
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile synthetic strategy to polysiloxanes containing sulfonyl side groups with high dielectric permittivity†
Simon J.
Dünki
ab,
Eduardo
Cuervo-Reyes
cd and
Dorina M.
Opris
*a
aSwiss Federal Laboratories for Materials Science and Technology Empa, Laboratory for Functional Polymers, Überlandstr. 129, CH-8600, Dübendorf, Switzerland. E-mail: dorina.opris@empa.ch
bÉcole Polytechnique Fédérale de Lausanne (EPFL), Institut des Matériaux, Station 12, CH 1015, Lausanne, Switzerland
cSwiss Federal Laboratories for Materials Science and Technology Empa, Laboratory of Materials for Energy Conversion, Überlandstr. 129, CH-8600, Dübendorf, Switzerland
dSwiss Federal Institute of Technology (ETH), CH-8093 Zürich, Switzerland
First published on 9th December 2016
Abstract
The chemical modification of polymers with lateral polar groups increases their dielectric permittivity above the glass transition temperature, making them attractive materials for dielectric elastomer actuators. Despite the large dipole moment of the sulfonyl moiety, its usefulness as a substituent in high permittivity polysiloxanes has not been explored so far. This work explores two post-polymerization synthetic strategies to reach such a goal, namely the oxidation of the thioether groups present in polysiloxanes which carry thioether side groups at every repeat unit and the modification of the vinyl groups of poly(methylvinylsiloxanes) with sulfonyl groups via thiol–ene chemistry. While both strategies in principle work, the oxidation of the thioether groups results in an undesired shortening of the polysiloxane chains. In contrast, the thiol–ene reactions give the target polymer in a clean and highly efficient process. For this reason the access to two sulfonyl containing thiols, to be employed in the thiol–ene reaction, was improved to the degree that they are now available on the 50 g scale as pure compounds. The sulfonyl content of the polysiloxanes was systematically varied by the use of two different thiols in the thiol–ene post-polymerization modification, one of which carried the sulfonyl group, the other a (dummy) butyl group instead. The prepared polymers were characterized by NMR, DSC, TGA, GPC, and impedance spectroscopy. All polymers show glass transition temperatures below room temperature. Dielectric permittivity measurements at room temperature show that the permittivity of the polymers at the frequency with minimal losses can be fine-tuned from about 5 up to 22.7. Because of their high dielectric permittivity, low glass transition temperatures, and easy and scalable synthesis from cheap materials, these novel polymers are attractive components for high permittivity elastomers to be employed in actuators, capacitors, and flexible electronics.
Introduction
Polysiloxanes have a backbone composed of oxygen and silicon atoms that carry two organic substituents.1 The most often used polysiloxane is the one that carries two methyl groups and is usually referred to as silicone. Silicones can be found in many applications that range from insulators, glues, implants to cosmetics.2 Due to their low Tg and excellent mechanical and dielectric properties, they came more and more into the focus of research and have been proposed for many innovative applications in stretchable electronics and devices.3 The most common method to synthesize polysiloxanes is either via ring-opening polymerization under anionic or cationic conditions or via condensation reactions.1 Polysiloxanes can carry reactive groups such as hydrosilane or vinyl which allow for the introduction of functional groups via post-polymerization modifications.4 By properly selecting the functional groups, polysiloxanes with tunable properties have been prepared.5,6 The possibility of increasing the dielectric permittivity by chemical modification with polar side groups has also been recognized.7–9 The high flexibility of the backbone ensures that the Tg of the modified polysiloxane is still sufficiently low to allow the formation of elastomers after cross-linking. Such polar groups are introduced to the polysiloxane chain by hydrosilylation,10 copper catalyzed azide–alkyne cycloaddition,11 or thiol–ene addition.12 Hydrosilylation is occasionally inefficient since the required Pt catalyst can be poisoned by sulfur or some organic groups.13 The azide–alkyne cycloaddition requires tedious synthetic steps of the starting precursors and necessitates a copper catalyst which is difficult to remove from the product.11We have recently used the thiol–ene reaction for the post-polymerization modification of polysiloxanes with polar nitrile side groups.12 Elastomers with a relative permittivity as high as 18 at high frequencies were achieved.14 Other dipoles such as 4-nitroaniline,15 chloropropyl,16 4-nitrobenzene,11 4-(4-nitrophenylazo)aniline,17 4-oxy-benzaldehyde,17 4-aminopyridine,17 Disperse Red 1,18 cyanopropyl,19 azide,20 and trifluoropropyl21,22 have also been explored, but to achieve polysiloxanes that reach a permittivity value above 10 at high frequencies is still challenging. An up to date review on this topic is available.23 The sulfonyl group has an attractively large dipole moment and should allow the formation of materials with increased dielectric permittivity.
Here, we report the synthesis of polysiloxanes containing sulfone side groups. Two synthetic strategies were explored, which rely either on the oxidation of the thioether side groups of a polysiloxane, or on the thiol–ene reaction of poly(methylvinylsiloxane) PV with thiols that carry the sulfonyl groups. The resulting polymers were characterized by 1H NMR, 13C NMR, FTIR, GPC, and DSC. Additionally, the dielectric properties of these polymers were investigated as a function of sulfonyl content and at frequencies from 0.1 Hz to 1 MHz.
Experimental section
Materials and characterization
Unless otherwise stated, all chemicals were reagent grade and used as received. 3-Sulfolene, toluene, CH2Cl2, CHCl3, Et2O, ethyl acetate (EtOAc), hexane (H) and acetone were purchased from VWR. Thioacetic acid, 2,2-dimethoxy-2-phenylacetophenone (DMPA), chlorotrimethylsilane (TMS-Cl), thionyl chloride (SOCl2), tetramethylammonium hydroxide (TMAH), triethylamine (Et3N), butanethiol, benzene, THF and MeOH were purchased from Sigma-Aldrich. Acetic acid (AcOH), hydrogen peroxide (H2O2), and LiAlH4 were purchased from Fluka (Aldrich). 1,3,5,7-Tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane (V4) was purchased from ABCR. The synthesis of PV (Mn = 96 kDa Mw = 225 kDa, PDI = 4.5) was done according to the literature.24A SwiftCure HL-250 UV lamp (∼35 mW cm−2) from Peschl Ultraviolet was used. 1H, 13C, and 29Si NMR spectra were recorded at 298 K on a Bruker Avance 400 NMR spectrometer using a 5 mm broadband inverse probe at 400.13 MHz, 100.61 MHz, and 79.46 MHz respectively. Chemical shifts (δ) in ppm are calibrated to residual solvent peaks (CDCl3: δ = 7.26 and 77.16 ppm, DMSO-d6: δ = 2.50 and 39.52 ppm). Size exclusion chromatograms were recorded with an Agilent 1100 Series HPLC (columns: serial coupled PSS SDV 5 μm, 100 Å and PSS SDV 5 μm, 1000 Å, detector: DAD, 235 nm and 360 nm; refractive index). THF was used as the mobile phase, PDMS standards were used for the calibration, and toluene as the internal standard. Elemental analysis (EA) was carried out on a LECO TruSpec Micro (C/H), LECO RO-478 (O) and LECO CHNS-932 (S) from LECO Instrumente GmbH. IR spectra were recorded on a Bruker Tensor 27 FT-IR with an ATR interface; peak intensity is given as weak (w), medium (m) or strong (s). Permittivity measurements were done in the frequency range of 0.01 Hz to 1 MHz using a Novocontrol Alpha-A Frequency Analyzer. The root mean square voltage of the probing AC electric signal applied to the samples was 1 V. The diameter of the electrodes was either 5 mm or 20 mm. Thermogravimetric analysis (TGA) was conducted with a Perkin Elmer TGA7 at a heating rate of 20 °C min−1 from 30 °C to 900 °C under a nitrogen or helium gas flow with a sample size of about 17 mg. Differential scanning calorimetry (DSC) investigations were undertaken on a Perkin Elmer Pyris Diamond DSC instrument. Two heating and one cooling steps with a heating and cooling rate of 20 °C min−1 in the temperature range of either −120 °C to 100 °C or −90 °C to 100 °C were conducted per measurement under a nitrogen flow (50 ml min−1). The second cooling step was considered for the evaluation of the Tg. About 10 mg of the sample was weighed in aluminum crucibles shut with pierced lids. Mass spectroscopy measurements were conducted on a Bruker Daltonics maXis ESI-QTOF.
