
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle lithium-ion conducting poly(tetrafluorostyrene sulfonate) – polyether block copolymer electrolytes†
Zhecheng
Shao
and
Patric
Jannasch
 *
*
Polymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden. E-mail: patric.jannasch@chem.lu.se; Fax: +46 46 222 40 12; Tel: +46 46 222 98 60
First published on 13th December 2016
Abstract
Solid single-ion conducting polymers continue to attract significant interest as electrolyte materials with great potential to improve safety and performance of energy storage devices. Still, their low conductivity is a significant hurdle presently preventing their application. Here, we report on highly conductive BAB triblock copolymers with A blocks of either poly(ethylene oxide) (PEO) or poly(ethylene oxide-co-propylene oxide) (PEOPO), and B blocks of poly(lithium 2,3,5,6-tetrafluorostyrene-4-sulfonate) (PPFSLi). The copolymers were readily synthesised by atom transfer radical polymerisation (ATRP) of 2,3,4,5,6-pentafluorostyrene from polyether macroinitiators, followed by quantitative thiolation using NaSH and subsequent oxidation to form the sulfonate anions. The copolymers possessed high thermal stability and their ionic content was conveniently controlled by the block ratio during the ATRP. Above the polyether melting point, PEO-based block copolymers with [O]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Li] = [18]:[1] showed the highest conductivity, close to 1.4 × 10−5 S cm−1 at 60 °C, while at lower temperatures, the PEOPO materials reached the highest conductivity, nearly 1.5 × 10−6 S cm−1 at 20 °C. The high conductivity of the former copolymer suggests weak interactions of the lithium ions with the pentafluorosulfonate anions in combination with a high degree of Li+ dissociation facilitated by PEO. The results of the present study demonstrate that well-designed block copolymers containing lithium pentafluorostyrene sulfonate units can approach the levels of conductivity required for high-temperature lithium battery applications.
:[Li] = [18]:[1] showed the highest conductivity, close to 1.4 × 10−5 S cm−1 at 60 °C, while at lower temperatures, the PEOPO materials reached the highest conductivity, nearly 1.5 × 10−6 S cm−1 at 20 °C. The high conductivity of the former copolymer suggests weak interactions of the lithium ions with the pentafluorosulfonate anions in combination with a high degree of Li+ dissociation facilitated by PEO. The results of the present study demonstrate that well-designed block copolymers containing lithium pentafluorostyrene sulfonate units can approach the levels of conductivity required for high-temperature lithium battery applications.
Introduction
Today portable devices such as laptops and cell phones rely heavily on lithium-ion batteries. These power sources offer significant advantages in terms of high specific energy and energy density compared to alternatives such as lead/acid or nickel metal hydride batteries.1,2 However, the flammability of the employed liquid electrolytes, commonly a lithium salt dissolved in a mixture of carbonates (e.g., ethylene carbonate and dimethyl carbonate), presents a significant drawback which causes safety issues and limits not only the operating temperature range, but also the battery lifetime because of their high reactivity with the electrode materials.1,2 Furthermore, the use of these liquid electrolytes prevents the utilization of lithium metal, which is widely considered to be the ultimate negative electrode material.1–3 Employing solid polymer electrolytes, consisting of a lithium salt dissolved in a polar polymer, would solve most of the safety issues associated with the liquid electrolytes.1–5 The most widely studied salt-in-polymer systems is based on poly(ethylene oxide) (PEO) and lithium bis(trifluorosulfonyl)imide (LiTFSI) salt. In these systems, the ionic conductivity is closely coupled with the relatively slow segmental mobility of the polymer in the amorphous state. Moreover, both the anion and Li+ are free and mobile which results in that only about 20% of the ionic current is carried by the Li+ ions.5 The mobility of both ions further leads to polarization effects which decrease the charge–discharge rates of the battery.5In solid single Li+-ion conducting polymer electrolytes, the anion is tethered to the polymer and ideally only the cation is mobile.5–22 This solves many of the inherent problems of liquid and salt-in-polymer electrolytes. However, the conductivity of single-ion conducting polymer electrolytes is usually very low, mainly because of the low level of ionic dissociation and slow dynamics of the polymer chains.5,6 This issue may be at least partly alleviated by careful polymer design and increased operation temperature. Several studies have demonstrated that the performance can be enhanced by preparing well-designed block copolymers in which anionic blocks are combined with conductive PEO blocks.5,6 For example, Mayes and coworkers have reported 1–2 orders of magnitude higher ion conductivities when the anions were placed in a separate block, spatially separated from the ion conducting PEO blocks, in comparison with the stoichiometrically equivalent materials where the anions were placed in the PEO blocks.7 This was explained by higher ion dissociation in the former copolymer due to migration of Li+ into the PEO phase. The results implied that the energy gained by Li+ solvation in PEO is sufficient to outweigh the electrostatic energy penalty for nanoscale separation of Li+ and the covalently bound anion.7 Most of the recent work on state-of-the-art single-ion conducting polymer electrolytes have been performed with styrenic and acrylate blocks functionalized with sulfonyl(trifluoromethanesulfonyl)imide lithium groups.14–22 In a recent study, Bouchet and co-workers reported on BAB triblock copolymers having a central B block of PEO and flanking A blocks of poly[4-styrenesulfonyl(trifluoromethanesulfonyl)imide].22 They reported a very high conductivity of 1.3 × 10−5 S cm−1 at 60 °C reached by a copolymer containing 20 wt% of the ionic block.
In the present work we have for the first time tethered lithium 2,3,5,6-tetrafluorobenzene sulfonate groups to polymers and investigated their properties as single Li+-ion conducting BAB block polymer electrolytes. Previously, Sanchez and co-workers have studied and compared lithium pentafluorobenzene sulfonate and LiTFSI salts dissolved in PEO.23 Although the latter polymer electrolyte was more conductive, the lithium pentafluorobenzene sulfonate system provided much higher cationic transference numbers, as well as high thermal and electrochemical stability. The BAB triblock copolymers of the present work were designed with B blocks of poly(lithium 2,3,5,6-tetrafluorostyrene-4-sulfonate) (PPFSLi) and A blocks of either PEO or poly(ethylene oxide-co-propylene oxide) (PEOPO). The triblock copolymers were synthesised by atom transfer radical polymerisation (ATRP) of 2,3,4,5,6-pentafluorostyrene from polyether macroinitiators, followed by thiolation using NaSH and subsequent oxidation with H2O2 to form the sulfonate anions. The ionic content ([O]:[Li]) was conveniently controlled by the block ratio targeted in the ATRP. The copolymers were evaluated as Li+-single ion conducting electrolytes with a focus on the molecular structure, thermal stability, phase behaviour and ionic conductivity.
