process which can be assigned to the uniform (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 micelles. Compared to this, the micellar aggregates have a stretching parameter 0.7 < β < 0.85 independent of T. The correlation time τ is related to the translational diffusion coefficient D by D = 1/(<τ>q2). The scattering vector q can be calculated by q = 4πnsin(θ)/λ. In our case n is the refractive index of water and 2θ is the scattering angle. The averaged correlation time is <τ> = τ/β × Γ(1/β), while Γ is the gamma function.
The expected linear variation of 1/<τ> with q2 was nicely fulfilled by our data (Fig. 5 and S15†), for both motions and at every measured temperature. By using the Stokes–Einstein relation, a hydrodynamic radius Rh = kBT/6ηπD can be calculated, with kB, T, and η as the Boltzmann constant, the absolute temperature, and the solvent viscosity at the used temperature, respectively. Note that the hydrodynamic radius of the uniform PDMAEMA-containing micelles is a factor 30 smaller than that of the aggregates at 20 °C (Fig. 5 and S14†).
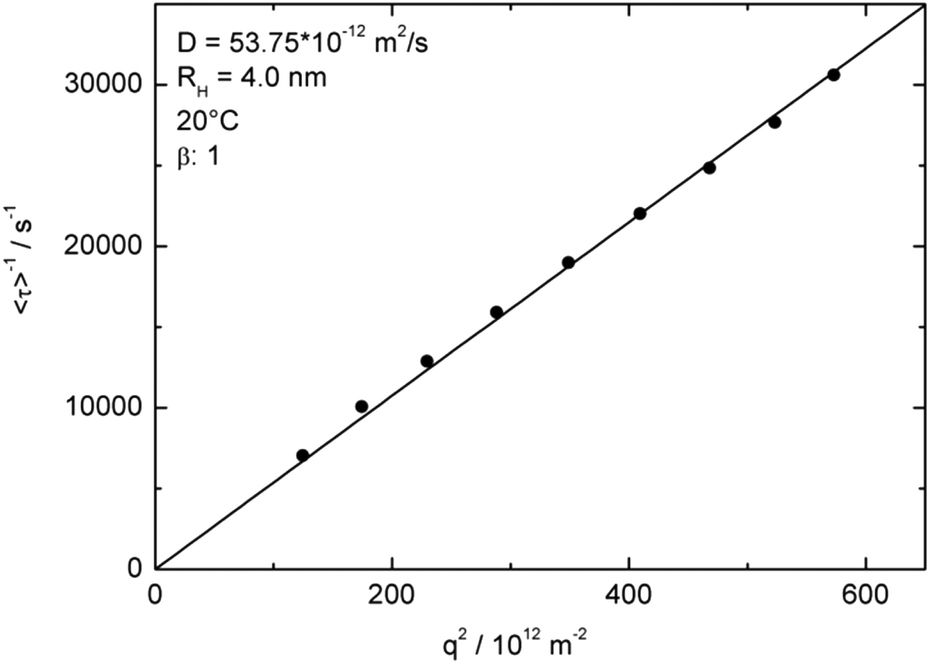 | ||
| Fig. 5 q 2 dependence of inverse relaxation times to determine the diffusion coefficient and the resulting hydrodynamic radii for the fast process of the uniform micelle motions measured at 20 °C. | ||
To investigate the temperature-responsiveness of the BCP micelles formed from (PMMA13-co-PDMAEMA22)-b-PDMAEMA132, the temperature was varied between 50 °C and 6 °C. Fig. S16† shows the behaviour of the inverse relaxation times scaled with the temperature dependent viscosity of the water63 and the temperature versus the squared scattering vector for different temperatures. The hydrodynamic radius of the aggregates at 40 °C was approximately 117 ± 40 nm (Fig. S15,† left). Compared to this, the uniform spherical micelles revealed a value of Rh of 4 ± 0.1 nm. When the solution was cooled down to 20 °C, the hydrodynamic radius, Rh, continuously increased by 10%. For temperatures below 20 °C and above 10 °C the radius was nearly constant and ∼18% larger than before at 40 °C. At 6 °C the radius of the aggregates shrank again (Fig. S5†). Although this repeated shrinkage of the aggregates could not be fully interpreted yet, for the proposed release studies the presence of a significant increase of the uniform micelles while decreasing the temperature below 30–35 °C is of utmost importance. It is noteworthy that for temperatures above 40 °C, the radius of the micelles increased dramatically and the polymers precipitated. Almost the same temperature-responsiveness of the micelles was found for the micellar aggregates. The chain radius grew by a factor of approximately 30%, when cooling the temperature from 40 °C to 30 °C. After that growth, the radius was constant until 15 °C and shrank again at lower temperatures. Fig. 6 shows and summarizes the temperature behaviour of the hydrodynamic radii for the fast process (BCP micelles) while the thermo-responsiveness of the aggregates is given as Fig. S17.† The observed temperature variation of the hydrodynamic radii was fully reversible.