3-Thioacetylsulfolane (2)
To a solution of 3-sulfolane (1) (30.9 g, 261 mmol, 1 eq.) in toluene (330 ml), thioacetic acid (44.0 g, 578 mmol, 2.2 eq.) and DMPA (1.4 g, 5.5 mmol, 0.02 eq.) were added and the reaction mixture was irradiated with a UV lamp for 7.5 min below 50 °C. The reaction mixture was cooled with an ice/salt bath to −10 °C and the precipitate was filtered and washed with cold toluene to afford 2 as a white solid (48.8 g, 251 mmol, 96%).
1H NMR (400 MHz, CDCl3, δ): 4.18–4.11 (m, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –S), 3.54 (dd, J = 13.5 Hz, 8.0 Hz, 1H, CH–C2–SO2), 3.28–3.21 (m, 1H, CH2–C2–SO2), 3.11 (ddd, J = 13.25 Hz, 7.6 Hz, 0.6 Hz, 1H, CH2–C2–SO2), 2.97 (dd, J = 13.5 Hz, 9.0 Hz, 1H, CH–C2–SO2), 2.63–2.55 (m, 1H, C2–CH2–SO2), 2.25–2.15 (m, 1H, C2–CH2–SO2); 13C NMR (100 MHz, CDCl3, δ): 194.0 (CO–S), 56.2 (CH–
–S), 3.54 (dd, J = 13.5 Hz, 8.0 Hz, 1H, CH–C2–SO2), 3.28–3.21 (m, 1H, CH2–C2–SO2), 3.11 (ddd, J = 13.25 Hz, 7.6 Hz, 0.6 Hz, 1H, CH2–C2–SO2), 2.97 (dd, J = 13.5 Hz, 9.0 Hz, 1H, CH–C2–SO2), 2.63–2.55 (m, 1H, C2–CH2–SO2), 2.25–2.15 (m, 1H, C2–CH2–SO2); 13C NMR (100 MHz, CDCl3, δ): 194.0 (CO–S), 56.2 (CH–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–SO2), 51.6 (CH2–H2–SO2), 37.7 (H–S), 30.6 (H3–CO), 29.2 (H2–CH2–SO2); MS: [M + Na]+ (C6H10O3S2Na; calc.: 216.9969; found: 216.9964); IR: 3015 (w), 2957 (w), 2922 (w), 1688 (s), 1459 (w), 1427 (w), 1407 (w), 1355 (w), 1318 (m), 1296 (s), 1267 (m), 1242 (w), 1205 (m), 1149 (w), 1122 (s), 1085 (m), 1058 (w), 965 (m), 901 (m), 864 (w), 775 (w), 737 (m), 681 (w), 624 (s), 570 (s), 534 (w), 513 (w), 453 (s), 406 (s); EA: Calcd: C 37.09, H 5.19, O 24.71, S 33.01; found C 37.02, H 5.02, O 24.64, S 33.10.
H2–SO2), 51.6 (CH2–H2–SO2), 37.7 (H–S), 30.6 (H3–CO), 29.2 (H2–CH2–SO2); MS: [M + Na]+ (C6H10O3S2Na; calc.: 216.9969; found: 216.9964); IR: 3015 (w), 2957 (w), 2922 (w), 1688 (s), 1459 (w), 1427 (w), 1407 (w), 1355 (w), 1318 (m), 1296 (s), 1267 (m), 1242 (w), 1205 (m), 1149 (w), 1122 (s), 1085 (m), 1058 (w), 965 (m), 901 (m), 864 (w), 775 (w), 737 (m), 681 (w), 624 (s), 570 (s), 534 (w), 513 (w), 453 (s), 406 (s); EA: Calcd: C 37.09, H 5.19, O 24.71, S 33.01; found C 37.02, H 5.02, O 24.64, S 33.10.
3-Mercaptosulfolane (3)
3-Thioacetylsulfolane (2) (80.1 g, 412 mol, 1 eq.) was dissolved in THF (350 ml) and MeOH (250 ml) at 40 °C. TMS-Cl (6.7 g, 6.2 mmol, 0.016 eq.) was added and the reaction mixture was stirred at 50 °C for 3 days. The solvent was evaporated under reduced pressure, the residue dissolved in CH2Cl2 and washed with sat. aq. NaHCO3. The aq. phase was vigorously extracted with CH2Cl2, the combined org. phases were dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The residue was distilled at 1.2 mbar and 143 °C to obtain compound 3 as a colorless liquid (55.3 g, 363 mmol, 88%).
1H NMR (400 MHz, CDCl3, δ): 3.64–3.56 (m, 1H, C–SH), 3.54 (dd, J = 13.3 Hz, 7.3 Hz, 1H, CH–C2–SO2), 3.37–3.31 (m, 1H, CH2–C2–SO2), 3.10 (dddd, J = 13.3 Hz, 9.8 Hz, 7.7 Hz, 0.3 Hz, 1H, CH2–C2–SO2), 2.98 (dd, J = 13.2 Hz, 9.6 Hz, 1H, CH–C2–SO2), 2.69–2.61 (m, 1H, CH2–C2–CH), 2.16 (ddt, J = 13.57 Hz, 8.23 Hz, 9.90 Hz, 1H, CH2–C2–CH), 2.02 (d, J = 7.4 Hz, 1H, S); 13C NMR (100 MHz, CDCl3, δ): 60.1 (CH–H2–SO2), 52.4 (CH2–H2–SO2), 34.0 (CH2–H2–CH), 33.8 (H–SH); MS: [M + Na]+ (C4H8O2S2Na; calc.: 174.9863; found: 174.9859); IR: 3006 (w), 2948 (w), 2558 (w), 1452 (w), 1411 (w), 1291 (s), 1267 (s), 1212 (m), 1166 (w), 1114 (s), 1057 (w), 985 (w), 902 (m), 840 (w), 762 (m), 727 (m), 678 (w), 568 (s), 452 (s), 406 (s); EA: Calcd: C 31.56, H 5.30, O 21.02, S 42.13; found C 31.42, H 5.32, O 21.25, S 42.29.
2-Chloroethyl methyl sulfide (5)
A solution of SOCl2 (153.5 g, 1.290 mol, 1.18 eq.) in dry CHCl3 (90 ml) was added dropwise to a solution of 2-(methylthio)-ethanol (101.1 g, 1.097 mol, 1 eq.) in dry CHCl3 (130 ml) over 3 h under argon. The released HCl gas was neutralized with aq. NaOH solution using a gas washing bottle. The reaction mixture was refluxed for 3.5 h and then cooled to RT. It was then washed with water (200 ml), sat. aq. NaHCO3 solution (200 ml) and brine (200 ml). The aqueous phases were extracted with CHCl3 (2 × 100 ml). The combined org. phases were dried over MgSO4, filtered and the solvent was removed under reduced pressure to obtain 5 as a yellowish liquid (106.2 g, 0.960 mol, 88%).