Experimental
Materials
Poly(ethylene oxide) (PEO, Sigma-Aldrich, Mn,SEC = 32.6 kg mol−1, PDI = 1.23), poly(ethylene oxide-co-propylene oxide) (PEOPO, Sigma-Aldrich, Mn,SEC = 12.4 kg mol−1, 75% EO, PDI = 1.21), α,α′-dibromo-p-xylene (Sigma-Aldrich, 98%), copper(I)bromide (Sigma-Aldrich, 98%), 2,2′-bipyridyl (bipy, Acros, 99+%), anhydrous potassium carbonate (Sigma-Aldrich, 99+%), 18-crown-6 (Sigma-Aldrich, 99+%), copper(0) powder (Sigma-Aldrich, 99.5%), sodium hydrogensulfide hydrate (Sigma-Aldrich, technical grade), glacial acetic acid (Acros, 99.5+%), hydrogen peroxide (Sigma-Aldrich, 30 wt% in H2O), hexane (Honeywell, 99%), diethyl ether (Honeywell, 99%), and N,N-dimethylacetamide (DMAc, Sigma-Aldrich, 99.8+%) were used as received. Tetrahydrofuran (THF, Honeywell, 99%) and o-xylene (Sigma-Aldrich, 99+%) were dried using molecular sieves (Acros, 4 Å 8–12 mesh). 2,3,4,5,6-Pentafluorostyrene (PFS) (Sigma-Aldrich, 99%) was passed through a column packed with alumina (Alfa Aesar, activated, basic, Brockmann Grade I, 58 Å), degassed and stored under an inert atmosphere. The ion exchange resin Amberlite IR-120 in its sodium form (Sigma-Aldrich) was appropriately conditioned before use.Preparation of polyether macroinitiators
The Br-PEO-Br and Br-PEOPO-Br macroinitiators were prepared by attaching benzylic bromide initiator groups at each chain end of PEO and PEOPO, respectively. Br-PEO-Br was prepared by mixing PEO (0.4 mmol, 14 g, 1 eq.), α,α′-dibromo-p-xylene (8 mmol, 2.1 g, 20 eq.), 18-crown-6 (4 mmol, 1.5 g, 10 eq.), anhydrous potassium carbonate (8 mmol, 1.1 g, 20 eq.) and THF (300 mL) in a dark brown round bottom flask under nitrogen protection. The mixture was kept under reflux for 4 days at 80 °C. Subsequently, the hot reaction mixture was filtered and the filtrate was cooled to −18 °C and recrystallized in THF (300 mL). The product was then dried under vacuum at 50 °C and collected as a white powder (yield: 95%). Br-PEOPO-Br was synthesized by a similar procedure. However, after the reflux the hot reaction mixture was filtered and the solvent of the filtrate was removed under reduced pressure. Next, the remaining material was first dried in a vacuum at 50 °C overnight, and then treated with water (200 mL) at 60 °C for 10 min, followed by removal of the insoluble solid by filtration. The product was a light yellow viscous liquid which was collected after freeze-drying and further dried in a vacuum at 50 °C (yield: 85%).Preparation of precursor triblock copolymers
The triblock copolymers combining PPFS blocks with PEO and PEOPO blocks, respectively, were prepared by using the polyether macroinitiators for ATRP of PFS, as illustrated in Scheme 1. For example, in the preparation of the PPFS-PEO block copolymers, Br-PEO-Br (0.1 mmol, 3.5 g, 1 eq.), copper(I) bromide (0.4 mmol, 58 mg, 4 eq.), copper(0) powder (0.28 mmol, 18 mg, 2.8 eq.) and 2,2′-bipyridyl (0.8 mmol, 126 mg, 8 eq.) were first dissolved in degassed o-xylene (10 mL) at 80 °C. The system was degassed three times under nitrogen protection before addition of PFS via a syringe (see Table 1 for amounts). Subsequently, the solution was kept at 110 °C for 48 h, before being quenched with THF (50 mL). It was then passed through a column packed with alumina, before precipitation of the copolymer in hexane (200 mL). Next, the PEO-PPFS product was collected as a white powder by filtration, washed with diethyl ether and dried in a vacuum at 50 °C for 48 h. The PEOPO-PPFS triblock copolymers were prepared in a similar way using Br-PEOPO-Br as the macroinitiator. After passing the reaction mixture through the alumina column, the solvent was removed under reduced pressure. Diethyl ether (50 mL) was used to wash the remaining viscous product twice. The product was obtained as a light yellow viscous liquid after drying in a vacuum at 50 °C for 48 h. The block copolymer samples were designated as PEO-PPFSx and PEOPO-PPFSx respectively, where x is the PFS content in wt%. | ||
| Scheme 1 Synthetic pathway to PEO-sPPFSLiy and PEOPO-sPPFSLiy triblock copolymers via the preparation of polyether macroinitiators, ATRP of PFS, followed by thiolation, oxidation and ion-exchange to form the sPPFSLi blocks [key: (i) α,α′-dibromo-p-xylene, K2CO3, 18-crown-6, THF, 80 °C, 96 h, (ii) PFS, CuBr, bipyr, o-xylene, 110 °C, 48 h, (iii) NaSH, DMAc, 20 °C, 8 h, (iv) H2O2, AcOH, 48 h, and (v) LiCl]. | ||
| Sample | [PFS] : [EO] in synthesisa | PPFS content (wt%) | Degree of polymerizationb of the PPFS blocks | PFS conversion (%) | PPFS block Mn,NMRb (kg mol−1) |
M n,NMR (kg mol−1) | M n,SEC (kg mol−1) | PDI MwMn−1 |
|---|---|---|---|---|---|---|---|---|
| a Molar ratio between [PFS] to [EO]. b Calculated from NMR data. | ||||||||
| PEO-PPFS5 | 10 | 5 | 9.5 | 71 | 1.8 | 34.4 | 34.7 | 1.29 |
| PEO-PPFS10 | 15 | 10 | 20 | 73 | 3.9 | 36.5 | 43.0 | 1.29 |
| PEO-PPFS20 | 25 | 20 | 45 | 72 | 8.7 | 41.3 | 45.7 | 1.30 |
| PEO-PPFS30 | 35 | 30 | 77 | 75 | 15.0 | 47.6 | 43.6 | 1.31 |
| PEO-PPFS43 | 50 | 43 | 136 | 69 | 26.4 | 59.0 | 53.9 | 1.33 |
| PEOPO-PPFS16 | 20 | 16 | 12 | 71 | 2.3 | 14.6 | 14.2 | 1.31 |
| PEOPO-PPFS21 | 25 | 21 | 16 | 68 | 3.2 | 15.5 | 15.1 | 1.32 |
| PEOPO-PPFS35 | 30 | 35 | 33 | 71 | 6.5 | 18.8 | 16.5 | 1.29 |
Preparation of single-ion conducting triblock copolymers