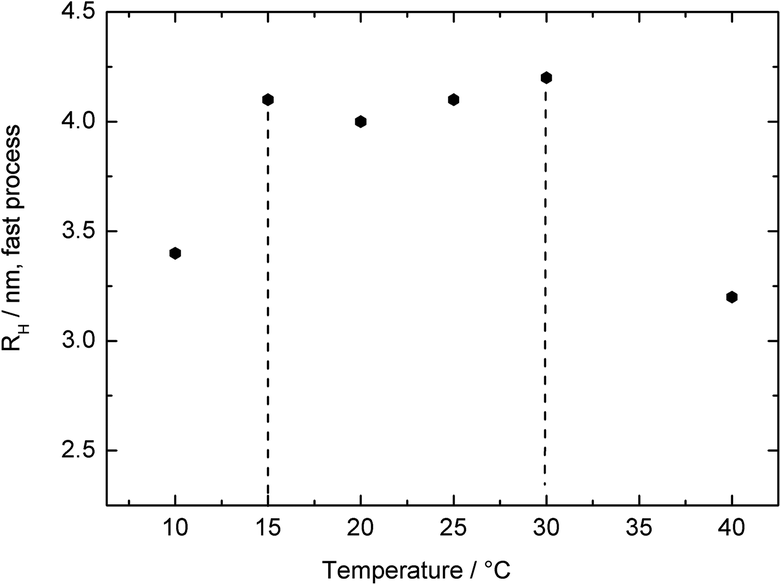 | ||
| Fig. 6 Temperature dependence of the hydrodynamic radii of the BCP micelles in water for the polymer (PMMA13-co-PDMAEMA22)-b-PDMAEMA132. | ||
As a summary of the DLS results, it can be concluded from Fig. 6 and S16† that the BCP aggregates as well as the uniform BCP micelles grow and shrink as a function of temperature. At temperatures above 20 °C there is a change in the solubility of polymers in water because it is above the first lower critical solution temperature. Between 20 °C and 10 °C it is a homogeneous solution and the polymer chains and the micelles are capable of swelling in water. This proves the suitability of the BCP micelle for further investigation as a thermo-responsive release platform, which will be described in the ensuing section.