1H NMR (400 MHz, CDCl3, δ): 3.65 (t, J = 7.9 Hz, 2H, Cl–C2), 2.84 (t, J = 7.9 Hz, 2H, C2–S), 2.16 (s, 3H, S–C3); 13C NMR (CDCl3, δ): 42.8 (Cl–H2), 36.3 (H2–S), 15.9 (S–H3).
2-Chloroethyl methyl sulfone (6)
To a solution of 2-chloroethyl methyl sulfide (5) (106.1 g, 0.959 mol, 1 eq.) in AcOH (240 ml), hydrogen peroxide solution (30% in H2O, 242.1 g, 2.135 mol, 2.2 eq.) was added dropwise so that the reaction mixture gently boiled. The reaction was further refluxed for 2 h and allowed to cool to RT. About 2/3 of the solvent was evaporated under reduced pressure. To the residue water (100 ml) was added followed by extraction with CH2Cl2 (4 × 200 ml). The org. layers were washed with sat. NaHCO3 solution (3 × 200 ml). The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure to obtain a yellowish liquid, which was negative in a peroxide test. Vacuum distillation (3 mbar, 110 °C) afforded 6 as a colorless liquid (87.82 g, 0.619 mol, 65%).
1H NMR (400 MHz, CDCl3, δ): 3.92 (t, J = 6.6 Hz, 2H, Cl–C2), 3.45 (t, J = 6.6 Hz, 2H, C2–SO2), 3.04 (s, 3H, SO2–C3); 13C NMR (100 MHz, CDCl3, δ): 57.1 (H2–SO2), 42.8 (SO2–H3), 36.2 (H2–Cl); MS: [M + H]+ (C3H8ClO2S; calc.: 142.9934; found: 142.9930); IR: 3022 (w), 2985 (w), 2932 (w), 1741 (w), 1629 (w), 1447 (w), 1412 (w), 1315 (m), 1285 (s), 1233 (w), 1206 (w), 1142 (s), 1119 (s), 1069 (w), 1036 (w), 968 (m), 951 (m), 917 (w), 883 (m), 803 (w), 742 (m), 701 (w), 677 (w), 639 (m), 510 (s), 496 (m), 487 (m), 468 (s), 434 (m); EA: Calcd: C 25.15, H 4.95, O 22.44, S 22.49, Cl 24.86; found C 25.15, H 4.97, O 22.73, S 22.20.
Methyl vinyl sulfone (7)
2-Chloroethyl methyl sulfone (6) (77.67 g, 0.547 mol, 1 eq.) was added dropwise to a solution of freshly distilled dry Et3N (53.40 g, 0.528 mol, 0.97 eq.) in dry Et2O (140 ml) over 2 h. The reaction mixture was stirred at RT until all the starting material was consumed. The precipitated salt was filtered, the filter cake was washed with acetone, and the solvent was evaporated under reduced pressure to obtain a brown liquid. The desired product 7 was obtained after vacuum distillation (0.1 mbar, 74 °C) as a colorless liquid (48.34 g, 0.455 mol, 83%).
1H NMR (400 MHz, CDCl3, δ): 6.72 (dd, J = 16.6 Hz, 9.9 Hz, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 6.45 (d, J = 16.6 Hz, 1H, CHC2), 6.14 (d, J = 9.9 Hz, 1H, CHC2), 2.95 (s, 3H, S–C3); 13C NMR (100 MHz, CDCl3, δ): 137.6 (H–SO2), 129.7 (H2), 42.4 (H3); MS: [M + H]+ (C3H7O2S; calc.: 107.0167; found: 107.0160); IR: 3107 (w), 3060 (w), 3022 (w), 2930 (w), 1615 (w), 1414 (w), 1389 (w), 1294 (s), 1127 (s), 957 (s), 783 (s), 685 (m), 624 (m), 512 (s), 482 (s), 447 (w); EA: Calcd: C 33.95, H 5.70, O 30.15, S 30.21; found C 33.17, H 6.24, O 30.82, S 29.67.
CH2), 6.45 (d, J = 16.6 Hz, 1H, CHC2), 6.14 (d, J = 9.9 Hz, 1H, CHC2), 2.95 (s, 3H, S–C3); 13C NMR (100 MHz, CDCl3, δ): 137.6 (H–SO2), 129.7 (H2), 42.4 (H3); MS: [M + H]+ (C3H7O2S; calc.: 107.0167; found: 107.0160); IR: 3107 (w), 3060 (w), 3022 (w), 2930 (w), 1615 (w), 1414 (w), 1389 (w), 1294 (s), 1127 (s), 957 (s), 783 (s), 685 (m), 624 (m), 512 (s), 482 (s), 447 (w); EA: Calcd: C 33.95, H 5.70, O 30.15, S 30.21; found C 33.17, H 6.24, O 30.82, S 29.67.
Thioacetic acid-S-[2-(methylsulfonyl)ethyl] ester (8)
To a solution of methyl vinyl sulfone (47.71 g, 449 mmol, 1 eq.) and thioacetic acid (41.35 g, 543 mmol, 1.2 eq.) in THF (550 ml), Et3N (54.51 g, 539 mmol, 1.2 eq.) was added dropwise at room temperature and allowed to stir for 2 h. Hydrochloric acid (2 mol l−1, 270 ml, 540 mmol, 1.2 eq.) was added to the reaction mixture and the solution was extracted with CH2Cl2 (5 × 200 ml). The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure. The product was recrystallized from EA/H; 3/2 as white crystals (55.62 g, 305 mmol). Purification by column chromatography (SiO2, EtOAc/H 1/1) of the residue obtained by evaporating the mother liquor afforded the additional product 8 as a white solid (22.16 g, 122 mmol). The overall yield of 8 was 95% (77.78 g, 427 mmol).
1H NMR (400 MHz, CDCl3, δ): 3.25 (s, 4H, S–C2–C2–SO2), 2.99 (s, 3H, C3–SO2), 2.38 (s, 3H, C3–CO); 13C NMR (100 MHz, CDCl3, δ): 195.1 (–O–S), 54.5 (–H2–SO2), 41.0 (H3–SO2), 30.7 (H3–CO), 22.3 (H2–S); MS: [M]+ (C5H10O3S2; calc.: 182.0071; found: 182.0056); IR: 3023 (w), 3001 (w), 2986 (w), 2941 (w), 1682 (s), 1646 (w), 1417 (m), 1354 (w), 1323 (m), 1299 (s), 1268 (s), 1225 (w), 1128 (s), 1111 (s), 1033 (w), 1005 (w), 969 (s), 954 (s), 932 (m), 777 (m), 747 (m), 738 (m), 704 (w), 633 (s), 518 (s), 492 (s); EA: Calcd: C 32.95, H 5.53, O 26.33, S 35.19; found C 32.81, H 5.42, O 26.54, S 34.89.
2-(Methylsulfonyl)-ethanethiol (9)
A solution of thioacetic acid-S-[2-(methylsulfonyl)ethyl] ester (8) (12.98 g, 71.2 mmol, 1 eq.) in dry THF (250 ml) was added dropwise to a suspension of LiAlH4 (3.48 g, 91.7 mmol, 1.3 eq.) in THF (160 ml) at 4 °C. After the addition, the reaction mixture was allowed to warm to RT and stirred for another 2 h. The reaction mixture was again cooled with an ice bath and the excess of LiAlH4 was quenched by slowly adding 10 ml water and hydrochloric acid (1 mol l−1, 120 ml). The phases were separated and the aqueous phase was extracted with CH2Cl2 (4 × 200 ml). The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure to obtain a yellowish oil. Purification by column chromatography (SiO2, EtOAc/H 2/1) afforded 9 as a colorless liquid (9.09 g, 64.8 mmol, 91%). Alternatively, 9 can be purified by vacuum distillation (123 °C at 0.3 mbar).