The PEO-PPFSx and PEOPO-PPFSx triblock copolymers were sulfonated by thiolation and oxidation of the corresponding non-ionic precursor copolymer (Scheme 1). The former copolymers were sulfonated by dissolving the PEO-PPFSx block copolymer (3 g) in DMAc (30 mL) at 90 °C, followed by cooling to room temperature before addition of sodium hydrogensulfide hydrate (2 g). The green-blue mixture was kept for 48 h and then acidified by slowly adding a portion of aqueous HCl solution (1 M, 30 mL) over a period of 30 min. Subsequently, all the solvents were removed by vacuum distillation and the remaining mixture was treated with DMAc (50 mL) at 90 °C for 30 min. The suspension was filtered and the clear filtrate was kept at −18 °C overnight. Next, the thiolated block copolymer intermediate was collected as a light yellow powder by filtration, washed with diethyl ether and air-dried. The thiolated block copolymer (2 g) was treated with glacial acetic acid (20 mL) and hydrogen peroxide solution (30%, 13 mL) at 50 °C for 48 h before bringing it to reflux for 1 h. All solvents were then removed under reduced pressure at 60 °C. Water was added to dissolve the remaining gel-like product and was later removed by freeze-drying to afford the sulfonated copolymer samples in the H+ form. Copolymers in the Li+ form were obtained by ion exchange to the lithium-ion form, and were further purified by dialysis (MWCO = 3500 Da) in deionized water for 48 h. The PEOPO-PPFSx copolymers were sulfonated using the same method, but applying a dialysis membrane with MWCO = 100–500 Da. The resulting sulfonated copolymers were designated as PEO-sPPFSLiy and PEOPO-sPPFSLiy, respectively, where y denotes the wt% of the sPPFSLi blocks. The structural data of these polymers are listed in Table 2.| Block copolymer | Block copolymer precursor | sPPFSLi content (wt%) | sPPFSLi block Mn (kg mol−1) | M n (kg mol−1) | [O]:[Li]a |
IECLi (mmol Li g−1) | [H2O] (ppm) |
|---|---|---|---|---|---|---|---|
| a Molar ratio of [O] to [Li]. | |||||||
| PEO-sPPFSLi13 | PEO-PPFS10 | 13 | 5.2 | 37.8 | 40 | 0.50 | 94 |
| PEO-sPPFSLi25 | PEO-PPFS20 | 25 | 11.8 | 44.4 | 18 | 0.95 | 82 |
| PEO-sPPFSLi37 | PEO-PPFS30 | 37 | 20.2 | 52.8 | 10 | 1.41 | 104 |
| PEOPO-sPPFSLi20 | PEOPO-PPFS16 | 20 | 3.1 | 15.4 | 21 | 0.76 | 125 |
| PEOPO-sPPFSLi26 | PEOPO-PPFS21 | 26 | 4.2 | 16.5 | 16 | 0.10 | 75 |
| PEOPO-sPPFSLi42 | PEOPO-PPFS35 | 42 | 8.6 | 20.9 | 8 | 1.6 | 110 |
Drying procedure and water determination
The PEO-sPPFSLiy and PEOPO-sPPFSLiy electrolyte samples were pre-dried in a vacuum oven at 50 °C overnight and further dried in the melt state during stirring at a high vacuum (<0.2 Pa) at 80 °C for 48 h, and then stored under an inert atmosphere. The water content of the dried samples was determined to be in the range of 75–110 ppm by Karl-Fischer titration using a TitroLine KF trace titrator.Measurements
All polymer samples were characterized by 1H NMR spectroscopy using a Bruker DRX400 spectrometer and DMSO-d6 or CDCl3 as a solvent. All the block copolymers were further characterized by 19F NMR spectroscopy.The molecular weights of the block copolymers were determined by size exclusion chromatography (SEC) employing a Viscotek GPCmax VE-2001 instrument. The samples are dissolved in chloroform and passed through a series of three Shodex columns (KF-805, -804, and -802.5) and a refractive index detector at room temperature. The elution rate was 1 mL min−1. Four PEO standards (Agilent) Mn = 100, 50, 12.6 and 4.25 kg mol−1 were used for calibration. The Mn values of the commercial PEO and PEOPO were determined to be 32.6 and 12.3 kg mol−1, respectively. The Mn values of the triblock copolymers were subsequently calculated using their PFS content determined by 1H NMR analysis.
Thermogravimetric analysis (TGA) was carried out using a TA instruments Q500 TGA analyzer. The thermal degradation was evaluated under nitrogen at a heating rate of 10 °C min−1 up to 600 °C. Prior to this heating ramp the samples were dried at 130 °C for 10 min. The degradation temperature (Td,95) was determined at the point where 95% of the sample mass remained. The thermal stability of the samples was further investigated using isothermal measurements at 60, 80, 100 and 120 °C over 10 h. Differential scanning calorimetry (DSC) was performed on a TA instruments Q2000 calorimeter at a heating rate of 10 °C min−1 under nitrogen. The melting temperature (Tm) was determined during the heating and the crystallization temperature (Tc) during the cooling scan.
In order to investigate the phase structure of the electrolytes, PEO-sPPFSLi21 was analyzed by small angle X-ray scattering (SAXS) in the q range between 0.14 and 7 nm−1, which corresponds to a d spacing between 0.9 and 44 nm. The sample was placed on a homemade sample stage in a SAXSLAB SAXS instrument, from JJ X-ray Systems ApS (Denmark) equipped with a Pilatus detector. The scattering experiments were performed using Cu Kα radiation with a wavelength of 1.542 Å generated within a high brilliance micro focus sealed tube with shaped multilayer optics operating at 50 kV and 60 mA.
The ionic conductivity of the electrolytes was evaluated by measuring the temperature dependence of impedance spectra during a heating–cooling–heating cycle in the region from 0 to 90 °C. Dried electrolyte samples with a diameter of 15 mm and a thickness of 107 μm were sandwiched between two gold-plated brass coin electrodes spaced by a PTFE ring spacer inside an Ar-filled glove box. The measurements were carried out using a computer controlled Novocontrol BDC40 high-resolution dielectric analyzer equipped with a Novocool cryostat unit. The samples were analyzed in the frequency range of 10−1–107 Hz at a 50 mV ac amplitude, and the conductivities were subsequently evaluated using the Novocontrol software WinDeta.