Stimuli-responsive release of the Ru(II) complex by external triggers
The stimuli-induced release of [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 was determined by atomic absorption spectrometry (AAS) for the purified micelles (see the Instrumentation section for detailed sample treatment). The micelles were loaded with the Ru(II) complex as already described above. Importantly, dialysis of the loaded micelles was carried out using the dialysis membranes Spectra/Por® Biotech CE. In our first attempts with other dialysis membranes, there was an observable color change of the dialysis tube due to the interaction of the released Ru(II) complex with the membrane material. Three different external triggers were applied in order to study the release capabilities of the herein investigated PDMAEMA-based BCPs. In Scheme S1 of the ESI,† the simplified mechanism for releasing the encapsulated Ru(II) complex is given. In our first attempts, the BCPs PMMA-b-PDMAEMA were used for encapsulation. As shown in Fig. 3A and B in the previous section, the incorporation could be confirmed by TEM measurements. However, it turned out that, even with using ultrasound for about 45 min, the BCP micelles did not release a detectable amount of ruthenium species (Fig. S18†). Only after a prolonged ultrasound time of 90 min, a tiny amount of Ru(II) complex was released to the aqueous environment. It has to be mentioned that the detection limit of the AAS setup was about 0.5 μM ruthenium.In order to study the influence on release properties, the molecular compositions of the BCPs were changed and MMA was copolymerized with DMAEMA in order to obtain (PMMA-co-PDMAEMA)-b-PDMAEMA. For simplification, we renamed (PMMA19-co-PDMAEMA22)-b-PDMAEMA191 to Copo1 and (PMMA13-co-PDMAEMA22)-b-PDMAEMA132 to Copo2. As a result, Copo2, which has a higher content of DMAEMA moieties in the copolymer segment, already showed a significant release of the Ru(II) complex at room temperature without an external stimulus (Fig. 7A, blank test). The explanation for this behaviour is as follows: Copo2 features a second LCST (compared to the pure PDMAEMA segment) approximately at room temperature. Therefore, a release of the Ru(II) complex occurred already at room temperature due to the more diluted chain segments. Compared to this, Copo1 features the second LCST at a lower temperature, i.e., at room temperature the core of the BCP micelle is collapsed hindering a significant release of the Ru(II) complex. Only by further decreasing the temperature below the LCST of Copo1, the statistical block segment PDMAEMA-co-PMMA is capable of releasing the payload. While lowering the temperature down to 3 °C, lowering the pH value of the aqueous environment or by application of ultrasound, an almost full release of the encapsulated Ru(II) complex was found. The absolute values for the Ru(II) complex loading are compiled in Table S4 in the ESI† nicely reflecting the release capabilities of the herein investigated BCP micelles upon treatment with the external stimuli.
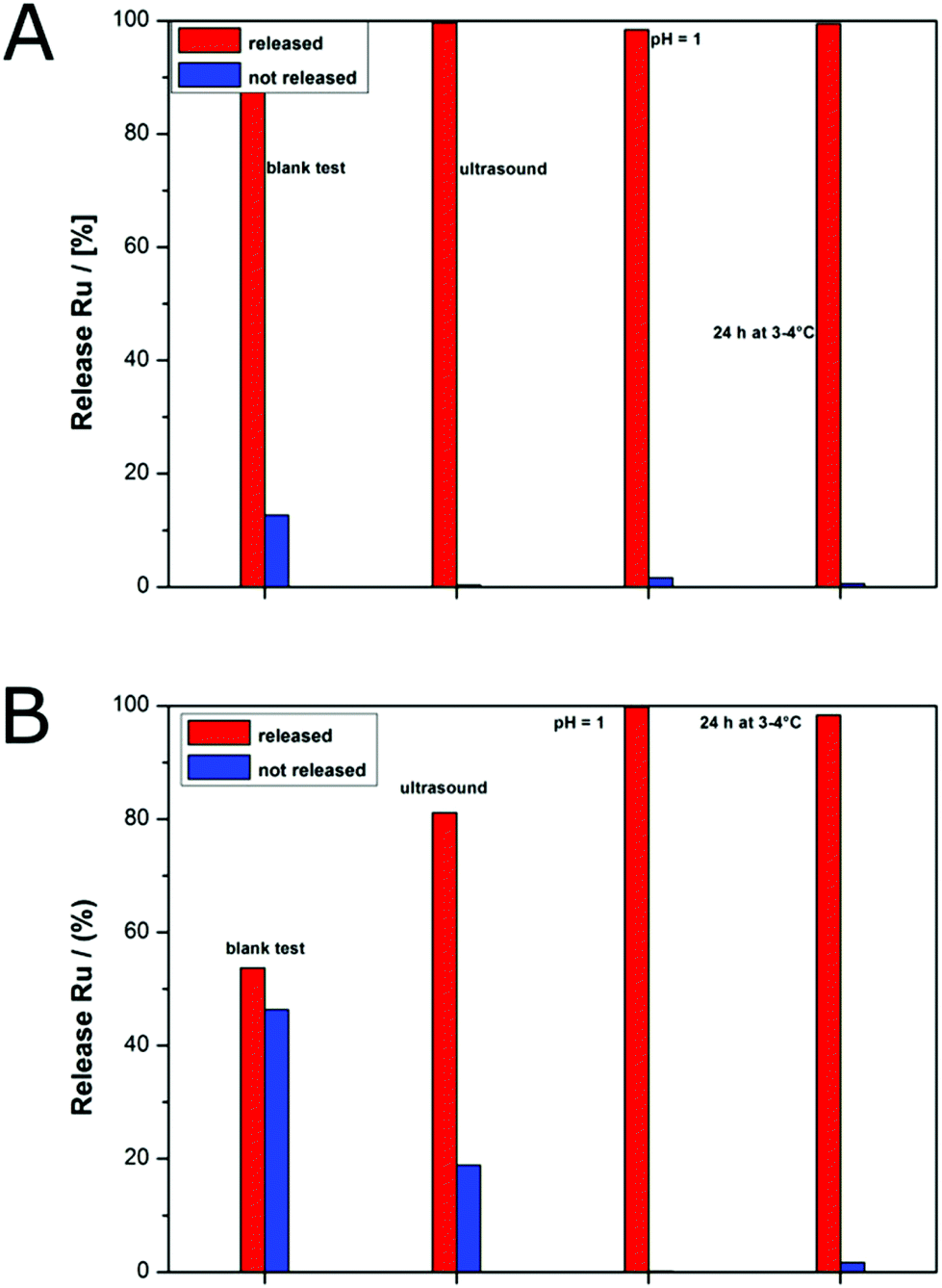 | ||
| Fig. 7 Release profiles for BCP micelles loaded with Ru(II) complex 7 based on Copo2 (A) and Copo1 (B) by using ultrasound, pH value change or storage at 3 °C as external triggers (see the Experimental section). The amount of the released ruthenium was determined by AAS. | ||