1H NMR (400 MHz, CDCl3, δ): 3.35–3.31 (m, 2H, C2–SO2), 3.03–2.98 (m, 5H, C3–SO2, C2–SH), 1.82 (t, J = 8.5 Hz, 1H, S); 13C NMR (100 MHz, CDCl3, δ): 58.3 (H2–SO2), 41.7 (SO2–H3), 17.3 (H2–SH); MS: [M]+ (C3H8O2S2 calc.: 139.9966; found: 139.9961); IR: 3011 (w), 2928 (w), 1412 (w), 1284 (s), 1268 (s), 1227 (w), 1117 (s), 1035 (w), 1013 (w), 959 (s), 861 (w), 780 (m), 751 (w), 641 (w), 509 (s), 494 (s), 465 (s), 432 (w); EA: Calcd: C 25.70, H 5.75, O 22.82, S 45.73; found C 25.72, H 5.80, O 23.09, S 45.46.
General procedure for the functionalization of polymethylvinyl siloxane
To a solution of poly(methylvinylsiloxane) (7.5 g, 87 mmol vinyl, 1 eq.) dissolved in distilled THF (200 ml), either 3, 9, butylthiol or a mixture of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :butanethiol or 9:butanethiol (130 mmol, 1.5 eq., see Table 1) was added and argon was bubbled through the solution for 10 min. Then, DMPA (0.22 g, 0.9 mmol, 0.01 eq.) was added. The flask was irradiated with UV light under vigorous stirring for 10 min. THF was partially removed under reduced pressure and the residue precipitated with MeOH. The precipitate was separated, dissolved in THF, and precipitated again with MeOH. This process was repeated several times until a satisfying purity was achieved. The polymers were dried under HV and elevated temperatures (15.8 g, 72%).
:butanethiol or 9:butanethiol (130 mmol, 1.5 eq., see Table 1) was added and argon was bubbled through the solution for 10 min. Then, DMPA (0.22 g, 0.9 mmol, 0.01 eq.) was added. The flask was irradiated with UV light under vigorous stirring for 10 min. THF was partially removed under reduced pressure and the residue precipitated with MeOH. The precipitate was separated, dissolved in THF, and precipitated again with MeOH. This process was repeated several times until a satisfying purity was achieved. The polymers were dried under HV and elevated temperatures (15.8 g, 72%).
| Sample | mol% sulfonyl | mol% sulfonyla | T g [°C] | ε′ @ 10 Hz | ε′ @ 104 Hz | σ [S cm−1] | Yield [%] | Vinyla [%] |
|---|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR (n/d = not detected). | ||||||||
| P1 | 0 | 0 | −99.9 | 5.0 | 4.9 | 4.2 × 10−13 | 93 | n/d |
| Pm(1:3) | 25 | 20.8 | −73.9 | 8.3 | 8.0 | 2.1 × 10−11 | 74 | n/d |
| Pm(1:1) | 50 | 50 | −34.5 | 11.5 | 10.7 | 1.8 × 10−11 | 72 | n/d |
| Pm(3:1) | 75 | 77.7 | −5.7 | 17.2 | 12.6 | 1.5 × 10−11 | 63 | <0.02 |
| Pm(1:0) | 100 | 100 | 9.3 | 11.9 | 7.4 | 2.2 × 10−13 | 67 | <0.2 |
| Pn(1:3) | 25 | 22.7 | −72.6 | 10.8 | 10.0 | 1.7 × 10−10 | 84 | <0.007 |
| Pn(1:1) | 50 | 54.6 | −43.1 | 16.1 | 15.4 | 3.2 × 10−10 | 89 | n/d |
| Pn(3:1) | 75 | 80.6 | −26.3 | 24.2 | 20.4 | 3.8 × 10−10 | 92 | <0.06 |
| Pn(1:0) | 100 | 100 | −19.2 | 27.7 | 22.7 | 3.5 × 10−10 | 88 | <0.4 |
P1
1H NMR (400 MHz, CDCl3, δ): 2.57–2.49 (m, 4H, C2–S–C2), 1.59–1.51 (m, 2H, C2–CH2–CH3), 1.45–1.36 (m, 2H, C2–CH3), 0.94–0.86 (m, 5H, Si–C2, CH2–C3), 0.14 (s, 3H, Si–CH3); 13C NMR (100 MHz, CDCl3, δ): 31.9 (S–H2–CH2–CH2), 31.8 (H2–CH2–CH3), 26.6 (Si–CH2–H2), 22.2 (H2–CH3), 18.5 (Si–H2), 13.9 (CH2–H3), 0.08 (Si–H3); 29Si NMR (79.5 MHz, CDCl3, δ): −24.1 (Si–CH3); IR: 2957 (w), 2930 (w), 2873 (w), 1464 (w), 1417 (w), 1378 (w), 1259 (m), 1224 (w), 1167 (m), 1085 (s), 1019 (s), 999 (s), 918 (w), 871 (w), 790 (s), 767 (s), 666 (w), 508 (w), 411 (s).
Pm(1:0)
1H NMR (400 MHz, DMSO-d6, δ): 3.65–3.52 (m, 2H, C–C2–SO2), 3.31–3.26 (m, 1H, CH2–C2–SO2), 3.16–3.08 (m, 1H, CH2–C2–SO2), 2.96 (dd, 1H, J = 12.2 Hz, 8.7 Hz, CH–C2–SO2), 2.65 (t, 1H, J = 8.4 Hz, C2–S–CH), 2.49–2.44 (m, 1H, C2–CH2–SO2), 2.05–1.95 (m, 1H, C2–CH2–SO2), 0.87 (t, 1H, J = 8.4 Hz, Si–C2), 0.17 (s, 3H, Si–CH3); 13C NMR (100 MHz, DMSO-d6, δ): 5.68 (CH–H2–SO2), 51.3 (CH2–H2–SO2), 38.6 (H–CH2–SO2), 29.3 (H2–CH2–SO2), 25.1 (Si–CH2–H2), 17.7 (Si–H2–CH2), 0.27 (Si–H3); 29Si NMR (79.5 MHz, DMSO-d6, δ): −23.5 (Si–CH3); IR: 3004 (w), 2953 (w), 2921 (w), 1449 (w), 1410 (w), 1295 (s), 1262 (s), 1208 (w), 1171 (m), 1115 (s), 1085 (s), 998 (s), 989 (m), 792 (s), 798 (s), 734 (s), 680 (m), 598 (w), 568 (s), 453 (s), 408 (s).
Pn(1:0)
1H NMR (400 MHz, DMSO-d6, δ): 3.37–3.32 (m, 2H, C2–SO2), 3.01 (s, 3H, C3–SO2), 2.88–2.84 (m, 2H, C2–CH2–SO2), 2.63 (t, 2H, J = 8.3 Hz, Si–CH2–C2), 0.88 (t, 2H, J = 8.3 Hz, Si–C2), 0.17 (s, 3H, Si–CH3); 13C NMR (100 MHz, DMSO-d6, δ): 53.7 (H2–SO2), 40.4 (H3–SO2), 25.7 (Si–CH2–H2), 23.4 (H2–CH2–SO2), 17.5 (H2–Si), −0.29 (H3–Si); 29Si NMR (79.5 MHz, DMSO-d6, δ): −23.5 (Si–CH3); IR: 3017 (w), 2957 (w), 2924 (w), 1413 (w), 1308 (s), 1282 (s), 1260 (s), 1218 (w), 1171 (w), 1138 (s), 1085 (s), 999 (s), 938 (m), 884 (w), 789 (s), 767 (s), 666 (m), 510 (s), 495 (s), 467 (s), 410 (s).