Results and discussion
Synthesis
Three PEO-sPPFSLiy and three PEOPO-sPPFSLiy triblock copolymers with different values of y, i.e., with different block lengths and ionic contents, were prepared according to Scheme 1. The precursor PEO and PEOPO blocks had Mn = 33 and 12 kg mol−1, respectively, and both had a quite narrow polydispersity index (PDI) with MwMn−1 = 1.23 and 1.21, respectively (Table 1). In the synthesis of the block copolymers, the ionic content (y) was targeted to obtain electrolyte materials with [O]:[Li] ratios in the range between ∼10 and 40. This corresponded to average degrees of polymerization between 10 and 136 of the individual PPFS blocks.
In order to form ATRP macroinitiators for polymerization of the PPFS blocks, the polyether precursors were chain-end functionalized with benzyl bromide groups via a K2CO3-mediated reaction with α,α′-dibromo-p-xylene in THF at 80 °C. An excess of dibromoxylene was used to ensure full functionalization and avoid chain extension reactions. Moreover, 18-crown-6 was used as a phase transfer catalyst to assist the solvation of potassium carbonate in THF. The successful reaction was confirmed by 1H NMR spectroscopy, which indicated the shifts of the benzylic protons at 5.16 and 5.28 ppm, respectively, with equal integrals within the error of the method (Fig. 1). Moreover, as seen in Table 1, Mn and MwMn−1 evaluated by SEC remained essentially the same after the functionalization, which excluded the occurrence of significant chain extension reactions.
 | ||
| Fig. 1 1H NMR spectra of (a) the Br-PEO-Br macroinitiator and (b) PEO-PPFS20, and 19F NMR spectra of (c) PEO-PPFS20 and (d) PEO-sPPFSLi25. | ||
The PEO-PPFSx and PEOPO-PPFSx triblock copolymers were prepared by ATRP of PFS from the polyether macroinitiators using the CuBr/bipy system at 110 °C in o-xylene.24–26 A catalytic amount of Cu(0) powder was used to regenerate Cu(I). The molar ratio of PhCH2Br:Cu(I)Br:bipy was kept at 1:2:4 because the typical ratio of 1:1:2 led to unrepeatable ATRP results, possibly a consequence of the rather high molecular weight of the macroinitiators. The PPFS block length and the block ratio, and subsequently the ionic content ([O]:[Li]), of the block copolymer electrolytes were controlled by the [PFS]:[EO] ratio employed in the ATRP step (Table 1). The PPFS content of each block copolymer was calculated by comparing the integrated 1H NMR signals of the polyether and PPFS blocks at 3.4–3.9 and 1.9–3.0 ppm, respectively (Fig. 1 and S1†).24 Then the degree of polymerization of the PPFS blocks was calculated by taking into account the PPFS content and the Mn value of the respective polyether precursor. Furthermore, as seen in Fig. 1c, the appearance of the expected signals from the PPFS fluorine atoms in the 19F NMR spectra (ortho-F at 143.2, meta-F at 161.3 and para-F at 154.3 ppm) indicated the success of the ATRP.24–26 As seen in Table 1, the Mn values of the block copolymers obtained by direct SEC analysis (SEC traces shown in Fig. S2†) were in good agreement with the values calculated by using the NMR data and the Mn value of the respective polyether precursor. Finally, a typical PDI value for ATRP products close to MwMn−1 = 1.3 was found for all the block copolymers.
The PPFS blocks were sulfonated by substitution of the fluorine atoms at the para-position using NaSH, followed by oxidation of the resulting thiol groups to form the sulfonate groups, as described by Kerres et al.27 These authors prepared proton conducting poly(2,3,5,6-tetrafluorostyrene-4-sulfonic acid) via emulsion polymerisation. The polymer was reported to have a pKa value close to −2 and a proton conductivity higher than the perfluorinated Nafion® membrane at reduced relative humidity at 160 °C, demonstrating the electron withdrawing power of the neighboring aromatic fluorine atoms.27 Using a similar synthetic pathway, PPFS has been previously phosphonated to form proton conducting polymers and membranes.26,28–30 To obtain the present sulfonated copolymers, the para-fluorine atoms of the PPFS blocks were first completely and selectively substituted by thiol groups via the reaction with an excess of sodium hydrogen sulfide in DMAc solutions of the block copolymers, essentially following the method reported by Kerres et al.27 The salt formed after sequential acidification in the present case was removed by hot filtration of diluted DMAc solutions of the thiolated block copolymers. Next, the thiol groups were oxidized to sulfonic acid groups using hydrogen peroxide in DMAc at room temperature. The 19F NMR spectra of the sulfonated block copolymers showed two broad signals corresponding to the ortho-F at 142.8 ppm and meta-F at 139.3 ppm, which confirmed the complete functionalization of the PPFS blocks (Fig. 1).27 The sulfonic acid protons of the block copolymers were exchanged to lithium ions, and the copolymers were further dialyzed against deionized water for two days to obtain salt-free PEO-sPPFSLiy and PEOPO-sPPFSLiy samples. Finally, the samples were dried under high vacuum at 80 °C.
During the course of the present work a total of five PEO-PPFS block copolymers, and accordingly five PEO-sPPFSLiy block copolymers, were prepared in total (Table 1). However, both PEO-sPPFSLi7 and PEO-sPPFSLi50, with the lowest and the highest sPPFSLi content, respectively, produced very low ionic conductivities (<10−10 S cm−1) and were therefore not investigated any further. At room-temperature, the PEO-sPPFSLiy and PEOPO-sPPFSLiy samples were light yellow soft solids and light brown pastes, respectively.
Thermal stability
The thermal decomposition of the ionic block copolymers, as well as the different precursor polymers, was studied by TGA under a nitrogen atmosphere. The neat PPFS, PEO and PEOPO showed Td,95 values of 370, 357 and 353 °C, respectively (Fig. S3†). The non-ionic block copolymers decomposed in two steps, seemingly corresponding to the decomposition of the two blocks. The values of Td,95 were noted between 352 and 361 °C for the PEO-PPFS series and between 334 to 345 °C for the PEOPO-PPFS series (Fig. S3†). In both cases, the values were close to the Td,95 of the neat PEO and PEOPO, respectively.As expected, the ionic block copolymers decomposed at significantly lower temperatures, between Td,95 = 200 and 264 °C, decreasing with the sPPFSLi content (Fig. 2). This can be attributed to the desulfonation of sPPFS moieties.27 The samples were also investigated under isothermal conditions under a nitrogen atmosphere. After 10 h, the samples kept at 60, 80 and 100 °C lost less than 0.02 wt%, while the samples kept at 120 °C lost less than 0.17 wt% (Fig. S4†). These results indicated that the block polymer electrolytes had sufficient thermal stability for high-temperature battery operation.