Compared to this, Copo1 with a lower amount of DMAEMA moieties in the copolymer segment revealed much less leakage of the BCP micelle and hence a significantly reduced amount of Ru(II) complex was released (Fig. 7B). BCP micelles based on Copo1 and after dialysis were subjected to ultrasound for 1 h. As a result, a residual amount of about 20% of the initial Ru(II) complex content was found, while about 80% could be released by ultrasound treatment. Micelles from the same batch loaded with exactly the same amount of Ru(II) complex showed an almost full release while changing the pH value to 1. Additionally, a full release of the payload could be accomplished by lowering the temperature down to 3 °C.
Conclusions
In the current work we present an efficient protocol for the preparation of poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) based multi-stimuli responsive block copolymers (BCPs) with poly(methyl methacrylate) (PMMA) via anionic polymerization protocols. The block copolymers were capable of micellation in water and used for the incorporation of a Ru(II) polypyridyl complex, i.e., [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2, which was previously shown to act as a potential photosensitizer in photodynamic therapy. The hydrophobic block segment was built by copolymerizing MMA with DMAEMA in order to enhance the Ru(II) complex release properties and to tailor the lower critical solution temperature (LCST) of the micelles. The thermo-responsiveness of the prepared BCP micelles and single polymer chains was investigated by temperature-dependent dynamic light scattering (DLS) experiments. These experiments revealed an increase of the hydrodynamic radii of the BCP micelles below a temperature of 25–30 °C. While loaded micelles derived from PMMA-b-PDMAEMA showed an excellent shielding effect upon ultrasound, i.e., no ruthenium release was accomplished, a good switchability for (PMMA19-co-PDMAEMA22)-b-PDMAEMA191 (Copo1) was observed. This strong shielding effect of BCPs featuring a pure PMMA segment was attributed to the distinct core/shell structure of the loaded BCP micelles compared to more homogeneous BCP micelles as shown by TEM investigations. The latter micelles feature the capability to efficiently release the Ru(II) complex upon ultrasound, by lowering the pH value, and by decreasing the temperature as determined and quantified by atomic absorption spectrometry (AAS) after applying external triggers and dialysis. This study will pave the way to addressable and highly functional polymer-based nanocarrier systems with unprecedented properties that are stable and can be employed in water. Potential applications are foreseen in a variety of disciplines, in particular in biomedicine and smart delivery systems.Experimental
Reagents
All solvents and reagents were purchased from Alfa Aesar, Sigma Aldrich, Fisher Scientific, ABCR and used as received unless stated otherwise. Deuterated solvents were additionally purchased from Deutero GmbH, Kastellaun, Germany. Tetrahydrofuran (THF) was distilled from sodium/benzophenone under reduced pressure (cryo-transfer) prior to the addition of 1,1-diphenylethylene and n-butyllithium (n-BuLi) followed by a second cryo-transfer. MMA and DMAEMA were first dried by stirring over calcium hydride (CaH2), distilled, followed by drying over trioctylaluminium and a second cryo-transfer prior to use. Lithium chloride (LiCl) was dissolved in a small amount of purified THF and placed into an ampule. After removing THF in a vacuum, the ampule was carefully heated out under high vacuum, refilled with nitrogen, and then stored in a glovebox. All syntheses were carried out under an atmosphere of nitrogen using Schlenk techniques or a glovebox equipped with a Coldwell apparatus. The Ru(II) complex was synthesized as reported previously.47Instrumentation