Results and discussion
Three different strategies can in principle be used to introduce sulfonyl groups on the polysiloxane chain. First, polymerization of cyclosiloxane monomers that carry polar sulfonyl groups can be used. However, such polymerizations are usually inefficient, i.e. the equilibrium of the polymerization is shifted to cyclic monomers. As a consequence the reaction yield is rather low.25 Furthermore, the molar mass of the formed polymer is usually much too low to ensure good elasticity after cross-linking.The second possibility is to start from a polysiloxane that carries thioether side groups which are oxidized to sulfones (Scheme 1). We have recently reported a polysiloxane that carries thioether side groups.12 It was prepared starting from a hydroxy end-terminated poly(methylvinylsiloxane) PV (Mn = 81 kDa, Mw = 190 kDa, PDI = 2.35) whose vinyl groups were reacted with butylthiol (BuSH) via thiol–ene addition.
 | ||
| Scheme 1 The synthesis of a polysiloxane containing butylether side groups P1 and its oxidation to P2. | ||
The oxidation of thioethers can be conducted with different oxidizing agents such as mCPBA,26 pure H2O2,27 H2O2 with various catalysts,28 KMnO4,29 or oxone.30 Typically, these oxidations are carried out under harsh, acidic conditions in aqueous media which may lead to polysiloxane depolymerization.
Therefore, mCPBA was chosen since it offers the mildest oxidation conditions and is soluble in organic solvents. Furthermore, Garin et al.26 already described the compatibility of mCPBA with polysiloxanes. Following the described procedure, we can confirm that the conversion of P1 to the corresponding sulfone P2 proceeds quantitatively. The 1H NMR spectra show a clear shift of the protons vicinal to the thioether group at 2.5 ppm to the characteristic region for sulfones around 3.0–2.9 ppm (Fig. S1†). Additionally, in the IR spectrum the absence of the peaks in the range around 1060–1040 cm−1 characteristic for partial oxidation to sulfoxide and the presence of the peaks at 1312 cm−1 and 1130 cm−1 characteristic for sulfones are clear indications that the reaction occurred (Fig. S2†). To verify whether the siloxane backbone withstood the oxidation conditions, the product was analyzed by GPC. As can be seen in Fig. S3,† the siloxane backbone was affected during this step. An attempt to suppress the depolymerization process by reducing the reaction time from 5 days to 65 min and by quenching with Na2S2O3 and neutralization with NaHCO3 solution was not successful. By conducting the reaction at −15 °C for 5 min, the depolymerization was partially suppressed, but still the molecular weight of the oxidized polymer was reduced to roughly a fifth of the starting polymer. Therefore, the oxidation of the polysiloxane modified with thioether side groups to sulfone turned out to be unsuitable for our purpose.
The third possibility to synthesize polysiloxanes with sulfonyl side groups is via a post-polymerization modification. For this an efficient reaction of functional side groups of a polysiloxane with appropriately functionalized moieties that contain sulfonyl groups has to be used. Sulfonyl groups were introduced via a hydrosilylation reaction of siloxane monomers (dichlorosilane31 or heptaalkylcyclotetrasiloxane32) or of a poly(methylhydrosiloxane).33 However, the reaction requires the use of the an expensive Pt catalyst, elevated temperatures, and prolonged reaction time. Additionally, this reaction is difficult to conduct to completion and the unreacted hydrosilyl groups slowly hydrolyze in time and unwanted gelation occurs. The thiol–ene reaction is among one of the most efficient reactions for the post-polymerization modifications. It proceeds within minutes under UV irradiation, reaches high conversion, is robust, and gives little side reactions.34 As starting materials polymers that carry either thiol groups or double bonds can be used. Unfortunately, commercial polysiloxanes containing thiols have either a low molar mass or a low content of thiols and the synthesis of polysiloxanes containing thiols of a controlled molar mass is challenging.35 Therefore, we used as starting materials PV and two sulfones that carry thiols which are easily accessible. The two thiols, 3-mercaptosulfolene (3) and 2-(methylsulfonyl)-ethanethiol (9) are easily accessible on a large scale (50 g) and are cheap (Scheme 2). The synthesis of 3 starts from 3-sulfolene (1) which was converted to thioester 2.36 Thioester 2 is known in the literature and was prepared via thermally induced thiol–ene reaction of 1 in the presence of thioacetic acid and AIBN, but the reaction yield was rather low. The reason behind the low reaction yield was the thermal instability of 1 at elevated temperatures. This problem was overcome by conducting the thiol–ene reaction under UV light with DMPA as an initiator and by keeping the reaction temperature below 50 °C. The reaction proceeded in a few minutes and the yield was increased from 30% with AIBN to 96% with DMPA. The cleavage of the acetyl group of 2 was evaluated under basic and acidic conditions. When a stoichiometric amount of butylamine was used, a quantitative turnover was observed, but the subsequent work up turned out to be challenging due to the similar polarity of the acetylated butylamine and the desired thiol. Although the reaction under acidic conditions was not complete, a slightly higher yield was achieved due to the straightforward workup.
 | ||
| Scheme 2 Synthesis of thiols 3 and 9. | ||
The second thiol selected, 2-(methylsulfonyl)-ethanethiol (9) was prepared starting from 2-(methylthio)-ethanol (4), which was quantitatively converted to 5 with SOCl2. The obtained thioether 5 was oxidized with H2O2 to the corresponding sulfoxide 6 with a moderate yield. 6 was then reacted with triethylamine to afford vinyl sulfone 7 in 83% yield. Thioester 8 was obtained from a subsequent Michael addition of 7 with thioacetic acid. The cleavage of the acetyl group of 8 under acidic conditions gave only moderate yield. The reduction with LiAlH4 left the sulfone group unaffected and the reaction yield increased to 91% (Scheme 2).
The thiol–ene reaction of 3 and 9 was conducted on polymethylvinyl siloxane PV (Scheme 3). This polymer was prepared by anionic polymerization with the tetramethylammonium hydroxide (TMAH) initiator under thermodynamic control (Mn = 96 kDa and Mw = 225 kDa).24 For the thiol–ene additions freshly distilled THF and the DMPA photoinitiator and an over stoichiometric amount of thiols to the vinyl groups were used to avoid side reactions. The sulfonyl content of the polysiloxanes was systematically varied by the use of two different thiols in the thiol–ene post-polymerization modification, one of which carried the sulfonyl group, the other a butyl group instead. Two series of polymers that differ by the type and the content of sulfonyl groups were prepared. Series Pm contains sulfolane side groups and series Pn contains methylsulfone side groups. We note that the sulfolane has a somewhat larger volume than the methylsulfone group and therefore may require more time to orient in an electric field, and thus may behave different under an electric field in spite of their rather similar dipole moment. To tune the amount of sulfonyl groups incorporated, PV was reacted either with 3 or 9 to create polymers Pm(1:0) or Pn(1:0) which carry at every repeat unit a polar sulfonyl group or with various ratios of 3 or 9 (x) and butanethiol (y) to create polymers Pm(x:y) and Pn(x:y).