 | ||
| Fig. 2 TGA traces of PEO-sPPFSLiy (a) and PEOPO-sPPFSLiy (b) block copolymers, as well as the respective homopolymers. | ||
Phase transitions
All the materials were analyzed by DSC in order to study their phase behavior, and the results are summarized in Table 3. As expected, the degree of crystallinity of PEOPO was significantly depressed in comparison with the PEO precursor. In addition, the former polymer crystallized at a much lower temperature (Tc = −43 °C) in comparison with PEO (Tc = 44 °C). After the formation of the PPFS blocks, the crystallization and crystallinity of the polyether blocks of all the PEO-PPFS triblock copolymers were very similar to the precursor (Fig. S5†). Still, the value of Tc increased slightly with the PPFS content (Table 3). These results indicated complete phase separation of the dissimilar blocks. In the PEOPO-PPFS triblock copolymers, a pronounced cold crystallization occurred at −55 °C during the heating scan of PEOPO-PPFS16 and PEOPO-PPFS21. For PEOPO-PPFS35, with the highest PPFS content, no crystallization was detected during cooling and the start of the cold crystallization was shifted to −40 °C. In addition, the heat of fusion of the PEOPO blocks decreased to only approximately half of that of the PEOPO precursor (Table 3). This may indicate a change in morphology where the polyether phase became less continuous as the PPFS content increased. The glass transition temperature (Tg) of PPFS is close to 110 °C,24 but was not observed in any of the present polyether-rich block copolymers.| Sample |
T
d,95a, °C |
T
cb, °C |
ΔHcc, J g−1 |
T
md, °C |
ΔHmc, J g−1 |
|---|---|---|---|---|---|
| a Decomposition temperature determined at a 5 wt% sample loss at 10 °C min−1. b Crystallization temperature. c Based on polyether weight. d Melting point; n.d. – not detected. | |||||
| PEO | 357 | 44 | 154 | 65 | 157 |
| PEOPO | 353 | −43 | 37 | 0 | 44 |
| PEO-PPFS10 | 352 | 46 | 148 | 65 | 161 |
| PEO-PPFS20 | 358 | 47 | 156 | 64 | 166 |
| PEO-PPFS30 | 361 | 49 | 157 | 63 | 160 |
| PEO-sPPFSLi13 | 263 | −1 | 92 | 37 | 132 |
| PEO-sPPFSLi25 | 217 | −10 | 85 | 36 | 103 |
| PEO-sPPFSLi37 | 200 | −21 | 13 | 42 | 18 |
| PEOPO-PPFS16 | 344 | −46 | 14 | −1 | 40 |
| PEOPO-PPFS21 | 345 | −45 | 19 | −1 | 45 |
| PEOPO-PPFS35 | 334 | n. d. | 2 | −10 | 22 |
| PEOPO-sPPFSLi20 | 264 | −40 | 9 | −7 | 31 |
| PEOPO-sPPFSLi26 | 236 | n. d. | n. d. | −9 | 23 |
| PEOPO-sPPFSLi42 | 240 | n. d. | n. d. | −7 | 12 |
The DSC cooling and heating traces of the ionic block copolymer electrolytes are displayed in Fig. 3. In relation to the PEO-PPFS samples, the Tc and Tm values of the PEO-sPPFSLiy materials were sharply depressed and only a very low level of PEO crystallinity remained in the sample with the highest ionic content, PEO-sPPFSLi37. On the other hand, this sample displayed a slightly higher Tm than the two samples with the lower ionic content. Moreover, the value of Tc decreased with the increasing ionic content, from −1 to −20 °C. The PEOPO-sPPFSLiy samples displayed very broad melting intervals between −40 and 10 °C, and only PEOPO-sPPFSLi20 showed signs of crystallization during cooling. The two samples with the lowest ionic contents showed cold crystallization between −35 and −50 °C, while no crystallization was detected for PEOPO-sPPFSLi42 with the highest ionic content.
 | ||
| Fig. 3 DSC traces of the block copolymers in the PEO-sPPFSLiy (a) and the PEOPO-sPPFSLiy (b) series (cooling trace: - - -; heating trace: —). | ||
In summary, the polyether crystallinity was significantly depressed by exchanging PEO with PEOPO in the block copolymer. The formation of the non-ionic block copolymers did not significantly influence the crystallinity, which suggested immiscibility of the dissimilar blocks. After sulfonation, the polyether crystallinity decreased sharply with the ionic content, most probably because of an extensive Li–O coordination. This indicated an increasing compatibility and miscibility between the ionic sPPFSLi and polyether blocks.
Ionic conductivity
The temperature dependence of the ion conductivity of the block copolymers in the PEO-sPPFSLiy and PEOPO-sPPFSLiy series was measured by electrochemical impedance spectroscopy. In these single-ion conducting triblock copolymers, the sulfonate anions were covalently attached to the sPPFSLi blocks, which were restricted exclusively to short-range segmental mobility. Presumably, only the Li+ cation has long-range mobility in the materials and the transport number is expected to be very close to unity.5,6 Furthermore, we expected that the dissociation of the lithium sulfonate groups would be greatly facilitated by their position on the strongly electron-withdrawing tetrafluorobenzene units.27 The Li+ conductivity of PEO electrolytes is normally coupled to the segmental mobility. Hence, any crystallinity is likely to decrease the level of conductivity. A further factor that is often ignored, and which can strongly influence the performance of single-ion conducting electrolytes, is the water content. As these materials are highly charged, they are generally extremely hygroscopic. The presence of trace amounts of water can significantly facilitate the dissociation of the lithium ion and therefore increase the conductivity. The ionic conductivity of two samples with different water contents (400 and 75 ppm water, respectively) is presented in Fig. S6.† As expected, the sample with the higher water content reached much higher conductivity than the one with lower water content by a factor of 2.5. In the present case the single-ion conducting electrolytes were dried to a water content of 75–100 ppm before characterization.The DC conductivity values of the samples were determined from the frequency-independent conductivity plateaus observed in the plots of AC conductivity versus frequency (Fig. S7†). The conductivity data measured during heating of the PEO-sPPFSLiy samples from 0 to 90 °C are displayed in Fig. 4a. As seen, the conductivity of the two samples with the lowest sPPFSLi contents increased sharply from low levels in the temperature range of 30 to 50 °C because of melting of the PEO blocks. Consequently, the conductivity of these samples increased by almost 4 orders of magnitude between 0 and 90 °C. PEO-sPPFSLi13 and PEO-sPPFSLi25 reached 2.2 × 10−6 S cm−1 and 3.0 × 10−5 S cm−1, respectively, at 90 °C. In contrast, sample PEO-sPPFSLi37 with the highest sPPFSLi content was seemingly not influenced by any PEO melting but still reached a rather moderate level of conductivity, 2.9 × 10−6 S cm−1 at 90 °C. The data below 50 °C were in line with the DSC results, which showed that the PEO crystallinity decreased with the sPPFSLi content (Fig. 3). Above 50 °C, the highest conductivity was reached by PEO-sPPFSLi25 with an [O]:[Li] ratio of ∼18, as seen in Fig. 5.