NMR spectra were recorded with a Bruker DRX 500 NMR or with a Bruker DRX 300 spectrometer working at 500 MHz or 300 MHz (1H NMR). NMR chemical shifts are referenced relative to tetramethylsilane. SEC measurements were performed with DMF as the mobile phase (flow rate 1 mL min−1) on a GRAM column set from PSS (GRAM 30, GRAM 1000, GRAM 1000) at 50 °C. Calibration was carried out using PMMA standards (from Polymer Standard Service, Mainz).For determining the thermal properties of the polymers differential scanning calorimetry (DSC) was performed with a Mettler Toledo DSC-1 in the temperature range from 0 °C to 140 °C with a heating rate of 10 K min−1. TEM experiments were carried out on a Zeiss EM 10 electron microscope operating at 60 kV. All shown images were recorded with a slow-scan CCD camera obtained from TRS (Tröndle) in bright field mode. Camera control was computer-aided using the ImageSP software from TRS. Dynamic light scattering (DLS) experiments were carried out with a setup based on a He–Ne laser (λ = 632.8 nm) as the light source at different temperatures. Polarization of the primary beam is determined by using a Glan–Thomson prism. The scattered beam polarization is analyzed in vertical–vertical geometry. The scattered intensity was detected with an optical fiber coupled to two avalanche photodiodes. The intensity autocorrelation functions, calculated with ALV 5000 software for both photodiodes, were measured in angular steps of 10°. All measurements were performed by using cylindrical cuvettes (Hellma) for the samples in a temperature-controlled index matching bath. The correlation time τ is related to the translational diffusion coefficient D and the scattering vector q as D = 1/(τq2). The expected linear variation of 1/τ with q2 is very nicely fulfilled by our data. By using the Stokes–Einstein relation, a hydrodynamic radius Rh = kBT/6ηπD can be calculated, with kB, T, and η as the Boltzmann constant, absolute temperature, and solvent viscosity. Dialysis experiments were performed using dialysis membranes (Spectra/Por® Biotech CE) against an excess of water (1500 mL for approx. 6 mL of Ru-loaded block copolymer micelles). After 24 hours the water is changed, which is repeated overall three times.
For ultrasound experiments, a Hielscher UP50H operating at 30 kHz and 50 W equipped with a Hielscher MS1 titanium sonotrode was used while cooling the samples. For determining the Ru content, atomic absorption spectrometry (AAS) was performed. For additional DLS and zetapotential measurements a Zetasizer Nano ZS90 equipped with a 4 mW 633 nm He–Ne laser was used.
For AAS measurements a contrAA 700 high-resolution continuum-source atomic absorption spectrometer (Analytik Jena AG) was used. Pure samples of the respective complex were used for preparing the standards and calibration was done in a matrix-matched manner (meaning all samples and standards were adjusted to the same polymer concentration by dilution with distilled water if necessary). Triton-X 100 (1%, 10 μL) as well as nitric acid (13%, 10 μL) were added to all probes (100 μL) as modifiers. The samples were injected (25 μL) into coated standard graphite tubes (Analytik Jena AG) and thermally processed as previously described in more detail.64 Ru was quantified at a wavelength of 349.90 nm.65 The mean integrated absorbances of triplicate injections were used throughout the study.
Anionic block copolymerization of methyl methacrylate and N,N-dimethylaminoethyl methacrylate
SEC: PMMA19-co-PDMAEMA22:
M n = 5400 g mol−1; Mw = 6000 g mol−1; Đ = 1.11.