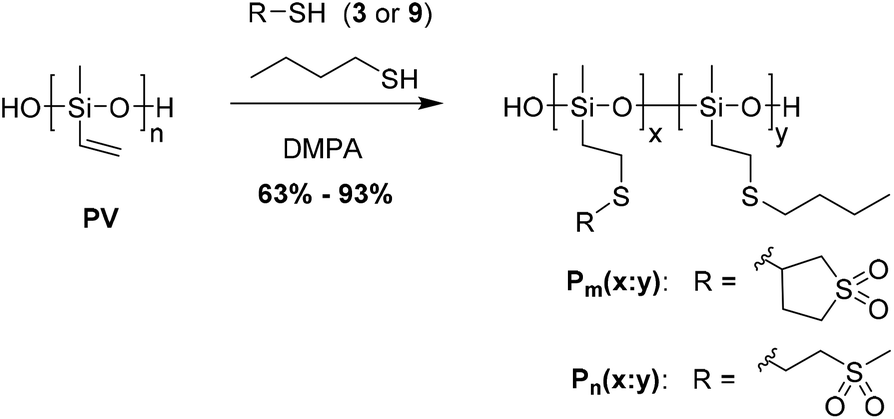 | ||
| Scheme 3 Synthesis of Pm(x:y) and Pn(x:y). | ||
Most of the polymers remained soluble in THF during the thiol–ene reaction. Polymers Pm(3:1), Pm(1:0) and Pn(1:0) that have a high content of sulfonyl groups precipitated during the thiol–ene reaction. Although this could affect the reaction, the 1H NMR spectra of the polymers show that the amount of the vinyl groups left unreacted was less than 0.4% (Fig. 1). Therefore it was concluded that the polymers started to precipitate when most of the vinyl groups were reacted. Table 1 shows the content of sulfonyl groups as found by 1H NMR. The reactivity of the three thiols used was quite similar since the content of the sulfonyl groups found by 1H NMR was very close to the prescribed one. While most of the prepared polymers were soluble in THF, the high polarity of the sulfonyl groups make polymers Pm(3:1), Pm(1:0) and Pn(1:0) insoluble in common organic solvents. They were only soluble in polar solvents such as DMSO and DMF, but insoluble in water. The polymers that were soluble in THF were purified by precipitation from THF with MeOH, while Pm(3:1), Pm(1:0) and Pn(1:0) were only kneaded in THF, leading to a good purity, but significant lower yields (Table 1).
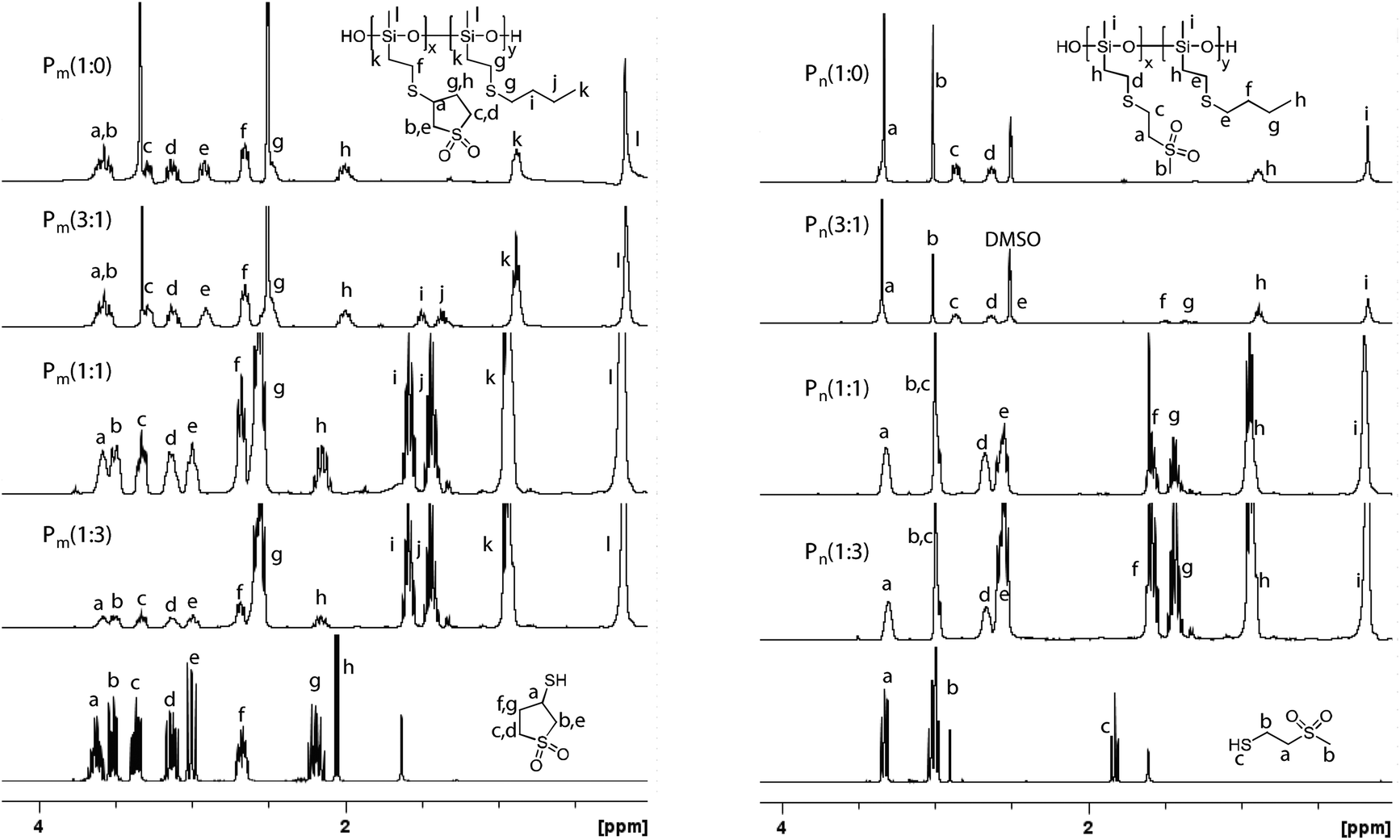 | ||
| Fig. 1 1H NMR spectra of Pm(x:y) (left) and Pn(x:y) (right). The precise assignment of the protons was possible. The spectra were recorded in CDCl3 or in DMSO-d6. | ||
The molecular weights of the prepared polymers and their weight distribution were investigated with GPC (Fig. 2). Due to the above-mentioned reduced solubility of Pm(3:1), Pm(1:0), and Pn(1:0) we were unable to obtain molecular weight data by GPC. All other samples were measured using THF as the mobile phase. Some of the prepared polymers show a bimodal distribution. This might be due to a covalent binding of two polymer chains as a side reaction during the radical mediated thiol–ene addition. A trend toward a slightly lower molecular weight with the increasing content of sulfonyl groups in the polymers might be a reflection of the poor solubility in THF of the modified polymers. The change in the polarity of the polymer might lead to a smaller hydrodynamic volume, ultimately reflected by the seemingly lower molecular weight. Another explanation could be that the polar polymers of a higher molar mass might be less soluble than the lower molar mass ones and thus may be filtered out before the separation on the GPC column.
 | ||
| Fig. 2 GPC elugrams of Pm(x:y) and Pn(x:y) in THF. Due to the insolubility of Pm(3:1), Pm(1:0) and Pn(1:0) in THF, no GPC was conducted on them. | ||
TGA measurements show good thermal stability of all polymers up to about 250 °C where a small amount of volatiles is removed. Most of the polymers start to degrade above 300 °C. The decomposition temperature decreased with the increasing amount of dipoles in the material (Fig. S4†). The amount of residue left for all polymers is less than 30%.