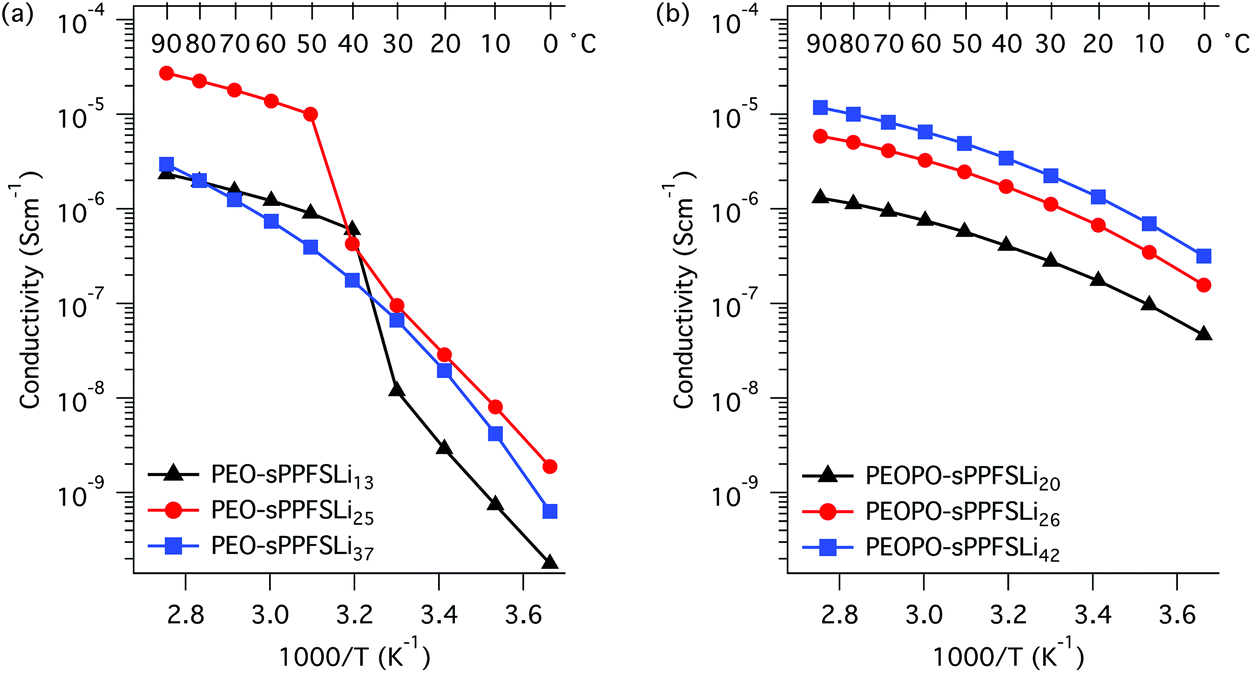 | ||
| Fig. 4 Arrhenius conductivity plots of the PEO-sPPFSLiy (a) and the PEOPO-sPPFSLiy (b) block copolymer electrolytes. | ||
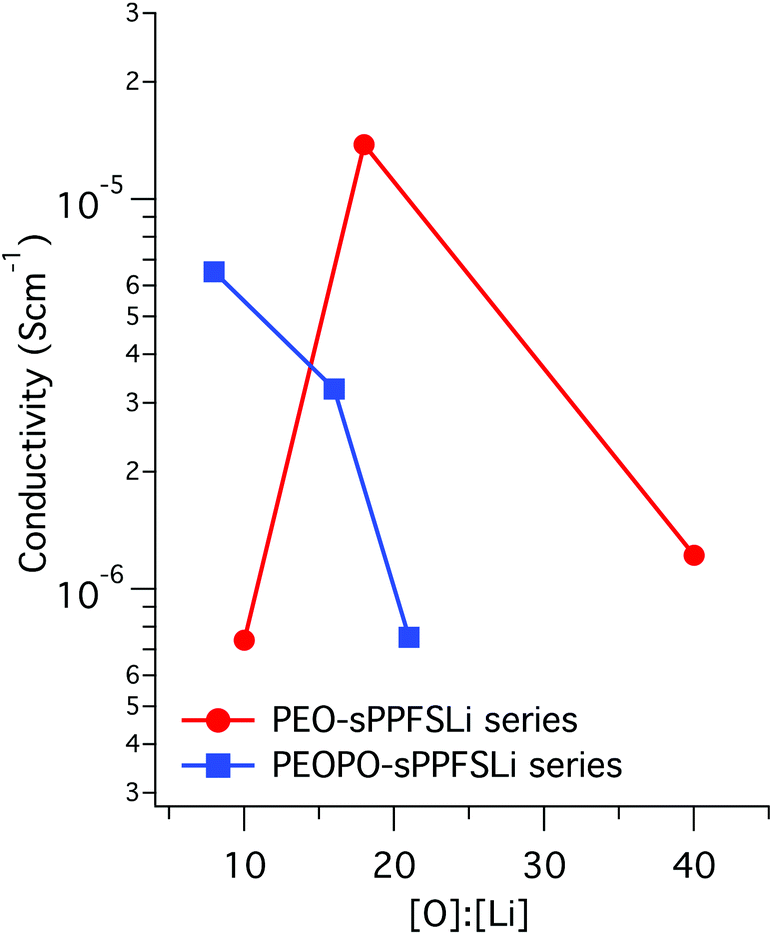 | ||
| Fig. 5 Conductivity versus [O]:[Li] ratio of the block copolymer electrolytes at 60 °C. | ||
The conductivity of the copolymers in the PEOPO-sPPFSLiy series is displayed in Fig. 4b. Because the low propensity of the PEOPO blocks to crystallize, as seen in Fig. 3b, the conductivity between 0 and 90 °C was not influenced by any polyether crystallinity and melting. Consequently, the conductivity of the PEOPO-sPPFSLiy series was significantly higher than that of the PEO-sPPFSLiy series below 50 °C, and reached above 4 × 10−7 and 1 × 10−6 S cm−1 at 0 and 20 °C, respectively. The conductivity of the PEOPO-sPPFSLiy series increased with the sPPFSLi content, and the maximum conductivity at 90 °C reached just above 1 × 10−5 S cm−1 for PEOPO-sPPFSLi42 with [O]:[Li] ∼ 8:1. This was a factor of 3 below the conductivity of PEO-sPPFSLi25. Consequently, no optimum [O] : [Li] ratio was found for the PEOPO-sPPFSLiy series (Fig. 5).