(PMMA19-co-PDMAEMA22)-b-PDMAEMA191:
M
n = 31800 g mol−1; Mw = 35800 g mol−1; Đ = 1.13.
1 H-NMR (300 MHz, CDCl3, 300 K): δ = 0.90–1.05 (m, H2,5); 1.81–2.01 (m, H1,4); 2.28 (m, H8); 2.55 (m, H7); 3.58 (m, H3); 4.05 (m, H6) ppm.
SEC: PMMA13-co-PDMAEMA22:
M n = 4700 g mol−1; Mw = 5300 g mol−1; Đ = 1.11.
(PMMA13-co-PDMAEMA22)-b-PDMAEMA132:
M
n = 26900 g mol−1; Mw = 29600 g mol−1; Đ = 1.10.
1 H-NMR (300 MHz, CDCl3, 300 K): δ = 0.90–1.05 (m, H2,5); 1.82–1.98 (m, H1,4); 2.28 (m, H8); 2.56 (m, H7); 3.58 (m, H3); 4.05 (m, H6) ppm.
TEM sample preparation for (PMMA-co-PDMAEMA)-b-PDMAEMA and micelle loading with [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2
The corresponding PMMA-co-PDMAEMA-b-PDMAEMA samples (50 mg) are dissolved in filtered (0.45 μm cut-off) acetone (0.5 mL) and water (5 mL, Milli-Q water) is added dropwise during constant and moderate stirring. For TEM investigations, the micellar solution is diluted with the 10-fold volume of filtered water and the solution is drop-cast on a carbon-coated copper grid, followed by drying under ambient conditions. For block copolymer micelle loading, the corresponding PMMA-co-PDMAEMA-b-PDMAEMA samples (50 mg) and [Ru(bipy)2-dppz-7-hydroxymethyl][PF6]2 (5 mg) are dissolved in filtered acetone (1 mL). In the next step, water (5 mL) is added dropwise during moderate stirring. An orange and stable solution is obtained. For TEM investigations, the micellar solution is diluted with the 10-fold volume of filtered water and the resulting solution is drop-cast on a carbon-coated copper grid, followed by drying under ambient conditions.Ruthenium release from (PDMAEMA-co-PMMA)-b-PDMAEMA micelles by external triggers
Acknowledgements
The authors thank Christian Rüttiger for additional TEM images. M. G. thanks the Fonds der Chemischen Industrie (FCI) for financial support of this work. M. G. and M. A. acknowledge the German Research Foundation (DFG GA 2169/1-1). This work has been additionally supported in the frame of the LOEWE project iNAPO by the Hessen State Ministry of Higher Education, Research and the Arts and in the frame of the Smart Inorganic Polymer EU network (COST CM10302, SIPS). J. E. would like to thank the German Academic Exchange Service (DAAD) for a Postdoctoral fellowship. G. G. thanks the Swiss National Science Foundation (Professorships No. PP00P2_133568 and PP00P2_157545), the University of Zurich and the Stiftung für wissenschaftliche Forschung of the University of Zurich (G. G). This work has received support under the program “Investissements d'Avenir” launched by the French Government and implemented by the ANR with the reference ANR-10-IDEX-0001-02 PSL (G. G.). C. M. thanks the Forschungskredit of the University of Zurich (Grant K-73532-01-01).References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nature, 2010, 9, 101–113 CrossRef PubMed.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- J. I. Clodt, V. Filiz, S. Rangou, K. Buhr, C. Abetz, D. Höche, J. Hahn, A. Jung and V. Abetz, Adv. Funct. Mater., 2013, 23, 731–738 CrossRef CAS.
- M. Gallei, S. Rangou, V. Filiz, K. Buhr, S. Bolmer, C. Abetz and V. Abetz, Macromol. Chem. Phys., 2013, 214, 1037–1046 CrossRef CAS.
- C. G. Schäfer, M. Gallei, J. T. Zahn, J. Engelhardt, G. P. Hellmann and M. Rehahn, Chem. Mater., 2013, 25, 2309–2318 CrossRef.
- C. G. Schäfer, C. Lederle, K. Zentel, B. Stühn and M. Gallei, Macromol. Rapid Commun., 2014, 35, 1852–1860 Search PubMed.