Differential scanning calorimetry (DSC) was also conducted on all polymers. Fig. 3 shows the influence different types and contents of sulfonyl groups has on the Tg. They show that the expected Tg shift to higher temperatures as the concentration of the sulfonyl groups is increased. The Tg of series Pn increased from about −99.9 °C to −19.2 °C, while that of series Pm increased up to about 9.3 °C. The lower Tg of Pn compared to Pm is an indication of a higher dipolar mobility of the methylsulfone group compared to sulfolane which is bulkier. Despite the tendency of sulfones to crystalize, no melting was observed for both series of polymers up to 200 °C.
 | ||
| Fig. 3 T g of polymers Pm(x:y) and Pn(x:y)versus mol% of sulfonyl groups. | ||
Frequency (ν) dependent impedance spectroscopy was used to investigate how the dielectric response is influenced by different types and contents of sulfonyl groups. Relative permittivity (ε′) and dielectric losses (ε′′) are shown in Fig. 4. We also show the real part (resistive component) of the frequency dependent conductivity (σ = 2πνε′′) and the loss-tangent tan(δ) = ε′′/ε′. The sharp increase in permittivity at low frequencies is due to electrode polarization, when ions can move and accumulate at the electrodes, and is therefore of no relevance for the present study. A common feature of all samples is the presence of ionic conductivity, which is evidenced by the straight line with the −1 slope at low frequencies in the log–log plot of ε′′ vs. ν. The conductivity of both series of polymers is rather low. For instance, series Pn has conductivities below 4.2 × 10−10 S cm−1, while series Pm displays values of less than 2.3 × 10−11 S cm−1. The other important characteristic of the spectra is the dipolar loss peak with a center in the MHz region for most samples (of which in most cases only the rising slope can be seen). One should keep in mind that ε′′ and ε′ are analytically connected by the Kramers–Kronig relations, which implies that as one increases the probing frequency crossing a loss peak, the relative permittivity will display a step-down. Therefore, an optimal working frequency, at which the permittivity is high and the losses are low, corresponds to the minimum of the loss tangent, where the contribution to the losses from the ionic conductivity has decreased enough and we have not yet entered in the region of high dipolar losses. This optimal frequency lies between 103 Hz and 104 Hz for most of our samples. We summarized permittivity values at 10 Hz and 10 kHz in Table 1. As expected, the permittivity increases in both series with the content of dipoles incorporated. The samples of series Pm exhibit a slightly lower permittivity compared to the samples of series Pn (Fig. 5). This was expected for the slightly lower dipole moment and a reduced dipole density in the polymers of series Pm, where the sulfolane moiety contains one carbon more compared to methylsulfone. Dipole size also plays the expected role in the dynamics; i.e., the characteristic frequency for the center of the dipolar loss is higher for the methylsulfone group (above 105 Hz) than for the sulfolane group on Pm. The highest permittivity value in this range was reached by sample Pn(1:0) with 22.7. The same sample displays a value of 27.7 at 10 Hz.
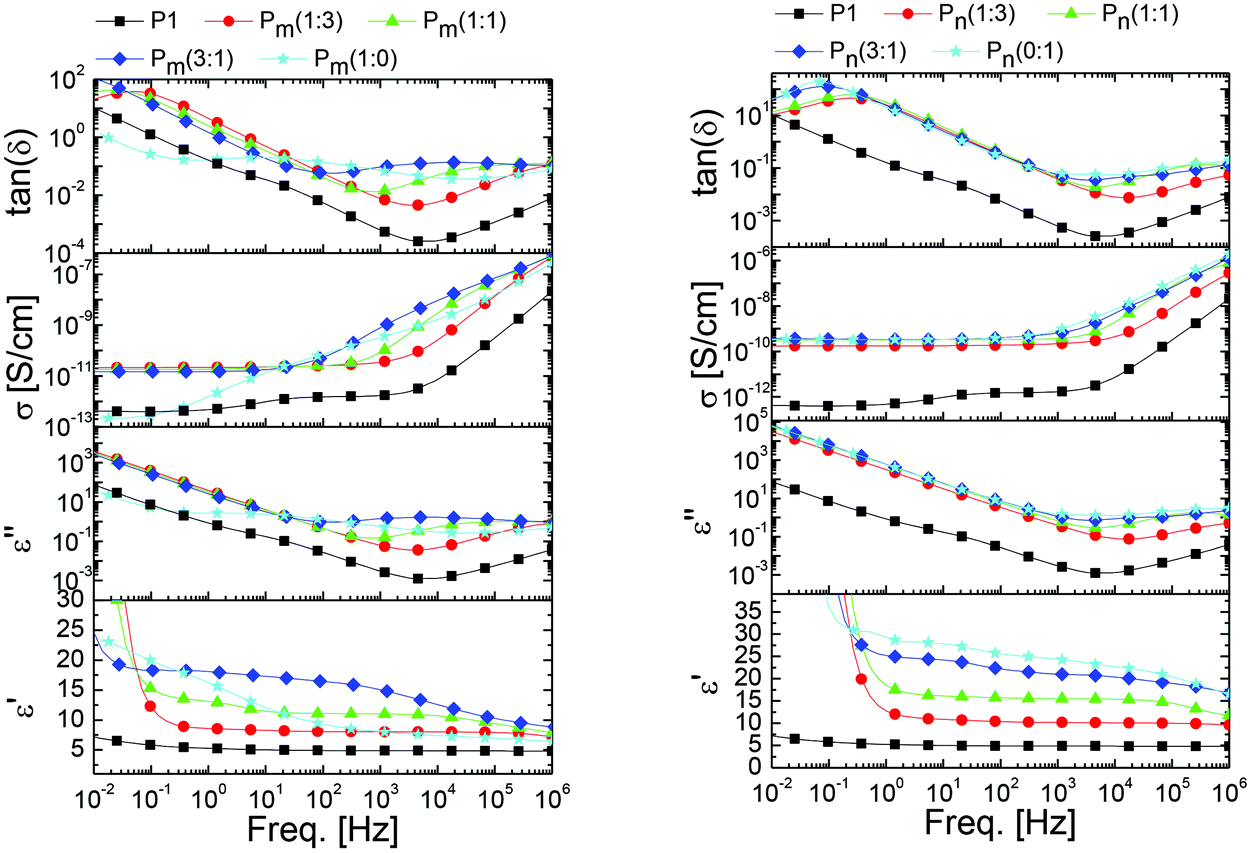 | ||
| Fig. 4 Room temperature dielectric permittivity (ε′), dielectric loss (ε′′), conductivity (σ), and loss-tangent tan(δ) vs. frequency for Pm(x:y) (left) and Pn(x:y) right. The plots for P1 are shown in left and right panels to ease the comparison. | ||
 | ||
| Fig. 5 Left axis: relative permittivity at the first minimum of the loss-tangent. Right axis: loss-tangent at its first minimum. Inset: frequency at the loss-tangent minimum. Squares and triangles correspond to Pm and Pn samples, respectively. | ||
For polymers that have a content of 50 mol% sulfonyl groups or lower, a clear permittivity plateau extends from 5 Hz to 10 kHz, which indicates that the contribution from dipolar losses can be neglected in this region. A higher content of dipolar groups leads to a broadening of the loss peak and its effect extends to lower frequencies. The rising slope of these broad peaks, which lie in the range 0 < α ≤ 1 and decreases as the dipole concentration increases, is a feature that has not been fully addressed in the past. It has been recently shown that the type of relaxation dynamics (i.e., how an induced polarization decays in time as the external field is switched off) is analytically related to this slope.37 Materials with α = 1 are well represented by Debye or Kohlrausch–Williams–Watt relaxation functions, featuring a standard (fast) exponential or stretched exponential time-decay. In these cases, there is an average relaxation time, well above which the system can be assumed to be at equilibrium. On the other hand, systems with α < 1 have a qualitatively different (slow) power-law relaxation, P(t) ∝ t−α. These dipolar systems return slowly to equilibrium, and the average relaxation time (which is by definition the integral over time of the relaxation function) diverges. In these cases the dipolar system is said to have a glassy dynamic. Thus, identifying the slope of the loss peak for functionalized elastomers is of fundamental and practical relevance. Glassy systems can maintain an induced polarization for a longer time but will show less intense, delayed and history dependent responses to varying fields. It should be noted that our impedance measurements were done above the structural–glass transition temperature and therefore the glassy dynamic of the sulfonyl groups sets in before the system becomes a structural glass. As expected, the glassy behavior of the dipoles is accentuated (and the exponent α decreases) at higher dipole concentrations, since dipoles are able to interact more and their movement becomes more correlated. In Fig. 5 we show the permittivity values at the frequency of the minimum loss-tangent, and the corresponding loss-tangent, and the frequency (in the small inset). With the increase in the content of sulfonyl groups, the Pn series delivers higher permittivity with less losses. The strong down-shifting and broadening of the loss peak in the Pm series, which is not seen for Pn samples, can be understood considering the proximity to Tg. The glass temperature is higher for the sulfolane containing polymers and the higher the dipole content, the closer the Tg gets to the room temperature. Dynamical correlations anticipating the glass transition are therefore expected to show stronger effects for the Pm series.