Because PEO melting and crystallization occurred during the heating and cooling of the PEO-sPPFSLiy samples, there was a significant hysteresis in the conductivity values measured in the temperature range between 10 and 50 °C (Fig. 6). Thus, the conductivity at 20 °C was measured to be 3 × 10−8 and 2 × 10−6 S cm−1 during heating and cooling, respectively. In contrast, no hysteresis was observed for the conductivity of the PEOPO-sPPFSLiy samples when measured during heating and cooling, and the data virtually overlapped (Fig. 6).
 | ||
| Fig. 6 Conductivity data measured during heating–cooling–heating scans of (a) PEO-sPPFSLi25 and (b) PEOPO-sPPFSLi26 in the temperature range of 0–90 °C. The large hysteresis seen between 10 and 50 °C for the former sample because of PEO crystallization and melting was completely absent for the PEOPO-based sample. | ||
Recently, Balsara et al. used small angle X-ray scattering (SAXS) to study PEO-poly(styrenesulfonyllithium(trifluoromethylsulfonyl)imide) diblock copolymers in which the PEO molecular weight was 5.0 kg mol−1, while that of the ionic block was varied between 2.0 and 7.5 kg mol−1.20 When the ionic block length was kept small, the block copolymers were microphase separated with a crystalline PEO-rich domain and a glassy phase domain containing ionic clusters. Above the PEO melting point, the Li+ ions were released from the clusters to form a homogeneously disordered morphology at which the conductivity increased abruptly by several orders of magnitude.20 When the molecular weight of the ionic block was above 5.4 kg mol−1, the material was disordered at all temperatures and there was no abrupt change in conductivity. These findings are very similar to the observations made in the present case. This prompted us to investigate our materials using SAXS to see if any order to disorder transitions could be identified. Measurements were performed both below and above the polyether melting points. However, no scattering maxima were observed in a representative sample (PEO-sPPFSLi25) at 23 °C (below Tm), as well as at 55 °C (above Tm) in the q-range corresponding to the d-spacings between 0.9 and 44 nm (Fig. S8†). The molecular weight of the PEO block was much higher (30 kg mol−1) in the present study compared to the study by Balsara et al. (5 kg mol−1). Hence, the scattering maxima may appear outside the q-range of our equipment.
The convex shape of the conductivity curves of the PEOPO-sPPFSLiy series, as well as of the PEO-sPPFSLiy series above the Tm of PEO, agrees well with the general temperature dependence observed for amorphous polymer electrolytes which may be described by using the Vogel–Tammann–Fulcher (VTF) equation:
| logσ = logσ0 − Ea/[R(T − T0)] | (1) |
σ0 values, which increased with the content of sPPFSLi in both series. The value Ea increased with the sPPFSLi content in both series. However, the increase was very weak in the case of the PEOPO-sPPFSLiy series and very strong for the PEO-sPPFSLiy series. The value of T0 was ∼15–28 K higher in the PEO-sPPFSLiy series compared to the PEOPO-sPPFSLiy series and showed only small variations within the respective series.
| Block copolymer | logσ0 |
E a (kJ mol−1) | T 0 (K) | R |
|---|---|---|---|---|
|
a Obtained by fitting measured conductivity data to the VTF equation: logσ = logσ0 − Ea/[R(T − T0)].
|
||||
| PEO-sPPFSLi13 | −4.7 | 1.03 | 232 | 0.99987 |
| PEO-sPPFSLi25 | −3.5 | 1.15 | 229 | 1 |
| PEO-sPPFSLi37 | −3.3 | 2.60 | 222 | 0.99995 |
| PEOPO-sPPFSLi20 | −4.8 | 1.48 | 204 | 0.99988 |
| PEOPO-sPPFSLi26 | −4.1 | 1.50 | 207 | 0.99987 |
| PEOPO-sPPFSLi42 | −3.8 | 1.51 | 207 | 0.99987 |
The conductivity reached by PEO-sPPFSLi25 with [O] : [Li] ∼ 20 was very high, e.g. 1.4 × 10−5 S cm−1 at 60 °C, and may be compared with similar state-of-the-art single-ion conducting block copolymers containing lithium sulfonyl(trifluoromethanesulfonyl)imide, known for its high degree of dissociation in solid polymer electrolytes.5,6 Bouchet and co-workers reported on the preparation and properties of BAB triblock copolymers having a center block of PEO (35 kDa) and flanking poly[4-styrenesulfonyl(trifluoromethanesulfonyl)imide] blocks in the lithium form.22 They reported a conductivity of 1.3 × 10−5 S cm−1 at 60 °C obtained with a block copolymer containing 20 wt% of the ionic block, corresponding to [O] : [Li] ∼ 30. This level of conductivity virtually coincides with that of the present materials. Jangu et al. prepared microphase separated BAB triblock copolymers by reversible addition–fragmentation chain transfer polymerization (RAFT) where the “soft” A blocks were statistical copolymers of di(ethylene glycol) methyl ether methacrylate and 4-styrenesulfonyl(trifluoromethanesulfonyl)imide, and the “hard” B blocks were polystyrene.17 These materials reached a conductivity close to 1 × 10−5 S cm−1 at 90 °C. Porcarelli and coworkers used RAFT to prepare AB diblock copolymers with poly(lithium 1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethylsulfonyl)imide) combined with poly(ethylene glycol) methyl ether methacrylate blocks.18 These soft polymer electrolytes reached a maximum conductivity of 1.2 × 10−5 S cm−1 at 55 °C.