- C. Rüttiger, V. Pfeifer, V. Rittscher, D. Stock, D. Scheid, S. Vowinkel, F. Roth, H. Didzoleit, B. Stühn, J. Elbert, E. Ionescu and M. Gallei, Polym. Chem., 2016, 7, 1129–1137 RSC.
- R. H. Staff, M. Gallei, K. Landfester and D. Crespy, Macromolecules, 2014, 47, 4876–4883 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS PubMed.
- R. Haag, Angew. Chem., Int. Ed., 2004, 43, 278–282 CrossRef CAS PubMed.
- C. de Las Heras Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- D. G. Shchukin and H. Möhwald, Science, 2013, 341, 1458–1459 CrossRef CAS PubMed.
- L. P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198–14205 CrossRef CAS PubMed.
- M. Marguet, L. Edembe and S. Lecommandoux, Angew. Chem., Int. Ed., 2012, 51, 1173–1176 CrossRef CAS PubMed.
- Y. Zhao, L.-P. Lv, S. Jiang, K. Landfester and D. Crespy, Polym. Chem., 2015, 6, 4197–4205 RSC.
- X. Huang and B. Voit, Polym. Chem., 2013, 4, 435–443 RSC.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042–9049 CrossRef CAS PubMed.
- S. Kubowicz, J. F. Baussard, J. F. Lutz, A. F. Thunemann, H. von Berlepsch and A. Laschewsky, Angew. Chem., Int. Ed., 2005, 44, 5262–5265 CrossRef CAS PubMed.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. Muller, Nature, 2013, 503, 247–251 Search PubMed.
- T. I. Löbling, O. Borisov, J. S. Haataja, O. Ikkala, A. H. Gröschel and A. H. Müller, Nat. Commun., 2016, 7, 12097 CrossRef PubMed.
- S. Behzadi, M. Gallei, J. Elbert, M. Appold, G. Glasser, K. Landfester and D. Crespy, Polym. Chem., 2016, 7, 3434–3443 RSC.
- T. P. Lodge, A. Rasdal, Z. Li and M. A. Hillmyer, J. Am. Chem. Soc., 2005, 127, 17608–17609 CrossRef CAS PubMed.
- F. L. Baines, N. C. Billingham and S. P. Armes, Macromolecules, 1996, 29, 3416–3420 CrossRef CAS.
- W. Xu, A. A. Steinschulte, F. A. Plamper, V. F. Korolovych and V. V. Tsukruk, Chem. Mater., 2016, 28, 975–985 CrossRef CAS.
- T. Thavanesan, C. Herbert and F. A. Plamper, Langmuir, 2014, 30, 5609–5619 CrossRef CAS PubMed.
- Y. Zhou, Principles and Applications of Therapeutic Ultrasound in Healthcare, CRC Press Taylor and Francis Group, Boca Raton, USA, 2016 Search PubMed.
- G. A. Husseini, D. A. Christensen, N. Y. Rapoport and W. G. Pitta, J. Controlled Release, 2002, 83, 303–305 CrossRef CAS PubMed.
- A. Marin, M. Muniruzzaman and N. Rapoport, J. Controlled Release, 2001, 75, 69–81 CrossRef CAS PubMed.
- S. Agarwal, Y. Zhang, S. Maji and A. Greiner, Mater. Today, 2012, 15, 388–393 CrossRef CAS.
- U. Rungsardthong, M. Deshpande, L. Bailey, M. Vamvakaki, S. P. Armes, M. C. Garnett and S. Stolnik, J. Controlled Release, 2001, 73, 359–380 CrossRef CAS PubMed.
- P. V. D. Wetering, J.-Y. Cherng, H. Talsma, D. J. A. Crommelin and W. E. Hennink, J. Controlled Release, 1998, 53, 145–153 CrossRef PubMed.
- I. Tabujew and K. Peneva, Functionalization of Cationic Polymers for Drug Delivery Applications, RSC Polymer Chemistry Series, 2014 Search PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2004, 1, 279–293 CrossRef CAS PubMed.
- F. S. De Rosa and M. V. L. B. Bentley, Pharm. Res., 2000, 17, 1447–1455 CrossRef CAS.