Summary and conclusions
We have shown that the thiol–ene reaction is efficient for the synthesis of polysiloxanes containing polar sulfonyl side groups. A quantitative conversion of the vinyl groups occurs within a few minutes. The content of sulfonyl groups was tuned by using different ratios of functional thiols 3 or 9 to butanethiol. The polysiloxane backbone imparts flexibility which is reflected in the Tg of the prepared polymers which is in all cases below room temperature. The Tg of Pn increased from −99.9 °C to −19.2 °C and for Pm, it reached a maximum value of 9.3 °C. While the polymers containing up to 75 mol% sulfonyl groups are soluble in THF, the solubility of the polymers that carry at every repeat unit a sulfonyl group was significantly reduced. They were only soluble in polar solvents such as DMSO and DMF, but were found to be insoluble in water. The permittivity of series Pn was tuned from 4.9 up to 22.7 at 104 Hz (the frequency at which the system has the lowest loss-tangent). The sample with the highest dipole content of series Pn showed conductivities of 10−10 S cm−1 while that of Pm showed conductivities below 10−11 S cm−1 which are still in the range of insulator materials. Due to their high permittivity, low Tg, and low conductivity, the reported polymers are promising materials for the synthesis of high permittivity elastomers which have high application potential in actuators, capacitors, and stretchable electronics.Authors contribution
S. J. D. performed the synthesis and the characterization of all materials. S. J. D., D. M. O and E. C.-R. wrote the manuscript with input from all authors. E. C.-R. and D. M. O. did the interpretation of the dielectric spectra. D. M. O. initiated the activity on polar silicones, designed the materials, and coordinated this research. All authors contributed to discussions and have given approval to the final version of the manuscript.Acknowledgements
We gratefully acknowledge the Swiss National Science Foundation (SNF132101, Swiss-Romanian Cooperation Program, grant no. IZERZO_142215/1 and 206021_150638/1) and the Swiss Federal Laboratories for Materials Science and Technology (Empa, Dübendorf) for financial support. We also acknowledge B. Fischer for DSC and TGA measurements and Prof. Nüesch for his kind support (both Empa).Notes and references
- W. Noll, Chemistry and Technology of Silicones, Academic Press, New York and London, 1968 Search PubMed.
- Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications, ed. R. G. Jones, W. Ando and J. Chojnowski, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000 Search PubMed.
- T. Sekitani and T. Someya, Adv. Mater., 2010, 22, 2228–2246 CrossRef CAS PubMed.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- G. J. J. Out, A. A. Turetskii and M. Moller, Macromol. Rapid Commun., 1995, 16, 107–112 CrossRef CAS.
- G. Agrawal, J. Wang, B. Brüster, X. Zhu, M. Möller and A. Pich, Soft Matter, 2013, 9, 5380–5390 RSC.
- C.-Y. Yang and G. E. Wnek, Polymer, 1992, 33, 4191 CrossRef CAS.
- Z. Zhu, A. G. Einset, C.-Y. Yang, W.-X. Chen and G. E. Wnek, Macromolecules, 1994, 27, 4076 CrossRef CAS.
- K. L. Wu, D. Y. Wan and Y. Y. Chiang, Gaofenri Tongxun, 1979, 1, 41 Search PubMed.
- C. Racles, M. Alexandru, A. Bele, V. E. Musteata, M. Cazacu and D. M. Opris, RSC Adv., 2014, 4, 37620–37628 RSC.
- F. B. Madsen, I. Javakhishvili, R. E. Jensen, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2014, 5, 7054–7061 RSC.
- S. J. Dünki, M. Tress, F. Kremer, S. Y. Ko, F. A. Nüesch, C.-D. Varganici, C. Racles and D. M. Opris, RSC Adv., 2015, 5, 50054–50062 RSC.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- S. J. Dünki, Y. S. Ko, F. A. Nüesch and D. M. Opris, Adv. Funct. Mater., 2015, 25, 2467–2475 CrossRef.
- B. Kussmaul, S. Risse, G. Kofod, R. Waché, M. Wegener, D. N. McCarthy, H. Krüger and R. Gerhard, Adv. Funct. Mater., 2011, 21, 4589–4594 CrossRef CAS.
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, RSC Adv., 2015, 5, 10254–10259 RSC.
- C. Racles, V. Cozan, A. Bele and M. Dascalu, Des. Monomers Polym., 2016, 19, 496–507 CrossRef CAS.
- L. Zhang, D. Wang, P. Hu, J.-W. Zha, F. Yous, S.-T. Li and Z. M. Dang, J. Mater. Chem. C, 2015, 3, 4883–4889 RSC.
- C. Racles, M. Cazacu, B. Fischer and D. M. Opris, Smart Mater. Struct., 2013, 22, 104004 CrossRef.
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polymer, 2014, 55, 6212–6219 CrossRef CAS.
- M. Dascalu, S. J. Dünki, J.-E. Q. Quinsaat, Y. S. Ko and D. M. Opris, RSC Adv., 2015, 5, 104516–104523 RSC.
- H. Böse, D. Uhl and R. Rabindranath, Proc. SPIE, 2012, 8340, 83402E–834021 CrossRef.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, Macromol. Rapid Commun., 2016, 37, 378–413 CrossRef CAS PubMed.
- Z. Leita, P. Hlozek, B. Bucek and M. Jelinek, J. Polym. Sci., Part C: Polym. Symp., 1967, 16, 669–677 CrossRef.
- S. J. Dünki, M. Dascalu, F. A. Nüesch and D. M. Opris, Proc. SPIE, 2016, 9798K, 1–12 Search PubMed.
- S. Garin, L. Lecamp, B. Youssef and C. Bunel, Eur. Polym. J., 1999, 35, 473–482 CrossRef CAS.
- F. Tormena, R. Rittner, L. C. Ducati and M. P. Freitas, J. Mol. Struct., 2006, 800, 45–50 CrossRef.
- K. Masayuki, I. Arsushi, T. Noguchi and J. Yamamoto, Synlett, 2010, 1557–1561 Search PubMed.
- K. Matsumoto, E. A. Costner, I. Nishimura, M. Ueda and C. G. Willson, Macromolecules, 2008, 41, 5674–5680 CrossRef CAS.
- J. Das, S. S. Bag and A. Basak, J. Org. Chem., 2016, 81, 4623–4632 CrossRef CAS PubMed.
- A. P. Purdy and A. Hwang, Polym. Mater. Sci. Eng., 2001, 84, 641 CAS.
- B. Kanner and B. Prokai, US4110271, Union Carbide Corporation, NY, 1978 Search PubMed.
- Y.-J. Wang, S. Liang, W. Liu, U. H. Choi and R. H. Colby, Polym. Prepr., 2009, 50(2), 866 CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- E. Perju, S. J. Dünki and D. M. Opris, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2940–2948 CrossRef CAS.
- B. Li, R. Buzon and B. Hritzko, Synlett, 2012, 131–133 CrossRef CAS.
- E. Cuervo-Reyes, Sci. Rep., 2016, 6, 29021 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, MS, EA, IR, and GPC elugrams. See DOI: 10.1039/c6py01917j |
| This journal is © The Royal Society of Chemistry 2017 |