The level of conductivity reached by the present single Li+-ion conducting block copolymers may also be compared with solid block copolymer electrolytes containing a free lithium salt. For example, Bouchet et al. studied PS-PEO-PS triblock copolymers doped with the LiTFSI salt.3 They found a conductivity of 5 × 10−4 S cm−1 at 60 °C for a block copolymer having a center PEO block of 35 kDa and with [O] : [Li] = 20. In addition, we have previously reported a similar level of conductivity (3 × 10−4 S cm−1 at 60 °C) for a PPFS-PEOPO-PPFS triblock copolymer having a central block of 12 kDa and with an LiTFSI concentration corresponding to [O] : [Li] = 20.25
Conclusions
Two series of BAB type triblock copolymers with fixed central blocks of either PEO or PEOPO flanked by two PPFSLi blocks were readily prepared by ATRP of PFS from the respective polyether macroinitiator, followed by quantitative substitution of the fluorine atoms at para-positions using NaSH. Complete oxidation of the resulting thiol groups gave the PPFSLi blocks after ion-exchange. The ionic content of the copolymers were directly linked to the relative block lengths, which was efficiently regulated by the relative amount of PFS to the macroinitiator used in the ATRP. The ionic triblock copolymers had high thermal stability with decomposition temperatures above 210 °C under a nitrogen atmosphere. As expected, the polyether crystallinity decreased with the increasing propylene oxide content and the decreasing [O] : [Li] ratio. Very high levels of ionic conductivity were measured for the two series. Above the polyether melting point, PEO-based block copolymers showed the highest conductivity, up to 1.4 × 10−5 S cm−1 at 60 °C, while below the same point, a PEOPO-material reached the highest conductivity, approximately 1.5 × 10−6 S cm−1 at 20 °C. The former conductivity is in level with that of state-of-the-art block copolymer electrolytes functionalised with sulfonyl(trifluoromethanesulfonyl)imide lithium groups and suggest weak interactions of the lithium ions with the pentafluorosulfonate anions in combination with their enhanced dissociation facilitated by PEO. The results also hint that the room-temperature conductivity of these electrolytes may be enhanced by decreasing the molecular weight of the PEO block to depress its crystallinity. The results of the present study demonstrate that favourably designed block copolymers containing lithium pentafluorostyrene sulfonate units can approach the levels of conductivity necessary for high-temperature lithium battery applications. However, to conclude about the practical applicability of the present materials requires further characterisation of the electrochemical properties, including electrochemical stability, which is the focus of our next study.Acknowledgements
We thank the Swedish Energy Agency for funding the project “High Temperature Lithium Batteries” (project 37722-1), and Patrik Johansson for helpful discussions.Notes and references
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- D. Devaux, D. Gle, T. N. T. Phan, D. Gigmes, E. Giroud, M. Deschamps, R. Denoyel and R. Bouchet, Chem. Mater., 2015, 27, 4682–4692 CrossRef CAS.
- P. G. Bruce and C. A. Vincent, J. Chem. Soc., Faraday Trans., 1993, 89, 3187–3203 RSC.
- L. Z. Long, S. J. Wang, M. Xiao and Y. Z. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 CAS.
- Z. G. Xue, D. He and X. L. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 CAS.
- S. W. Ryu, P. E. Trapa, S. C. Olugebefola, J. A. Gonzalez-Leon, D. R. Sadoway and A. M. Mayes, J. Electrochem. Soc., 2005, 152, A158–A163 CrossRef CAS.
- E. Tsuchida, N. Kobayashi and H. Ohno, Macromolecules, 1988, 21, 96–100 CrossRef CAS.
- D. Benrabah, S. Sylla, F. Alloin, J. Y. Sanchez and M. Armand, Electrochim. Acta, 1995, 40, 2259–2264 CrossRef CAS.
- N. Matsumi, K. Sugai, M. Miyake and H. Ohno Ohno, Macromolecules, 2006, 39, 6924–6927 CrossRef CAS.
- S. C. Dou, S. H. Zhang, R. J. Klein, J. Runt and R. H. Colby, Chem. Mater., 2006, 18, 4288–4295 CrossRef CAS.
- L. M. Bronstein, R. L. Karlinsey, B. Stein, Z. Yi, J. Carini and J. W. Zwanziger, Chem. Mater., 2006, 18, 708–715 CrossRef CAS.
- H. Mazor, D. Goodnitsky, E. Peled, W. Wieczorek and B. Scrosati, J. Power Sources, 2008, 178, 736–743 CrossRef CAS.
- H. R. Allcock, D. T. Welna and A. E. Maher, Solid State Ionics, 2006, 177, 741–747 CrossRef CAS.
- R. Meziane, J. P. Bonnet, M. Courty, K. Djellab and M. Armand, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS.
- S. W. Feng, D. Y. Shi, F. Liu, L. P. Zheng, J. Nie, W. F. Feng, X. J. Huang, M. Armand and Z. B. Zhoua, Electrochim. Acta, 2013, 93, 254–263 CrossRef CAS.
- C. Jangu, A. M. Savage, Z. Y. Zhang, A. R. Schultz, L. A. Madsen, F. L. Beyer and T. E. Long, Macromolecules, 2015, 48, 4520–4528 CrossRef CAS.
- L. Porcarelli, A. S. Shaplov, M. Salsamendi, J. R. Nair, Y. S. Vygodskii, D. Mecerreyes and C. Gerbaldi, ACS Appl. Mater. Interfaces, 2016, 8, 10350–10359 CAS.
- S. Inceoglu, A. A. Rojas, D. Devaux, X. C. Chen, G. M. Stone and N. P. Balsara, ACS Macro Lett., 2014, 3, 510–514 CrossRef CAS.
- A. A. Rojas, S. Inceoglu, N. G. Mackay, J. L. Thelen, D. Devaux, G. M. Stone and N. P. Balsara, Macromolecules, 2015, 48, 6589–6595 CrossRef CAS.
- Q. Ma, H. Zhang, C. W. Zhou, L. P. Zheng, P. F. Cheng, J. Nie, W. F. Feng, Y. S. Hu, H. Li, X. J. Huang, L. Q. Chen, M. Armand and Z. B. Zhou, Angew. Chem., Int. Ed., 2016, 55, 2521–2525 CrossRef CAS PubMed.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- E. Paillard, F. Toulgoat, C. Iojoiu, F. Alloin, J. Guindet, M. Medebielle, B. Langlois and J. Y. Sanchez, J. Fluorine Chem., 2012, 134, 72–76 CrossRef CAS.
- K. Jankova and S. Hvilsted, Macromolecules, 2003, 36, 1753–1758 CrossRef CAS.
- K. Jankova, P. Jannasch and S. Hvilsted, J. Mater. Chem., 2004, 14, 2902–2908 RSC.
- Z. C. Shao, A. Sannigrahi and P. Jannasch, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4657–4666 CAS.
- V. Atanasov, M. Burger, S. Lyonnard, L. Porcar and J. Kerres, Solid State Ionics, 2013, 252, 75–83 CrossRef CAS.
- V. Atanasov and J. Kerres, Macromolecules, 2011, 44, 6416–6423 CrossRef CAS.
- I. Dimitrov, S. Takamuku, K. Jankova, P. Jannasch and S. Hvilsted, Macromol. Rapid Commun., 2012, 33, 1368–1374 CrossRef CAS PubMed.
- I. Dimitrov, S. Takamuku, K. Jankova, P. Jannasch and S. Hvilsted, J. Membr. Sci., 2014, 450, 362–368 CrossRef CAS.
- H. Z. Vogel, Z. Phys., 1921, 22, 645 CAS.
- G. Tammann and G. Z. Hesse, Anorg. Allg. Chem., 1926, 156, 245 CrossRef.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339 CrossRef CAS.
- I. J. A. Mertens, M. Wubbenhorst, W. D. Oosterbaan, L. W. Jenneskens and J. Van Turnhout, Macromolecules, 1999, 32, 3314 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01910b |
| This journal is © The Royal Society of Chemistry 2017 |