- T. Debele, S. Peng and H.-C. Tsai, Int. J. Mol. Sci., 2015, 16, 22094 CrossRef CAS PubMed.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126 CrossRef CAS PubMed.
- G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. A. McFarland, Coord. Chem. Rev., 2015, 282–283, 127–138 CrossRef CAS.
- S. Bonnet, Comments Inorg. Chem., 2015, 35, 179–213 CrossRef CAS.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- G. Li, L. Sun, L. Ji and H. Chao, Dalton Trans., 2016, 45, 13261–13276 RSC.
- http://www.stockhouse.com/news/press-releases/2015/01/15/theralase-identifies-lead-photo-dynamic-compound-for-human-clinical-trials .
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem. – Eur. J., 2014, 20, 14421–14436 CrossRef CAS PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef CAS PubMed.
- V. Pierroz, R. Rubbiani, C. Gentili, M. Patra, C. Mari, G. Gasser and S. Ferrari, Chem. Sci., 2016, 7, 6115–6124 RSC.
- A. Frei, R. Rubbiani, S. Tubafard, O. Blacque, P. Anstaett, A. Felgenträger, T. Maisch, L. Spiccia and G. Gasser, J. Med. Chem., 2014, 57, 7280–7292 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, Y. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044–4056 RSC.
- T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari and G. Gasser, Angew. Chem., Int. Ed., 2014, 53, 2960–2963 CrossRef CAS PubMed.
- C. S. Jin and G. Zheng, Lasers Surg. Med., 2011, 43, 734–748 CrossRef PubMed.
- E. Paszko, C. Ehrhardt, M. O. Senge, D. P. Kelleher and J. V. Reynolds, Photodiagn. Photodyn. Ther., 2011, 8, 14–29 CrossRef CAS PubMed.
- E. B. Gyenge, S. Hiestand, S. Graefe, H. Walt and C. Maake, Photodiagn. Photodyn. Ther., 2011, 8, 86–96 CrossRef PubMed.
- J. Svensson, A. Johansson, S. Gräfe, B. Gitter, T. Trebst, N. Bendsoe, S. Andersson-Engels and K. Svanberg, Photochem. Photobiol., 2007, 83, 1211–1219 CrossRef CAS PubMed.
- G. Bœuf, G. V. Roullin, J. Moreau, L. Van Gulick, N. Zambrano Pineda, C. Terryn, D. Ploton, M. C. Andry, F. Chuburu, S. Dukic, M. Molinari and G. Lemercier, ChemPlusChem, 2014, 79, 171–180 CrossRef.
- A. Arce, F. Fornasiero, O. Rodríguez, C. J. Radke and J. M. Prausnitz, Phys. Chem. Chem. Phys., 2004, 6, 103–108 RSC.
- M. Gallei, R. Klein and M. Rehahn, Macromolecules, 2010, 43, 1844–1854 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. Morsbach, J. Elbert, C. Rüttiger, S. Winzen, H. Frey and M. Gallei, Macromolecules, 2016, 49, 3406–3414 CrossRef CAS.
- J. Morsbach, A. Natalello, J. Elbert, S. Winzen, A. Kroeger, H. Frey and M. Gallei, Organometallics, 2013, 32, 6033–6039 CrossRef CAS.
- J. Kestin, M. Sokolov and W. W. Wakeham, J. Phys. Chem., 1978, 7, 941–948 CAS.
- J. Skiba, C. Schmidt, P. Lippmann, P. Ensslen, H.-A. Wagenknecht, R. Czerwieniec, F. Bandl, I. Ott, T. Bernaś, B. Krawczyk, D. Szczukocki and K. Kowalski, Eur. J. Inorg. Chem., 2016 DOI:10.1002/ejic.201600281.
- S. Schäfer, I. Ott, R. Gust and W. S. Sheldrick, Eur. J. Inorg. Chem., 2007, 2007, 3034–3046 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Data on additional SEC, DSC, NMR, turbidimetry measurements, TEM, DLS, release mechanism, and conditions for release studies. See DOI: 10.1039/c6py02026g |
| ‡ Present address: Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, 02139, USA. |
| This journal is © The Royal Society of Chemistry 2017 |